
Abstract
Catalytic enantioselective Strecker reactions on an achiral substrate using sub-stoichiometric amounts of a chiral catalyst represent an evolving key strategy for the effective synthesis of α-amino nitriles. We herein disclosed the set-up of a flow-based methodology for enantioselective Strecker, employing ethyl cyanoformate as a relatively safe cyanide source, a cinchona-based catalyst, and methanol as additive. A thorough exploration of key parameters allowed the identification of the most efficient reagent mixing mode, the optimum solvent for the flow synthesis, minimum catalyst loading, additive, temperature, and residence time. The newly developed method allows straightforward reaction channeling towards the fast and complete formation of the α-amino nitrile products, thus reducing the yield drop due to indolenine degradation during long-lasting batch-wise reactions. Moreover, we herein provide preliminary hints for sustainability, by proposing a simple procedure for catalyst recycling, thus opening the way for further optimization of the proposed methodology.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The condensation of aldehydes with ammonia and hydrogen cyanide to provide α-amino nitriles followed by hydrolysis of the nitrile groups is the first-born method for the de novo synthesis of α-amino acids[1].
This reaction, known as the “Strecker synthesis” represents a powerful tool in the preparation of both biogenic and non-natural α-amino acids and related structural templates, such as hydantoins[2]. Its multicomponent, i.e., three-component nature implies the possibility of a rapid, modular, and diversity-oriented assembly of complex α-amino acid derivatives whose preparation would otherwise be challenging, if not impossible by other methods. Moreover, the simplicity of the reaction protocol as well as the likely availability of starting materials under prebiotic conditions strongly suggest the involvement of Strecker amino acid synthesis within chemical origin of life theories[3, 4]. Accordingly, α-amino nitriles are regarded as conceivable prebiotic precursors to nicotinic acids, and nucleic acids[5,6,7,8].α-Amino nitrile containing compounds have a profound impact on bio-chemical sciences, owing to their unique capacity of generating molecular diversity for diverse application, ranging from the total synthesis of complex alkaloids to the preparation of N-heterocycles and α-amino nitrile containing drugs[5]. Moreover, in peptide research, sterically hindered and α,α-disubstituted amino acids, derived from suitably functionalized α-amino nitriles, have been extensively applied to induce restricted conformations, resulting in well-defined peptide/pseudopeptide secondary structures[3, 9].
The cyanomethylene amino substructure is embedded in structurally diverse naturally occurring alkaloids, such as the tetrahydroisoquinoline anti-tumor antibiotic saframycin A (1)[10,11,12], phthalascidin 650 (2)[13, 14], and girgensohnine (3) [15] (Fig. 1). A cyclic α-amino nitrile substructure, namely a pyrrolidine-2-carbonitrile skeleton is also included in the dipeptidyl peptidase-4 (DPP-4) inhibitor class of anti-hyperglycemic drugs anagliptin (4), vildagliptin (5) and saxagliptin (6) (Fig. 1)[16, 17].

Representative biologically active α-amino nitrile-containing natural products (1–3) and synthetic drugs (4–6)
The Strecker synthesis, as well as its modified version involving the direct hydrocyanation of imines, remain relevant synthetic tools for the preparation of several drugs, such as anti-platelet agent clopidogrel (7, Plavix) [18], opioid analgesic drugs (e.g., carfentanil (8)) [19,20,21], the anti HCV drug boceprevir (9) [22] or the anti-HIV agent DPC 083 (10) (Fig. 2) [23]. Compounds derived from Strecker-type protocols are also key intermediates in the synthesis of pharmaceutically relevant indole alkaloids like reserpine (11) [24], hirsutine (12) [25] or eburnamonine (13) [26] (Fig. 2).

Selected drugs and natural products prepared by Strecker-type reactions (7–10) or Strecker intermediates (11–13)
Prompted by the escalating demand for enantioenriched α-amino acids and derivatives for a range of applications, many researchers at both academic and industrial level, have found a huge interest in the development of enantioselective Strecker reaction protocols. [27,28,29].
In this context, catalytic enantioselective Strecker reactions on an achiral substrate using sub-stoichiometric amounts of a chiral catalyst represent a key evolving strategy, with respect to the elder chiral auxiliary-assisted Strecker protocols and became the method-of-choice in the synthesis of chiral nonracemic amino nitriles [30].
The first example of a catalytic asymmetric Strecker reaction appeared in 1996 [1]; since then, several catalytic options based on hydrogen bonding activation, Lewis base activation, and Lewis acid-Lewis base bifunctional activation have been developed, mainly aiming at transforming acyclic (E)-aldimines [31,32,33,34,35,36,37,38] and ketimines [39,40,41,42] into optically active α-amino nitriles.
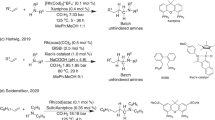
In stark contrast, much less attention has been paid to cyclic (Z)-aldimines in catalytic asymmetric Strecker reactions. In 2000, Jacobsen and coworkers disclosed the asymmetric hydrocyanation of 3,4-dihydroisoquinoline in the presence of a chiral 1,2-trans-diaminocyclohexane-based urea/Schiff base as the catalyst (Scheme 1(a)) [43]. Later on, asymmetric cyanation of 3,4-dihydroisoquinoline derivatives was reported in the presence of trimethylsilyl or acetyl cyanides [44, 45]. Tian and co-workers were the first to develop a chiral thiourea-catalyzed asymmetric cyanation of cyclic (Z)-aldimines, namely 3H-indoles, employing ethyl cyanoformate as an alternative cyanide source (Scheme 1(b)) [46]. Although the reaction proceeded smoothly and provided 2-cyanoindolines with high enantioselectivity, it required long reaction times of around 48–72 h at 10 °C [46].

Exemplificative catalytic asymmetric Strecker reactions on cyclic (Z)-aldimines
As a multicomponent reaction, the Strecker synthesis already incorporates many requirements for green chemistry. Paradoxically, despite its extensive applications both in academia and industry, the use of highly toxic cyanides sources and toxic solvents, go against green chemistry principles, and poses serious risks to human health and the environment. In order to address these essential concerns, many research efforts have been devoted towards more sustainable Strecker variants [3].
A plethora of alternative methodologies have been proposed lately, ranging from the use of organic solvents and alternative cyanide sources [47], to the use of Lewis acid catalysts [48,49,50], guanidine HCl [51], ionic liquids [52, 53], catalyst-free [54] and Strecker protocols employing elevated pressures [55]. Despite these developments, the reaction still suffers from significant drawbacks including protracted reaction times, an excess of the cyanide source and variable yields, which preclude the use of Strecker protocols on a production scale. In addition, a major shortcoming of the multi-component Strecker reaction is the competing cyanohydrin formation [56].
In recent years the use of continuous flow reactors has captured the attention of the modern-day synthetic chemists, with increasing uptake of the technologies as the systems have become commercially available, de facto affordable and widely applicable thanks to extensive developmental work including in-line analysis tools. With respect to conventional batch-wise protocols, continuous flow may enable several advantages in terms of process reproducibility and scalability, handling of toxic/hazardous chemicals.
The first heterogeneously catalyzed Strecker protocols in continuous flow appeared in literature in 2008 and were conducted employing polymer-supported (ethylenediaminetetraacetic acid)ruthenium(III) chloride or polymer-bound scandium(III) bis(trifluoromethanesulfonate) as the Lewis acid catalysts and trimethylsilyl cyanide as the cyanide source, with the aim of providing a simple and efficient methodology for the synthesis of aromatic and aliphatic α-aminonitriles and as a means of increasing the throughput of the system [57].
In 2014 a continuous flow oxidative cyanation protocol was disclosed, where α-amino nitriles were obtained in good to excellent yields using a variable-temperature continuous-flow LED- photoreactor for singlet oxygen generation, and trimethylsilyl cyanide served as an in-situ imine trap [58].
However, to the best of our knowledge, the only asymmetric flow-based protocol for the three component Strecker reaction was reported in 2012 and employed a heterogeneous self-supported chiral titanium cluster catalyst and trimethylsilyl cyanide as the cyanide source, although the enantioselective imine-cyanation/ Strecker reaction was described only on acyclic imine substrates [59].
In our quest towards the development of eco-friendly protocols for indole- and indoline-based privileged scaffolds [60], we recently proposed flow-aided methodologies for the telescoped synthesis on indoline-based structural templates and their peptidomimetic derivatives [61, 62] as well as mechanochemical procedures for sustainable Fischer and interrupted Fischer reactions [63].
Capitalizing on the acquired experience in dealing with metastable indolenine systems and prompted by the fact that catalytic asymmetric Strecker reactions with cyclic imines are barely studied, we decided to start filling this void of knowledge by implementing a convenient flow-based protocol, for the organocatalyzed asymmetric cyanation of cyclic (Z)-aldimines.
We herein disclose the set-up of a flow-based methodology for enantioselective Strecker, employing ethyl cyanoformate as a relatively safe cyanide source, a cinchona-based catalyst, i.e., (8R, 9R)-14a, (8R, 9S)-14a and (8R, 9S)-14b (Fig. 3) and methanol as additive. To the best of our knowledge, this is the first continuous flow protocol for the asymmetric synthesis of indolenines. A thorough exploration of key parameters allowed the identification of the most convenient reaction conditions, thus allowing the identification of the most efficient reagent mixing mode, the optimum solvent for the flow synthesis, minimum catalyst loading, additive, temperature, and residence time. The newly developed method allows straightforward reaction channeling towards the fast and complete formation of the α-aminonitrile products, thus reducing the yield drop due to indolenine degradation during long-lasting batch-wise reactions. Moreover, preliminary hints for sustainability are indicated, by proposing a simple procedure for catalyst recycling, thus opening the way for further optimization of the proposed methodology.

Cinchona-based catalysts (8R, 9R)-14a, (8R, 9S)-14a and (8R, 9S)-14b used in the present work
Results and discussion
Basic screening of reaction conditions in batch
A first investigation in batch carried out on model α,α′-disubstituted carbaldehydes was intended to reproduce reaction conditions from previous literature reports and to set up the starting point for the implementation of the reaction in segmented continuous flow mode. To this aim the spiro-cyclohexyl indolenine 15a was selected as the starting point for our experiments, since i) a batchwise asymmetric Strecker methodology using a cinchona-based catalyst was reported on this substrate [46]; ii) spirocyclic indolenines are generally more prone to undergo the concomitant 1,2-migration path towards the corresponding indole derivatives; a good outcome for a Strecker reaction on these substrates would easily guarantee a good performance on more tractable cyclic and acyclic aldimines; and iii) the employment of a spiroindolenine as a sterically rather hindered cyclic imine featuring a quite extreme steric congestion around the ‘target’ imine carbon atom, would provide critical hints regarding the accessibility by the cyanide component.
As summarized in Table 1, we applied the classical reaction conditions in batch, employing spiroindolenine 15a and ethyl cyanoformate 16 as a relatively safe cyanide source in absence (Table 1, entry 1) or in the presence of 10 mol% (Table 1, entry 2) or 20 mol% (Table 1, entry 3) of catalyst (8R, 9R)-14a and molecular sieves. As per previous report, the reaction was conducted in dry 1,2-dichloroethane at 10 °C, with the addition of 1.2 equivalents of methanol to the mixture. After 72 h of reaction at 10 °C we could observe almost complete conversion of the cyclic imine material into the corresponding α-amino nitrile 17. Regarding the reactions performed in the presence of the asymmetric catalyst (Table 1, entries 2 and 3) we registered in both cases a comparable outcome in terms enantiomeric ratios, namely 70:30 and 77:23, respectively, as determined by chiral HPLC analysis. Although sub-optimal in terms of reaction enantioselectivity, the conditions were projected into a segmented flow chemistry protocol for further optimization under conditions allowing for superior control.
Development of reactions conditions in segmented continuous flow mode
We aimed at elaborating optimised and robust flow conditions for the planned asymmetric Strecker reaction by systematically screening of flow reaction conditions. In line with our driving idea that flow chemistry applications should display a sustainable character not only with reference to implemented reaction conditions, but also in terms of the employed flow chemistry platforms, our flow system was realized in the previously described [61], comparably cost-effective fashion. In particular, an HPLC piston pump capable of sustaining reliably low flow rates was connected to a six-port Rheodyne injector for meso-scale preparative HPLCs, that was equipped with a 1 mL volume sample loop made from PTFE tubing and an injection port for conventional disposable syringes. Connection to a self-made reactor of 25 mL or 31 mL volume made from PTFE tubing, was realized via a simple T-piece. The self-made reactor was immersed in a water bath for the moderate cooling needed for reaction implementation. To maintain control of the system, a 7 bar back-pressure regulator was placed at the end of the line.
Anticipating the possibility of an upstream modification to combine the Strecker protocol with our previously developed flow-based synthesis of indolenine scaffolds [61], but also to adhere even more to green chemistry principles, screening of solvents suitable for both reactions was done first. In line with our batch investigation, we initially tested the reaction outcome using 1,2-DCE as the solvent (Table 2, entry 1).
Spiroindolenine 15a and ethyl cyanoformate 16 in the presence of catalyst (8R, 9R)-14a (10% mol) and 1.1 equivalents of MeOH (1 mL total solvent volume, 0.08 m concentration in 15a) were loaded into a sample loop. Injecting this solution with an overall flow rate of 0.2 mL/min into the tubular flow reactor (25 mL reactor volume) held at different temperatures caused the reaction mixture to produce the desired product upon 125 min of residence time. The exiting stream was collected in a flask for further purification. Although the above-mentioned conditions generated compound ( +)-17 in good yields (87%), the outcome in terms of enantiomeric ratio (e.r.) was not completely satisfying, since compound ( +)-17 was obtained with a 75:25. Leaving unaltered the reaction conditions and molar ratio of catalyst, dichloromethane was tested as solvent (Table 2, entry 2). In this case, an important yield drop of more than 30% was registered, accompanied, however, by a slight improvement in terms of e.r. Next attempts were mainly focused at improving the reaction eco-compatibility, by replacing halogenated solvents with ethyl acetate (Table 2, entry 3) and cyclopentyl methyl ether (Table 2, entry 4). In both cases, the reaction yields dramatically dropped, thus discouraging subsequent evaluation in terms of product enantiopurity.
We thus opted for transferring the best solvent conditions among those tested within this small preliminary screening into a more elaborated screening of catalyst nature and amount, in order to set an efficient asymmetric catalytic protocol for the Strecker reaction in flow on cyclic (Z)-aldimines. This investigation was also flanked by the parallel fine-tuning of the amount of 16 employed for the reaction, as reported in Table 3. The increase of catalyst (8R, 9R)-14a loading from 10 mol% up to 20 mol%, while providing only a slight increase of the yield, guaranteed a robust improvement in terms of e.r. (75:25 vs. 91:9, Table 2, entry 1 and Table 3, entry 1). The use of a higher amount of catalyst (40 mol%), quite predictably led to an optimum e.r. of 95:5, while inducing a slight yield drop by 10% (Table 3, entry 2). We also attempted running the reaction at 0 °C, using 10 mol% of catalyst (8R, 9R)-14a, and found a satisfying e.r. of 93:7, but lower yield of 70% (Table 3, entry 3).
Next, we increased the excess of 16 from 1.1 to 1.3 equivalents (Table 3, entry 4), which proved to be a successful move in terms of both reaction yield (93%) and e.r. (95:5). In these conditions, halving the catalyst loading was again of adverse effect for the e.r. (90:10, Table 3, entry 5).
As a control for the power with which the chiral catalyst induces stereoinformation, we assessed cinchona catalyst (8R, 9S)-14a, featuring opposite chirality with respect to (8R, 9R)-14a to the C9. The experiment substantially confirmed the results obtained with (8R, 9R)-14a under otherwise identical conditions, while chiral HPLC analysis confirmed the generation of (–)-17 (Table 3, entry 6). Pressurizing the system to only 1.8 bar, and working at both at 0 °C and 10 °C (Table 3, entries 7 and 8, respectively), led to decreased yields and enantiomeric ratios. Finally, we performed an experiment with squaramide-based catalyst (8R, 9S)-14b at 10 mol%, which provided compound (-)-17 with a totally superimposable outcome with respect the use of (8R, 9S)-14a under identical conditions (Table 3, entry 9).
Starting from the winning configuration of our flow path, we felt to assess the nature of the protic additive. Accordingly, as the last task, we assessed a small set of different alcohols.
In general, the reaction rate was greatly improved in the presence of a protic additive, although none of the tested alcohol could outperform methanol. Accordingly, performing the reaction in absence of methanol led to a dramatic yield drop, although the reaction kept its enantioselective outcome (Table 4, entry 2). In another attempt, we replaced methanol with ethanol; in this case, the reaction provided moderate yields (65%), accompanied by a significant drop in the enantiomeric ratio to 67:33 (Table 4, entry 3). Last, the use of isopropanol as the protic additive led to an even worse yield (44%), although flanked by a better performance in terms of enantiomeric ratio (86:14) (Table 4, entry 4).
With the functional set-up in hands, we investigated the scope of the reaction by using different indolenines as substrates (Table 5). First, the tetrahydropyran-fused spiroindolenine 15b, afforded the corresponding (Z)-aldimine ( +)-18 in good yield and excellent e.r. (Table 5, entry 3). The N-Boc-piperidine containing spiroindolenine 15c required the addition of 20 mol% of cinchona catalyst to generate compound ( +)-19 (Table 5, entry 4), since the use of 10 mol% unfortunately gave very poor results in term of e.r. (data not shown). The Cbz-protecting group in 15d only provided a moderate e.r. for compound ( +)-20 (Table 5, entry 5) while the process demonstrated to be amenable for 3,3-diethylindolenine 15e, providing compound ( +)-21 in excellent e.r. (Table 5, entry 6). Under the same conditions, we also tried to replace the classic phenylhydrazine with a derivative bearing a methyl group at 2-position (15f). The flow protocol provided compound ( +)-22 in excellent yield; however, the outcome in terms of e.r. was disappointing (Table 5, entry 7). In this latter case, the poor e.r. registered might be ascribable to a problematic coordination of the cinchona catalyst due to the steric obstruction caused by the presence of the methyl group, as shown in Scheme 2.

Proposed mechanism for the enantioselective Strecker on cyclic (Z)-aldimines using ethyl cyanoformate
In agreement with literature, a plausible reaction mechanism is represented in Scheme 2. The thiourea catalyst is responsible for the pre-orientation and the activation of substrates acting as bifunctional organocatalyst. It is reasonable that ethyl cyanoformate in the presence of an alcohol (R-OH) generates HCN during the enantioselective Strecker reaction. Therefore, an ammonium salt derived from the dihydroquinine-derived thiourea and HCN can be formed. The cyanide anion can be activated by the thiourea moiety of the catalyst, while the cyclic imine is activated upon formation of a hydrogen bond with the ammonium salt. The cyanide is directed to the Si-face of the imine, thus accounting for the observed enantioselectivity.
With these conditions in hands, our next goal was to further enhance sustainability of the proposed protocol. Although it was not possible to identify a better performing solvent other than 1,2-dichloroethane, the developed methodology displays an improved sustainability profile with respect to its batch counterpart. First of all, the drastic reduction of reaction time (125 min in flow vs. 72 h in batch) represents a robust development. The operational simplicity represents a further benefit, since the reaction implementation in flow did not require additional efforts for realizing a dry system, such as the use of activated molecular sieves, which is instead a crucial precaution when performing the reaction in traditional batch mode. The flow system delivered a space–time-yield (STY) of approx. 0.24 g L−1 h−1 for the model substrate, which is a significant intensification compared to 0.0208 g L−1 h−1 of batch counterpart process.
As a last means to further improve sustainability was thus identified the possibility of recovering the cinchona-based catalyst. In a first trial, given the basic functionalities embedded in the cinchona catalyst, we tried to employ the acidic resin Amberlyst 15 to catch, and then release again, the catalyst, as reported in the literature [64]. We set up this protocol by extending our flow sequence through a column reactor filled with sand containing 1 mmol Amberlyst 15. However, in the collected stream, no traces of product were detected, since the acidic resin also held back the 2-cyanospiroindoline product, alongside the cinchona catalyst. In another attempt, we moved to bromo-tris(triphenylphosphine)copper(I), used as ligand for the thiourea functionality as reported in the literature, but again we obtained a disappointing outcome [65]. Better results were obtained when employing just silica gel and sand in the column reactor (Scheme 3). This resulted in an in-line separation with the initially collected stream only containing the reaction product. Once all the product fractions were collected, the catalyst could be easily released by passing ethyl acetate and methanol through the column reactor. The entire amount of the employed cinchona catalyst was recovered, and the purity was up to 95%, as determined by NMR analysis (see Supporting Information for details). The catalyst recovering allowed to decrease the reaction Process Mass Intensity (PMI) from 2.56 g g−1 to 2.25 g g−1.

Proposed protocol for the recovering of the catalyst
Conclusions
We have developed a flow-based methodology for the enantioselective Strecker reaction. The newly disclosed protocol employs ethyl cyanoformate as a relatively safe cyanide source. The enantioselective outcome was guaranteed by the use of a thiourea cinchona-based catalyst, while the methanol was the best performing alcohol additive. A thorough exploration of key parameters allowed the identification of the most convenient reaction conditions, in terms of reagent mixing mode, solvent, catalyst, and alcohol additive, temperature and residence time. To the best of our knowledge, this is the first continuous flow protocol for the asymmetric Strecker on cyclic (Z)-aldimines. This newly developed method guarantees effective reaction channeling towards the fast and complete formation of the α-aminonitrile products, thus reducing the yield drop due to indolenine degradation during long-lasting batch-wise reactions. We have also provided preliminary evidence for developing a sustainable flow-based protocol, by setting up a simple ed efficient protocol for cinchona catalyst recycling. Further optimization of the methodology and scope broadening for the proposed methodology are currently ongoing in our laboratories.
Data availability
Experimental procedures, chiral HPLC analyses and spectroscopic data are provided in the supporting information file.
References
Iyer MS, Gigstad KM, Namdev ND, Lipton M (1996) Asymmetric catalysis of the Strecker amino acid synthesis by a cyclic dipeptide. J Am Chem Soc 118:4910–4911. https://doi.org/10.1021/ja952686e
Monteiro JL, Pieber B, Corea G, Kappe CO (2016) Continuous Synthesis of Hydantoins: Intensifying the Bucherer-Bergs Reaction. Synlett 27:83–87. https://doi.org/10.1055/s-0035-1560317
Masamba W (2021) Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. Molecules 2:1707. https://doi.org/10.3390/molecules26061707
Ashe K, Fernandez-Garcia C, Corpinot MK, Coggins AJ, Bucar DK, Powner MW (2019) Selective prebiotic synthesis of phosphoroaminonitriles and aminothioamides in neutral water. Commun Chem 2:23. https://doi.org/10.1038/s42004-019-0124-5
Kouznetsov VV, Galvis CEP (2018) Strecker reaction and alpha-amino nitriles: Recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 74:773–810. https://doi.org/10.1016/j.tet.2018.01.005
Cleaves HJ, Chalmers JH, Lazcano A, Miller SL, Bada JL (2008) A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Orig Life Evol Biosph 38:105–115. https://doi.org/10.1007/s11084-007-9120-3
Krishnamurthy R, Snieckus V (2017) Prebiotic Organic Chemistry and Chemical pre-Biology: Speaking to the Synthetic Organic Chemists. Synlett 28:27–29. https://doi.org/10.1055/s-0036-1589831
Parker ET, Zhou M, Burton AS, Glavin DP, Dworkin JP, Krishnamurthy R, Fernandez FM, Bada JL (2014) A plausible simultaneous synthesis of amino acids and simple peptides on the primordial Earth. Angew Chem Int Ed Engl 53:8132–8136. https://doi.org/10.1002/anie.201403683
Ohfune Y, Shinada T (2003) Asymmetric Strecker route toward the synthesis of biologically active alpha, alpha-disubstituted alpha-amino acids. Bull Chem Soc Jpn 76:1115–1129. https://doi.org/10.1246/bcsj.76.1115
Arai T, Takahashi K, Ishiguro K, Yazawa K (1980) Increased production of saframycin A and isolation of saframycin S. J Antibiot (Tokyo) 33:951–960. https://doi.org/10.7164/antibiotics.33.951
Fukuyama T, Yang L, Ajeck KL, Sachleben RA (1990) Total Synthesis of (+/-)-Saframycin-A. J Am Chem Soc 112:3712–3713. https://doi.org/10.1021/ja00165a095
Dong W, Liu W, Liao X, Guan B, Chen S, Liu Z (2011) Asymmetric total synthesis of (-)-saframycin A from L-tyrosine. J Org Chem 76:5363–5368
Martinez EJ, Owa T, Schreiber SL, Corey EJ (1999) Phthalascidin, a synthetic antitumor agent with potency and mode of action comparable to ecteinascidin 743. Proc Natl Acad Sci U S A 96:3496–3501. https://doi.org/10.1073/pnas.96.7.3496
Cuevas C, Perez M, Martin MJ, Chicharro JL, Fernandez-Rivas C, Flores M, Francesch A, Gallego P, Zarzuelo M, de La Calle F, Garcia J, Polanco C, Rodriguez I, Manzanares I (2000) Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org Lett 2:2545–2548. https://doi.org/10.1021/ol0062502
Mendez LYV, Kouznetsov VV (2013) First Girgensohnine Analogs Prepared Through InCl3-catalyzed Strecker Reaction and their Bioprospection. Curr Org Synth 10:969–973. https://doi.org/10.2174/157017941006140206105449
Juillerat-Jeanneret L (2014) Dipeptidyl peptidase IV and its inhibitors: therapeutics for type 2 diabetes and what else? J Med Chem 57:2197–2212. https://doi.org/10.1021/jm400658e
Scheen AJ (2015) A review of gliptins for 2014. Expert Opin Pharmacother 16:43–62. https://doi.org/10.1517/14656566.2015.978289
Wang LX, Shen JF, Tang Y, Chen Y, Wang W, Cai ZG, Du ZJ (2007) Synthetic improvements in the preparation of clopidogrel. Org Process Res Dev 11:487–489. https://doi.org/10.1021/op700025d
Feldman PL, Brackeen MF (1990) A Novel Route to the 4-Anilido-4-(Methoxycarbonyl)Piperidine Class of Analgetics. J Org Chem 55:4207–4209. https://doi.org/10.1021/jo00300a047
Feldman PL, James MK, Brackeen MF, Bilotta JM, Schuster SV, Lahey AP, Lutz MW, Johnson MR, Leighton HJ (1991) Design, Synthesis, and Pharmacological Evaluation of Ultrashort-Acting to Long-Acting Opioid Analgesics. J Med Chem 34:2202–2208. https://doi.org/10.1021/jm00111a041
Walz AJ, Hsu FL (2014) Synthesis of 4-anilinopiperidine methyl esters, intermediates in the production of carfentanil, sufentanil, and remifentanil. Tetrahedron Lett 55:501–502. https://doi.org/10.1016/j.tetlet.2013.11.058
Arasappan A, Venkatraman S, Padilla AI, Wu WL, Meng T, Jin Y, Wong J, Prongay A, Girijavallabhan V, Njoroge FG (2007) Practical and efficient method for amino acid derivatives containing beta-quaternary center: application toward synthesis of hepatitis C virus NS3 serine protease inhibitors. Tetrahedron Lett 48:6343–6347. https://doi.org/10.1016/j.tetlet.2007.07.002
Zhang FG, Zhu XY, Li S, Nie J, Ma JA (2012) Highly enantioselective organocatalytic Strecker reaction of cyclic N-acyl trifluoromethylketimines: synthesis of anti-HIV drug DPC 083. Chem Commun (Camb) 48:11552–11554. https://doi.org/10.1039/C2CC36307K
Stork G (1989) The Stereospecific Synthesis of Reserpine. Pure Appl Chem 61:439–442
Lounasmaa M, Miettinen J, Hanhinen P, Jokela R (1997) Short synthesis of (+/-)-hirsutine: Direct addition of dimethyl malonate anion to a 1,4-conjugate iminium salt of appropriate 3-ethylindolo[2,3-a]quinolizidine. Tetrahedron Lett 38:1455–1458. https://doi.org/10.1016/S0040-4039(97)00056-7
Goes AD, Ferroud C, Santamaria J (1995) Regioselective Single-Electron Transfer Photocatalytic Synthesis of 2-Cyano-3-Ethylidenepiperidines - New Route to the Total Synthesis of (+/-)-Cis-Eburnamonine. Tetrahedron Lett 36:2235–2238. https://doi.org/10.1016/0040-4039(95)00233-3
Najera C, Sansano JM (2007) Catalytic asymmetric synthesis of alpha-amino acids. Chem Rev 107:4584–4671. https://doi.org/10.1021/cr050580o
Elango R, Ball RO (2009) Pencharz PB Amino acid requirements in humans: with a special emphasis on the metabolic availability of amino acids. Amino Acids 37(1):19–27. https://doi.org/10.1007/s00726-009-0234-y
Wang J, Liu X, Feng X (2011) Asymmetric strecker reactions. Chem Rev 111:6947–6983. https://doi.org/10.1021/cr200057t
Cai XH, Xie B (2014) Recent advances in asymmetric Strecker reactions. Arkivoc 205–248. https://doi.org/10.3998/ark.5550190.p008.487
Pan SC, List B (2008) The catalytic acylcyanation of imines. Chemistry-an Asian Journal 3:430–437. https://doi.org/10.1002/asia.200700327
Wunnemann S, Frohlich R, Hoppe D (2008) Asymmetric Strecker reaction of N-benzlhydrylimines utilising new tropos biphenyldiol-based ligands. Eur J Org Chem 2008:684–692. https://doi.org/10.1002/ejoc.200700763
Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN (2009) Scaleable catalytic asymmetric Strecker syntheses of unnatural alpha-amino acids. Nature 461:968-U223. https://doi.org/10.1038/nature08484
Karimi B, Maleki A (2009) Catalytic asymmetric Strecker hydrocyanation of imines using Yb(OTf)(3)-pybox catalysts. Chem Commun 5180–5182. https://doi.org/10.1039/B908854G
Hatano M, Hattori Y, Furuya Y, Ishihara K (2009) Chiral Lanthanum(III)-Binaphthyldisulfonate Complexes for Catalytic Enantioselective Strecker Reaction. Org Lett 11:2321–2324. https://doi.org/10.1021/ol900680f
Seayad AM, Ramalingam B, Yoshinaga K, Nagata T, Chai CLL (2010) Highly Enantioselective Titanium-Catalyzed Cyanation of Imines at Room Temperature. Org Lett 12:264–267. https://doi.org/10.1021/ol902540h
Kaur P, Pindi S, Wever W, Rajale T, Li GG (2010) Asymmetric catalytic Strecker reaction of N-phosphonyl imines with Et2AlCN using amino alcohols and BINOLs as catalysts. Chem Commun 46:4330–4332. https://doi.org/10.1039/C0CC00287A
Kaur P, Pindi S, Wever W, Rajale T, Li GG (2010) Asymmetric Catalytic N-Phosphonyl Imine Chemistry: The Use of Primary Free Amino Acids and Et2AlCN for Asymmetric Catalytic Strecker Reaction. J Org Chem 75:5144–5150. https://doi.org/10.1021/jo100865q
Chen YJ, Chen C (2008) Enantioselective Strecker-type reaction of phosphinoyl ketimines catalyzed by a chiral Zr-bipyridyldiol catalyst. Tetrahedron-Asymmetry 19:2201–2209. https://doi.org/10.1016/j.tetasy.2008.09.011
Enders D, Gottfried K, Raabe G (2010) Organocatalytic Enantioselective Strecker Synthesis of alpha-Quaternary alpha-Trifluoromethyl Amino Acids. Adv Synth Catal 352:3147–3152. https://doi.org/10.1002/adsc.201000666
Zhang GW, Zheng DH, Nie J, Wang T, Ma JA (2010) Bronsted acid-catalyzed efficient Strecker reaction of ketones, amines and trimethylsilyl cyanide. Org Biomol Chem 8:1399–1405. https://doi.org/10.1039/B924272D
Liu YL, Shi TD, Zhou F, Zhao XL, Wang X, Zhou J (2011) Organocatalytic Asymmetric Strecker Reaction of Di- and Trifluoromethyl Ketoimines. Remarkable Fluorine Effect. Org Lett 13:3826–3829. https://doi.org/10.1021/ol201316z
Sigman MS, Vachal P, Jacobsen EN (2000) A general catalyst for the asymmetric Strecker reaction. Angew Chem-Int Ed 39:1279–1281. https://doi.org/10.1002/(sici)1521-3773(20000403)39:7%3c1279::aid-anie1279%3e3.0.co;2-u
Becker C, Hoben C, Kunz H (2007) Enantioselective organocatalysis of strecker and Mannich reactions based on carbohydrates. Adv Synth Catal 349:417–424. https://doi.org/10.1002/adsc.200600370
Kanemitsu T, Toyoshima E, Miyazaki M, Nagata K, Itoh T (2010) Asymmetric Acyl-Strecker Reaction Promoted by Novel Thiourea Organocatalyst. Heterocycles 81:2781–2792
Shao YD, Tian SK (2012) A highly enantioselective catalytic Strecker reaction of cyclic (Z)-aldimines. Chem Commun 48:4899–4901. https://doi.org/10.1039/C2CC31001E
Suginome M, Yamamoto A, Ito Y (2002) Bis(dialkylamino)cyanoboranes: highly efficient reagents for the Strecker-type aminative cyanation of aldehydes and ketones. Chem Commun 1392–1393. https://doi.org/10.1039/B203645B
Kobayashi S, Ishitani H (1999) Catalytic enantioselective addition to imines. Chem Rev 99:1069–1094. https://doi.org/10.1021/cr980414z
Das B, Ramu R, Ravikanth B, Reddy KR (2006) Studies on novel synthetic methodologies, part 67. (Bromodimethyl)sulfonium bromide catalyzed one-pot synthesis of alpha-aminonitriles. Synthesis-Stuttgart 1419–1422. https://doi.org/10.1002/chin.200636084
Kobayashi S, Ishitani H, Ueno M (1997) Facile synthesis of alpha-amino nitriles using lanthanide triflate as a Lewis acid catalyst. Synlett 115–116. https://doi.org/10.1002/chin.199723087
Heydari A, Arefi A, Khaksar S, Shiroodi RK (2007) Guanidine hydrochloride: An active and simple catalyst for Strecker type reaction. J Mol Catalys a-Chem 271:142–144. https://doi.org/10.1016/j.molcata.2007.02.046
Yadav JS, Reddy BVS, Eshwaraiah B, Srinivas M, Vishnumurthy P (2003) Three-component coupling reactions in ionic liquids: a facile synthesis of alpha-aminonitriles. New J Chem 27:462–465. https://doi.org/10.1039/B208844B
Mojtahedi MM, Abaee MS, Abbasi H (2006) Environmentally friendly room temperature Strecker reaction: One-pot synthesis of alpha-aminonitriles in ionic liquid. J Iran Chem Soc 3:93–97. https://doi.org/10.1007/BF03245797
Martinez R, Ramon DJ, Yus M (2005) Catalyst-free multicomponent Strecker reaction in acetonitrile. Tetrahedron Lett 46:8471–8474. https://doi.org/10.1016/j.tetlet.2005.10.020
Matsumoto K, Kim JC, Iida H, Hamana H, Kumamoto K, Kotsuki H, Jenner G (2005) Multicomponent Strecker reaction under high pressure. Helv Chim Acta 88:1734–1753. https://doi.org/10.1002/hlca.200590136
Atherton JH, Blacker J, Crampton MR, Grosjean C (2004) The Strecker reaction: kinetic and equilibrium studies of cyanide addition to iminium ions. Org Biomol Chem 2:2567–2571. https://doi.org/10.1039/B407853E
Wiles C, Watts P (2008) Evaluation of the Heterogeneously Catalyzed Strecker Reaction Conducted Under Continuous Flow. Eur J Org Chem 2008:5597–5613. https://doi.org/10.1002/ejoc.200800751
Ushakov DB, Gilmore K, Kopetzki D, McQuade DT, Seeberger PH (2014) Continuous-Flow Oxidative Cyanation of Primary and Secondary Amines Using Singlet Oxygen. Angew Chem-Int Ed 53:557–561. https://doi.org/10.1002/anie.201307778
Seayad AM, Ramalingam B, Chai CLL, Li CZ, Garland MV, Yoshinaga K (2012) Self-Supported Chiral Titanium Cluster (SCTC) as a Robust Catalyst for the Asymmetric Cyanation of Imines under Batch and Continuous Flow at Room Temperature. Chem-a Eur J 18:5693–5700. https://doi.org/10.1002/chem.201200528
Alfano AI, Brindisi M, Lange H (2021) Flow synthesis approaches to privileged scaffolds - recent routes reviewed for green and sustainable aspects. Green Chem 23:2233–2292. https://doi.org/10.1039/D0GC03883K
Alfano AI, Zampella A, Novellino E, Brindisi M, Lange H (2020) Harnessing interrupted Fischer in continuous flow: sustainable synthesis of (spiro)indolenine and (spiro)indoline privileged scaffolds. React Chem Eng 5:2091–2100. https://doi.org/10.1039/D0RE00329H
Alfano AI, Buommino E, Ferraro MG, Irace C, Zampella A, Lange H, Brindisi M (2021) Coupling Interrupted Fischer and Multicomponent Joullie-Ugi to Chase Chemical Diversity: from Batch to Sustainable Flow Synthesis of Peptidomimetics. ChemMedChem 16:3795–3809. https://doi.org/10.1002/cmdc.202100474
Porcheddu A, Mocci R, Brindisi M, Cuccu F, Fattuoni C, Delogu F, Colacino E, D’Auria MV (2022) Mechanochemical Fischer indolisation: an eco-friendly design for a timeless reaction. Green Chem 24:4859–4869. https://doi.org/10.1039/D2GC00724J
Carlone A, Bernardi L, McCormack P, Warr T, Oruganti S, Cobley CJ (2021) Asymmetric Organocatalysis and Continuous Chemistry for an Efficient and Cost-Competitive Process to Pregabalin. Org Process Res Dev 25:2795–2805. https://doi.org/10.1021/acs.oprd.1c00394
Saeed A, Larik FA, Jabeen F, Mehfooz H, Ghumro SA, El-Seedi HR, Ali M, Channar PA, Ashra H (2018) Synthesis, Antibacterial and Antileishmanial Activity, Cytotoxicity, and Molecular Docking of New Heteroleptic Copper(I) Complexes with Thiourea Ligands and Triphenylphosphine. Russ J Gen Chem 88:541–550. https://doi.org/10.1134/S1070363218030246
Acknowledgements
M. B. and A. I. A. would like to thank the Department of Excellence of Pharmacy (2023–2027), University of Naples Federico II. M. B., H. L. and A. I. A. acknowledge the MIUR grant “Department of Excellence of Pharmacy (2018-2022), University of Naples Federico II. Part of this research was supported by EU funding within the MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases to M.B. (Project no. PE00000007, INF-ACT).
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
A. I. A. performed all the experiments and contributed to manuscript preparation; A.S. performed the chiral HPLC analysis and the e.r. determination; A. C. designed the chiral HPLC experiments and analyzed the data; H. L. conceptualized the flow experiments and edited the manuscript; M. B. conceptualized the idea, supervised the experiments, analyzed the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
There are no conflicts to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alfano, A.I., Sorato, A., Ciogli, A. et al. Enantioselective catalytic Strecker reaction on cyclic (Z)-aldimines in flow: reaction optimization and sustainability aspects. J Flow Chem 14, 197–210 (2024). https://doi.org/10.1007/s41981-023-00279-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41981-023-00279-9