
Abstract
Calcium titanate-hydroxyapatite composites (CT-HAp) were synthesized using a conventional sintering process. The calcium source was derived from cockle shell waste. The combustion synthesis of CT (CaTiO3) produced fine, irregular, sub-micron size particles. The precipitation synthesis of HAp (Ca5(PO4)3OH) produced rod-like nanoparticles. The obtained powders were used to fabricate CT-HAp bioceramic composites sintered at 1250 °C for 2 h. The phase formation, microstructure, and physical and mechanical properties of HAp and various CT-HAp composites were investigated. A CT-HAp composite with 8%wt of CT, having a density of 3.00 ± 0.01 g/cm3 and a grain size of 0.90 ± 0.06 μm, produced the highest values for flexural strength, Vickers hardness and fracture toughness: 56.67 ± 1.122 MPa, 6.03 ± 0.02 GPa, and 7.77 MPa.m1/2, respectively. The chemical solubility of the CT-HAp composites was very low, showing an average weight loss of 0.24% in a simulated body fluid solution. The properties of the CT-HAp composites were controlled by CT content and porosity. In-vitro biological studies demonstrated the growth and proliferation of MC3T3-E1 osteoblast cells on all composites after three days of culture. The highest cell viability was exhibited on a CT-HAp composite with a CT content of 8%wt. The expression of alkaline phosphatase (ALP) activity on the composite surface promoted cellular osteoinduction, indicating the potential of the developed composite for bone formation and repair.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Calcium phosphate-based materials, especially hydroxyapatite (HAp)-based composites, are used in orthopedics and dentistry to repair or replace human bone and teeth [1] but the applications of these biomaterials are limited by the brittle nature of HAp. This limitation can be mitigated by the use of reinforcing materials, which are commonly metal oxides such as zirconium oxide (ZrO2), aluminium oxide (Al2O3) and titanium oxide (TiO2) [2].
An alternative approach that incorporates calcium titanate (CT, CaTiO3) particles with HAp (Ca5(PO4)3OH) has produced positive results in different processing conditions that have improved other material properties such as density, modulus, strength, hardness and fracture toughness as well as brittleness [1, 3,4,5,6,7,8,9,10,11,12,13]. Furthermore, CT improves the biocompatibility of the composite, producing a positive surface charge which resembles the surface electrical property of natural bone [14]. CT is a good substrate for apatite growth, and promotes the osseointegration that plays an important role in connecting the surfaces of the living bone and the implant. CT hydrolyzes in simulated body fluid (SBF), producing numerous Ti–OH groups. During the incubation process, contact between the implant surface and osteoblast cells is enhanced, and the exchange of Ca2+ and (PO4)3− ions is activated. In addition, TiO32− from CT attracts calcium and phosphorus ions from surrounding bone tissue and body fluids to promote nucleation. CT also enhances interfacial bonding within bioceramics. These physical and chemical changes have been shown to induce apatite formation, and enhance regeneration and integration during repairing bone defect rehabilitation [1, 3,4,5,6,7,8,9,10,11,12,13].
The cockle is an edible marine bivalve mollusk that it has an economic value as seafood and is widely consumed in Southeast Asia. In 2021, the Department of Fisheries, Ministry of Agriculture and Cooperatives reported that there were 1,637 cockle farms in Thailand that produced 33,526.27 tons of cockles. A large volume of waste cockle shells from processing is produced, causing an environmental problem. Recycling this waste could contribute to the more sustainable development of marine resources. Previous studies demonstrated that cockle shells contain calcium carbonate (CaCO3) in the crystal form of aragonite, consisting of 97% calcium (Ca) [15,16,17]. The proportion of CaO in commercial calcium carbonate is 99.10%wt [18], and the yield of CaO powder obtained from cockle shell after calcination is higher than that yielded by mussel and oyster shells [19]. Furthermore, cockle shells are used as a raw material in the synthesis of HAp by various methods [20,21,22,23,24,25,26,27,28]. However, the only shell that has been used to produce CT is the scallop shell [29]. Therefore, cockle shell is a potentially attractive, untapped, calcium source for the preparation of CT as well as HAp.
The present work aims to fabricate CT-HAp composites and improve their properties for biomedical applications. CT was synthesized by an energy-saving combustion synthesis method and HAp was prepared by a precipitation process. Reactant powders for both components were produced from waste cockle shell. The phase formation and morphology of the reactant powders and the fabricated CT-HAp composites were investigated. The CT-HAp composites were characterized and discussed in terms of physical, mechanical and biological properties.
Experimental procedures
Preparation of powders as a calcium source
Raw cockle shell for the present work was supplied by a seafood farm in Samut Songkhram Province on the Gulf of Thailand. X-ray fluorescence (XRF) analysis of the chemical composition of the cockle shell (Table 1) showed that CaO was a major component in coexistence with other trace oxides.
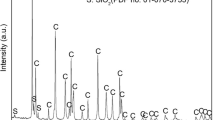
The XRF analysis was confirmed by X-ray diffraction (XRD) analysis (Fig. 1), which indicated that the cockle shell was mainly composed of CaCO3 and contained aragonite as a major crystalline form along with a minor phase of calcite. The XRD analysis detected SiO2, but not Na2O, Fe2O3 and SrO that were detected by XRF analysis. Although Na2O and SiO2 were both detected in trace amounts, only the peak of SiO2 was detected by XRD. Possibly the crystallinity of SiO2 was more perfect than the crystallinity of Na2O.

XRD pattern of raw waste cockle shell used in this work
Since biomaterials are implanted in the living body, any toxic impurities must be identified. Therefore, arsenic (As), cadmium (Cd) and lead (Pb) contents were determined by inductively coupled mass spectrometry (ICP-MS), and mercury (Hg) content was determined by direct mercury analysis. No Hg and Cd were found, and although As (0.117 ± 0.012 ppm) and Pb (< 0.100 ppm) were detected, the detected levels were below the maximum limit permitted by the relevant standards (ASTM F1185-88 and ISO 3779-1:2008) (Table 2). Therefore, the raw cockle shell was considered suitable for use in biomedical applications.
Raw cockle shell was washed several times with distilled water and then immersed in hydrogen peroxide (H2O2, 30%, Chem-Supply) solution for 24 h to eliminate soluble inorganic contaminants. The cleaned cockle shell was dried in an oven at 110 °C for 24 h. The dry cockle shell was then preheated at 200 °C to increase brittleness and simplify preliminary crushing into a coarse powder with an adapted crushing machine. The obtained coarse powder was milled for 1 h into a finer powder using a planetary ball mill (Fritsch GMBH, Pulverisette 6) with a silicon nitride (Si3N4) ball and jar at a rotation speed of 350 rpm. The milled powder was passed through a 325 mesh with a vibrating sieve machine (Nitto, ANF-30) to obtain particles of less than 45 μm. This powder was used as the calcium source for the synthesis of CT. Some of the milled and sieved cockle shell powder was then heated in air in a muffle furnace (Carbolite, RHF 16/8) at 900 °C for 2 h to transform CaCO3 into CaO as a source of calcium for the synthesis of HAp. The calcined CaO powder was analyzed by XRD (Fig. 2). The minor phase of calcium hydroxide (Ca(OH)2) present in the sample was due to the absorption of moisture from the air [30].

XRD pattern of cockle shell powder calcined at 900 °C
Preparation of reactant powders
Combustion synthesis of calcium titanate (CT) powder
Cockle shell powder with a particle size of 45 μm was mixed to homogeneity with anatase titanium dioxide powder (A-TiO2, 99.5%, < 45 μm, Degussa AG), by ball milling for 2 h using zirconia (ZrO2) balls and a nylon bottle. The molar ratio of cockle shell powder to A-TiO2 was 1.2: 1. In order to activate the solid-state reaction, the mixed powder was high-energy milled at 400 rpm for 30 min. The high-energy milled powder was then mixed at a molar ratio of 1:2 with magnesium powder fuel (Mg, 99%, ~ 45 μm, Riedel-Detlaen) by ball milling for 60 min. Using a hydraulic press (Huat Seng, 1939-15 T), 20 g of the obtained reactant powder were uniaxially pressed at 5 psi (0.03 MPa) into a cylindrical steel mold with a diameter of 25.4 mm. To provide the high heat necessary for combustion, a 5 g thermite compact was prepared from iron oxide (Fe2O3, 99%, < 5 μm, Sigma-Aldrich) and aluminium (Al, 93%, 45 μm, Himedia Laboratories) powders mixed at a molar ratio of 1: 2. The thermite compact was placed on top of the reactant compact and ignited. The combustion synthesis of CT took place under a controlled Ar atmosphere at a pressure of 0.5 MPa. The as-combusted product was separated from the by-product of thermite (Fe + Al2O3) and mechanically pestled into friable powder in an alumina (Al2O3) mortar. The by-product of magnesium oxide (MgO) was then leached from the powder product in 2 M hydrochloric acid (HCl, 37%, RCI Labscan) solution. Leaching was performed under moderate magnetic stirring for 180 min with a powder product-HCl solution ratio of 10 g: 100 mL. The as-leached sediment was captured with filter paper and washed several times with deionized (DI) water to neutralize pH. The washed as-leached powder was dried at 80 °C for 24 h and CT powder was obtained. The experimental procedures were adapted from and followed previous works [31, 32].
Precipitation of hydroxyapatite (HAp) powder
As-prepared CaO powder was suspended in DI water at a solid–liquid ratio of 20 g: 500 mL under constant magnetic stirring at 90 °C for 1 h. A suspension of 0.5 M Ca(OH)2 was obtained with a pH of 13.0. The pH of the suspension was then adjusted to 7.0 with orthophosphoric acid (H3PO4, 85%, RCI Labscan) solution under constant stirring at 90 °C for 1 h. After cooling the mixture down to room temperature, ammonium hydroxide (NH4OH, 30%, J.T.Baker) solution was added to the suspension under stirring to achieve a pH of 10.0. The suspension was then aged for 48 h at room temperature in a covered beaker while precipitation occurred. The precipitated powder was recovered with a filter paper attached to a suction pump. The recovered powder was washed repeatedly with DI water to remove the NH4+ ion until pH was 7.0. The powder was then dried in an oven at 80 °C for 24 h, ground, and finally calcined in air at 700 °C for 2 h at a heating rate of 5 °C/min.
Conventional solid-state sintering of CT-HAp composites
Various contents of CT powder (0–20%wt) were ball milled with HAp powder for 24 h in a wet medium (ethanol, 99.9%, RCI Labscan) using ZrO2 milling balls. Wet milling was used to ensure a homogenous mixture of the two different ceramic powders. Ethanol was removed by drying the powder mixture in an oven at 80 °C. The dried powder was mechanically ground in an agate mortar and 0.5 g of the powder was mixed with 0.1 mL of 3 wt% polyvinyl alcohol (PVA, 99%, Gohsenol) solution. The obtained mixture was uniaxially pressed (Krumor, HP-900) at 50 MPa to form a disc-shaped green pellet 15 mm in diameter and approximately 1.5 mm thick. The green composite pellet was sintered by a conventional solid-state route in air in a muffle furnace (Carbolite, RHF 16/8). The PVA binder was eliminated by heating for 1 h at 500 °C, and the composite pellet was further heated at 5 °C/min to 1250 °C and held at this temperature for 2 h. Sintering at 1250 °C was reported in a previous study to produce HAp ceramics with acceptable properties and without any phase transformation into tricalcium phosphate [33]. The sintering profile is depicted in Fig. 3.

Temperature profile for the fabrication of CT-HAp composites by conventional solid-state sintering
Materials characterization
The chemical composition of raw cockle shell was analyzed using XRF (PANalytical, Zetium). Different phases in the raw materials, reactant powders and sintered composites were identified by XRD analysis (PANalytical, Empyrean), carried out using Cu-Ka radiation. The crystal structures of identified phases were analyzed and matched by HighScore Plus software version 4.7. The heavy metal content of raw cockle shell was determined by ICP-MS (PerkinElmer, NexION2000). The elements present in the SBF solution that was used to test chemical solubility were identified by inductively coupled plasma optical emission spectroscopy (ICP-OES, PerkinElmer, AVIO 500). Hg content in raw cockle shell was determined by direct mercury analysis (NIC, MA3000). A scanning electron microscope (SEM, FEI, Quanta 400) was used to study the morphology and measure the particle size of as-prepared CT and HAp powders and to observe the microstructure of sintered composites. The average grain size of sintered composite samples was measured from SEM images by the mean linear intercept method, using ImageJ software version 1.53e. Prior to SEM observation, samples were sputter-coated (SPI, 12157Q-AB) with a thin layer of gold to enhance electrical conductivity. The particle size of as-prepared HAp was precisely evaluated by transmission electron microscopy (TEM, JEOL, JSM-2010). Energy dispersive X-Ray spectroscopy (EDX, X-Max, Oxford) was used to analyze the elemental distribution in sintered composites and to measure the content of Ca and P in HAp.
Materials properties
Physical properties
Ceramic composites were then hydrostatically weighed with a precision analytical balance (Sartorious, B5A224S-CW). In accordance with the ASTM C373-72 standard, CT-HAp specimens were first soaked in boiling distilled water for 5 h and then left immersed in distilled water at room temperature for 8 h. The sintered density and apparent porosity of ceramic composites were calculated using Eqs. (1) and (2) [33]:
where ρsintered is the sintered density and Papp is the apparent porosity; W0, W1, and W2 are the specimen dry weight, weight in water of the water-saturated specimen, and weight in air of the water-saturated specimen, respectively, and ρwater is the density of water (1 g/cm3). The linear shrinkage of the ceramic composites was calculated using Eq. (3) [33]:
where Db and Da denote the diameter of specimens before and after sintering.
Mechanical properties
The strength of CT-HAp composites was tested by determining biaxial flexure in accordance with the ISO/DIS 6872: 2013(E) standards, using a universal testing machine (Shimadzu, AGX-V). Disc-shaped specimens were prepared to the stipulated minimum thickness of 1.2 ± 2 mm and diameter of 12 mm. Specimens were placed on three hardened steel ball supports and the load was applied by a flat punch with a diameter of 1.5 mm at a crosshead speed of 0.5 mm/min. Biaxial flexural strength was calculated using Eq. (4) [33]:
where σ is biaxial flexural strength, P is the load to failure and b is the specimen thickness at the fracture origin. X and Y were determined using the following formulas [33]:
where, ν is Poisson’s ratio (0.25 for ceramic), r1 is the radius of the support circle (1.6 mm), r2 is the radius of the loaded area (0.8 mm) and r3 is the radius of the specimen.
The hardness of samples was determined using the Vickers micro-hardness test (Innovatest, NOVA 130/240) by indenting a diamond pyramid tip on a smooth polished surface with a load of 1 kgf (9.81 N) and dwell time of 10 s. The obtained Vickers hardness numbers were converted to GPa in SI units. In addition, the lengths of cracks emanating from the vertices of the diamond indentation were scaled by SEM and used to estimate the fracture toughness (KIC) of specimens in radial-median and any other crack systems, using Eqs. (5) and (6) [34]:
where, KIC is fracture toughness, F is the applied load (N), E is Young's modulus (GPa), HV is Vickers hardness (GPa), c is the longest lateral extension of a crack from the centre of the indentation (m), and a is the half of the indentation diagonal (m). The Young's modulus (assumed E≈Ec) of the particulate composite was estimated from the rule of mixture in Eq. (7) [35]:
where, Em and Ep denote the elastic modulus for the matrix (HAp = 79 GPa) and reinforcing material (CT = 161 GPa), respectively, and Vm and Vp denote the volume fractions of the matrix and reinforced particle, respectively.
Hardness and fracture toughness were used to determine the brittleness (B) of specimens, defined as [6]:
Chemical solubility
The chemical solubility of the CT-HAp composites was investigated in a simulated human extracellular fluid using a solution of SBF. The SBF solution was prepared according to the method of Kokubo and Takadama [36] by mixing sodium chloride (NaCl, 7.996 g), sodium bicarbonate (NaHCO3, 0.350 g), potassium chloride (KCl, 0.224 g), potassium biphosphate trihydrate (K2HPO4.3H2O, 0.228 g), magnesium chloride hexahydrate (MgCl2.6H2O, 0.305 g), calcium chloride (CaCl2, 0.278 g), sodium sulfate (Na2SO4, 0.071 g), tris(hydroxymethyl)aminomethane ((CH2OH)3CNH2, 6.057 g), 1 M hydrochloric acid solution (HCl, 1 43 mL), and DI water. The pH of the SBF solution was adjusted to 7.2. Samples for analysis were soaked in 500 mL of SBF solution for 7, 15, 21, and 28 days at a controlled temperature of 37 ℃ in a universal oven (Memmert, UN 55 PA 115 V). After soaking, the pH of samples was measured with a pH meter (Thermo-Orion, A111). Samples were then washed thoroughly with DI water and dried in an oven at 60 ℃ for 24 h. The chemical solubility of the ceramic composites was estimated from the weight remaining [37], calculated based on the weight before (W1) and after (W2) soaking, using Eq. (9):
Furthermore, Ca, P and Ti in the SBF solution were determined by ICP-OES and the chemical composition of sample surfaces was investigated by EDX analysis.
In-vitro biological properties
Cell culture
MC3T3-E1 osteoblastic cells from the American Type Culture Collection (ATCC) were cultured in 75 cm2 cell culture flasks containing alpha minimum essential medium (Alpha MEM, Gibco). A standard incubator (Eppendorf, Cellxpert) was operated at 37 °C with 5% CO2 and 95% relative humidity. The Alpha MEM was supplemented with a solution of 10% fetal bovine serum (FBS), 100 I.U./mL penicillin and 100 μg/mL streptomycin. The medium was replaced every 2–3 days, until cell densification remained at 80–90% for 3 days.
WST-1 assay to measure cell viability
The viability of cultured MC3T3-E1 cells was investigated on HAp and CT-HAp composite samples. Samples were placed in 24-well plates and cells were seeded (using Alpha MEM), as described in the previous subsection. After incubation, the medium was aspirated, composite samples were washed with phosphate buffered saline (PBS) solution, and 2 mL of 0.25% trypsin–EDTA solution were added to each well. The plates were then placed in the incubator for 5 min at 37 °C in 5% CO2. After that, 10 mL of medium were twice added by pipette into a 50 mL test tube, and centrifuged for 5 min (TOMY, MDX-310). Four milliliters of medium were added. Then, 10 μL of cells were sampled, stained with trypan blue dye (C34H28N6O14S4, Thermo Fisher Scientific) and micropipetted into a hemocytometer. Cytotoxicity was evaluated by counting viable cells under a phase contrast microscope (Olympus, CKK53). The number of cells was determined using Eq. (10) [38]:
*Dilution factor is usually 2.
In addition, approximately 1.5 × 104 MC3T3E-1 cells, counted with the hemocytometer, were seeded on composite samples, which were then washed twice with 1 mL of PBS solution containing 500 μL of Alpha MEM. The cells in each well were treated with 50 μL of WST-1 solution and incubated for 2 h. The optical density (OD) or absorbance of the solution was measured at 450 nm using an automated microplate reader (BioTek, Epoch). The percentage of cell viability was calculated from Eq. (11) [39]:
where, OD450 sample is the optical density obtained in the presence of WST-1 and OD450 control is the optical density obtained in the absence of WST-1, measured at 450 nm.
Osteogenic induction and alkaline phosphatase staining
MC3T3E-1 cells were seeded in wells of a 24-well plate at a density of 1.5 × 104 cells per sample and incubated for two days in a complete growth medium (CGM), which was supplemented with 0.1 μM dexamethasone (Sigma-Aldrich), 50 μM L-ascorbic acid (Sigma-Aldrich), and 10 mM β-glycerolphosphate disodium salt hydrate (Sigma-Aldrich). Subsequently, the cells were osteogenically induced for 24 h using osteogenic induction medium (ODM). The induced cells were then cultured in CGM for an additional seven days. Alkaline phosphatase (ALP) staining was performed as previously described [40]. Briefly, the cells were fixed with 70% ethanol for 30 min and stained with a 0.2% (v/v) solution of 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) (Sigma-Aldrich). The intensity of ALP staining was assessed using a stereomicroscope (Nikon, SM2745T). The number of blue-stained pixels was analyzed as a ratio to the total sample surface area. The percentage changes in ALP activity was calculated by comparison to the sample without CT (0 wt%).
Cell fixation for SEM observation
Osteoblastic cell growths on composite samples were washed with 0.1 M PBS solution, to remove soluble proteins in the medium. Cells were fixed with 2.5% glutaraldehyde (C5H8O2, Sigma-Aldrich) diluted in 0.2 M PBS at room temperature for 1–2 h, and then washed 2–3 times with 0.1 M PBS solution for 1–2 min, then with distilled water for 1–2 min. Cells adhering to surfaces were dehydrated using a series of ethanol solutions (50, 60, 70, 80 and 90%). Adhering cells were soaked twice in each concentration for 15 min and then for 30 min in 100% ethanol solution. Finally, dehydration was completed in a critical-point dryer (CPD, Toursimis, Autosamdri 931).
Results and discussion
Characteristics of as-prepared reactant powders for CT-HAp composites
The XRD pattern of CT powder (Fig. 4(a)) indicated a major phase of orthorhombic CaTiO3 (space group: Pbnm) and a secondary phase of titanium dioxide (TiO2) at a very low concentration. The XRD pattern of HAp powder (Fig. 4(b)) showed only a pure Ca5(PO4)3OH phase (space group: P63/m) with a hexagonal crystal system. The crystal system and crystalline parameters of CaTiO3 and Ca5(PO4)3OH phases were referenced in the powder diffraction file databases (according to the referenced PDF numbers).

XRD pattern of reactant powders (a) calcium titanate and (b) hydroxyapatite
The SEM image of CT powder (Fig. 5(a)) revealed irregular agglomerations with a particle size of less than 1 μm. The HAp powder (Fig. 5(b)) comprised clusters of small particles. TEM observation confirmed HAp particles were rod-like with a particle size in the nanometer range (Fig. 5(c) and (d)). The Ca/P ratio of hydroxyapatite used in this work, determined by EDX, was close to the Ca/P ratio of adult human bone (1.71) [41].

The micrographs show SEM images of calcium titanate (a) and hydroxyapatite (b), and TEM images (c) and (d) of hydroxyapatite particles at different magnifications
Characteristics and properties of conventional-sintered CT-HAp composites
HAp and CT-HAp composite samples, sintered at 1250 °C for 2 h, were analyzed using XRD. The XRD pattern of the HAp sample (0%wt) (Fig. 6) presented single peaks of HAp with the chemical formula Ca5(PO4)3OH. The XRD patterns of CT-HAp composites (2–20%wt of CT) presented major peaks of HAp and minor peaks of CT. The intensity of the CT peaks (~ 47.5°) increased with increments of CT content. These findings confirmed that HAp and CT-HAp composites had been synthesized without any phase dissociation or new phase formation. Unreacted TiO2, originally present in CT powder (Fig. 4(a)), was presented in very low amounts compared with HAp and CT, and therefore the peak of TiO2 was not recorded.

XRD patterns of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
SEM images of HAp and CT-HAp composites showed that the surface microstructure of all specimens (Fig. 7) comprised a bimodal grain size distribution of HAp particles smaller than 2 μm. The microstructures were homogenous and CT particles were evenly distributed in the HAp matrix at CT contents of 2 to 8%wt. In addition, the average grain size of the CT-HAp composites decreased from 1.14 ± 0.20 to 0.90 ± 0.06 μm with increments of CT content from 2 to 8%wt. The reduction of grain size was due to the effective inhibition of grain boundaries by CT particles. At 10%wt of CT and above, the microstructure of the CT-HAp composites became less homogenous, developing many pores, including large pores, and CT particles tended to agglomerate more and adhere to HAp. Also, more abnormal grain growth was observed and the average grain size increased from 0.98 ± 0.10 to 1.44 ± 0.35 μm as CT contents increased from 10 to 20%wt.

SEM images of the surface morphology of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
In addition, SEM images of cross-sections of HAp and the composites (Fig. 8) showed that composites with higher CT contents, in addition to increasing agglomeration of CT particles, exhibited greater porosity, displaying pores clustered around agglomerated particles of CT. Dubey et al. [6] in a study of CT-HAp composites containing high amounts of CT, reported that the number of CT/CT contacts increased with increments of CT content. In the present work, CT-HAp composites containing more than 8%wt of CT exhibited increased agglomerations of CT. Also, this result might be affected by contacts between CT particles. In another work [1] more clusters of CT particles were observed at high CT contents.

SEM cross-sectional images of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
To identify phase distributions, SEM-EDX elemental mapping was performed of HAp, CT and CT-HAp composites containing 10 and 20%wt of CT. HAp presented Ca, P and O (Fig. 9(a)), while CT and both CT-HAp composites presented Ca, P, Ti and O (Fig. 9(b) and (c)). CT has a higher density than HAp, and thus was visible as a brighter area. To confirm the findings, two different areas on the surface of CT-HAp with 20%wt of CT were scanned and EDX spectra were recorded (Fig. 9(d)). The darker area (marked *) presented Ca, O, and P with a small amount of Ti, indicating hydroxyapatite as the major phase. In contrast, the brighter area (marked •) revealed the presence of Ca, Ti and O contents of 22.82, 20.84 and 56.34 at%, respectively, which were close to the relevant references [42] shown in the enclosed table. This result indicated that the composition of the brighter area of the composite was CT.

Typical SEM-EDX mapping of hydroxyapatite (a) 0%wt calcium titanate, and calcium titanate-hydroxyapatite composites with calcium titanate contents of (b) 10%wt and (c) 20%wt, and (d) magnified SEM-EDX point scan of a calcium titanate-hydroxyapatite composite with 20%wt of calcium titanate
Physical and mechanical properties of conventional-sintered CT-HAp composites
The sintered density and apparent porosity of the synthesized materials were plotted as functions of CT content (Fig. 10). The density of HAp in this work was 2.97 ± 0.08 g/cm3 (94% of theoretical density). The density of CT-HAp composites with less than 8%wt of CT slightly increased as a function of CT content from 2.98 at 0wt% to 3.01 ± 0.02 g/cm3 at 6%wt of CT. The increasing density of CT-HAp composites with less than 8%wt of CT was in agreement with the rule of mixture for composites and the greater theoretical density of CT (3.98 g/cm3) than HA (3.16 g/cm3) [1, 35] but the density of 8%wt CT-HAp was very slightly lower at 3.00 ± 0.01 g/cm3. Above 8%wt of CT, the density of the composite slightly decreased, reaching its lowest value of 2.91 ± 0.02 g/cm3 at 20%wt of CT. The reduced density of the composite at CT contents of 8wt% and above was related to the increased porosity of the material. CT was denser than HAp, therefore, when CT particles were added to HAp powder in the consolidation process, the compressibility of the green sample was reduced and the porosity of the obtained CT-HAp composites increased as a function of CT content. Also, the higher melting point of CT particles (> 1900 °C for CT and ≈ 1500–1600 °C for HAp) [42,43,44], higher sintering temperature (1450 °C) [3] and great resistance to deformation (E = 161 MPa) [45] inhibited microstructural rearrangement during sintering. The reduced deformation and diffusion of the particles further promoted porosity, and higher contents of CT possibly increased the porosity of the composite due to the more uniform grain growth.

Sintered density and apparent porosity of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
The linear shrinkage of samples (Fig. 11) decreased with increments of CT content. The reduction in shrinkage of the composite was mainly due to the non-densifying inclusion, namely CT, introduced into the HAp matrix. When the amount of CT in the composite was high, the contact between CT particles was excessive and led to the development of pores that hindered densification [6,7,8].

Linear shrinkage of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
The biaxial flexural strength of the CT-HAp composite was affected by CT content. At CT contents from 2 to 8%wt, the flexural strength of the composite increased (Fig. 12). The flexural strength of 8%wt CT-HAp was 56.67 ± 1.12 MPa, compared to 45.56 ± 2.71 MPa for pure HAp-an increase of 24%. In contrast, flexural strength decreased at CT contents from 10 to 20%wt. CT has greater flexural strength than HAp [7, 8] and therefore, CT-HAp composites with an optimal microstructure and specific mechanical and physical properties can be achieved by controlling the CT content. In the discussion of microstructural evolution in “Characteristics of as-prepared reactant powders for CT-HAp composites” section, the grain size, porosity, pore size and pore distribution were related to CT content (Figs. 7 and 8). Composites containing 8%wt of CT or less displayed smaller grain sizes than composites containing 10%wt of CT or more, as well as lower porosity and smaller pores that were more homogenously distributed in the HAp matrix.

Biaxial fractural strength of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
Figure 13 shows the microhardness of HAp and the CT-HAp composites. The hardness of the composite increased with increments of CT content from 2 to 8%wt. The 8%wtCT-HAp composite exhibited the greatest hardness and the microhardness of composites with 10wt% or more of CT was considerably lower, showing a similar trend to flexural strength (Fig. 12). The results indicated that the mechanical properties of the CT-HAp composite (including hardness) depended on the CT content. Ramírez et al., [45] suggested that the formation of hard CT particles (HV≈5 GPa) supported the reduction of HAp deformation. The hardness of the composite, like its flexural strength, was significantly impacted by porosity.

The microhardness of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents
Figure 14(a) shows a sketch of a radial-median crack and the imprint of the diamond tip on the specimen surface after Vickers hardness testing. SEM images (Fig. 14(b)–(e)) show the impact areas on HAp and CT-HAp composite samples after Vickers hardness testing. The images on the right-hand side are at higher magnification and show the opening of the crack after unloading. The cracks formed on the surface of pure HAp and 2 and 8%wtCT-HAp (Fig. 14(b)–(d)) were of the radial-median type. The cracks diffused along the specimen surface, and the Vickers indent also caused regions of plastic deformation due to residual stresses at the point of the indent [46, 47]. However, no crack system could be clearly identified on the 20%wtCT-HAp composite (Fig. 14(e)), and the residual imprint was larger than the imprint in the other composites. The SEM micrograph of the indented surface of the pure HAp sample (Fig. 14(b)) shows a mainly transgranular mechanism of fracture, but the fracture mechanism observed in all the composites (Fig. 14(c)–(e)) was less transgranular and the crack openings were narrower. The 2 and 8%wtCT-HAp composites exhibited greater fracture toughness (KIC) than the HAp sample, with values of 5.75 and 7.77 MPa.m1/2, respectively, compared to 4.66 MPa.m1/2. This enhanced toughness contributed to the resistance to crack-growth of the composites. Moreover, to confirm the resistance to cracking, the brittleness (B) of the materials was determined by considering hardness and KIC. The calculated B values of HAp and 2 and 8%wtCT-HAp composites were 0.94, 0.88 and 0.75 μm−1/2, respectively. According to a previous study [48], ceramics with low B and high KIC values were less brittle because the concentration of stresses was insufficient to cause cracking. The 8%wt-CT-HAp sample demonstrated the largest KIC. Crack propagation in this composite was restricted by homogenously dispersed CT agglomerations in the HAp matrix, while the crack opening was very narrow (Fig. 15). This finding was consistent with the finding of J.G. Miranda-Hernandez et al. [49], who reported that fracture toughness was increased by the presence of CT. On the other hand, the 20%wt-CT-HAp sample had a lower KIC (2.11 MPa.m1/2) and higher B (2.35 μm−1/2) than the other samples. These poor properties were attributed to high porosity, low hardness and uneven distribution of CT particles.

The sketch (a) shows the imprint of the diamond tip and a radial-median crack formed after hardness testing. Images (b), (c), (d), and (e) show residual imprints (left) with crack openings (right) of hydroxyapatite and calcium titanate-hydroxyapatite composites with calcium titanate contents of 2, 8 and 20%wt

SEM-EDX mapping of calcium titanate-hydroxyapatite composite with 8%wt of calcium titanate
Chemical solubility of CT-HAp composites
Figure 16 shows the weight remaining of HAp and CT-HAp composites soaked in SBF solution for various time periods. It can be seen from Fig. 16 that weight was not lost in a consistent pattern. After 28 days in SBF solution, the remaining weight of some samples was 99.97 ± 0.11% of the initial weight. The very low weight loss (< 0.50%) of the samples was due to the high crystallinity and strong negative surface potential produced by sintering [50]. Also, the hexagonal HAp obtained in this work is highly stable in biological environments. The results indicated that CT-HAp composites degrade slowly and may be best used to repair bone defects which need a slow degradation rate. However, certain samples, 8%wt CT-HAp, for example, showed a greater average weight loss than others in the case of 8%wt CT-HAp, 0.32% compared to 0.08–0.20%. It is possible that the as-sintered sample set contained more surface defects such as micro-cracks, small pores and roughness [51, 52], which increased solubility by increasing the area exposed to SBF [53, 54].

The weight remaining of hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents soaked in SBF solution for different time periods
Table 3 shows concentrations of Ca2+ and PO43− ions found in SBF solution after soaking HAp and CT-HAp samples for various time periods. The Ti4+ ion was not detected in any condition. It can be seen that concentrations of Ca2+ and PO43− ions were lower after 7 days of soaking compared to the concentration of both ions found in fresh SBF. Concentrations were higher at 15 days and higher still at 21 days, increasing from 67.75 ± 1.1.13 to 78.51 ± 0.28 ppm for Ca2+, and from 51.82 ± 1.21 to 59.15 ± 0.09 for PO43−. Concentrations of both ions were lower at 28 days than at 21 days since the ions precipitated onto the samples. Indeed, the dissolution of Ca2+ from the CT-HAp composite promoted the exchange of ions on its surface. The exchange between Ca2+ ion from the composite and PO43− ion from the SBF solution induced the surface precipitation of apatite [50, 55, 56].
The elemental compositions of sample surfaces after soaking in SBF solution were determined by EDX analysis. The results showed that Ca and P concentrations and the Ca/P ratio on sample surfaces tended to increase with soaking time (Table 4). However, little fluctuation was noticed in the case of Ti. The increases in Ca and P concentrations were mainly attributed to the precipitation of titanate (TiO32−) and phosphate to form apatite by a dissolution-precipitation reaction [57, 58]. When the samples were soaked in SBF solution, the negatively charged OH− and HPO42− ions combined with positively charged Ca2+ ions to form Ca-rich calcium phosphate. With the accumulation of Ca ions, the surfaces were positively charged, and PO43− ions could easily attach [59]. The Ca/P ratio was up to 2.26 times higher than the theoretical value (1.67) of HAp due to the initial high Ca concentration in both CT and HAp. Furthermore, the large Ca content was ascribed to the initial adsorption of Ca ions from CaCl2 in SBF solution through ionic interaction with negatively charged phosphate groups. The high Ca/P ratio on the sample surface was a result of bioactivity that was proposed as a positive development [60]. In addition, minor elements such as Mg, Na, Cl and K, were found on the specimen surface in some conditions. These elements were present in the fresh SBF solution and could precipitate on sample surfaces as Mg(OH)2 and NaCl [61, 62].
Dubey et al. [4] proposed that bone formation on the negatively charged surface of HAp (negative hydroxyl and phosphate groups) [11] was enhanced by osteoconductivity and that the accumulation of positively charged Ca2+ ions depended on electrical conductivity. Other studies of CT-HAp composites [7, 63] reported that the higher electrical conductivity of CT particles (3.2 × 10–5 S/m compared to 2.4 × 10–16 S/m for HAp) enhanced the electrical conductivity of the composite. The enhancement of electrical conductivity has been attributed to ionic polarization caused by the displacement between Ti3+ and O2− in CT [4] and supports the osseointegration and osteoconductivity of bone tissue. Therefore, CT-HAp composites that contain the optimal CT content can promote electrical conductivity to generate better bioactive properties than are exhibited by pure HAp.
In-vitro biological properties of CT-HAp composites
WST-1 assay results were obtained after growing MC3T3-E1 osteoblast cells on CT-HAp composites for three days. The 8%wtCT-HAp composite produced the highest cell viability (Fig. 17). Perhaps the microstructure of this composite was more compatible for cellular survival. SEM observation of 8%wtCT-HAp revealed a better distribution of CT particles in the HAp matrix. Other factors also influenced cell viability. The grain size and porosity of the composites were involved. The 8%wtCT-HAp composite had a small grain size and low porosity compared to 10–20%wtCT-HAp composites. Contact between the osteoblastic cells and the material surface was better and the levels of calcium and phosphate ions for apatite formation were higher [11, 12]. Moreover, the surface roughness of the material was correlated with the enhancement of polar components and cell attachment [64]. The surface morphology of 8%wtCT-HAp was perhaps more suitable for cell adherence and therefore cell survival.

Cell viability of MC3T3-E1 osteoblast cells after three days growing on hydroxyapatite and calcium titanate-hydroxyapatite composites with different calcium titanate contents. Results were analyzed by a one-way ANOVA and Scheffe’s post-hoc test. P values < 0.05 were considered statistically significant. *denotes a statistically significant difference with respect to control (0%wt of calcium titanate)
SEM images of osteoblast cells cultured on the sintered materials showed that all CT-HAp composites and pure HAp (0% wt) exhibited good cell attachment, without any sign of cytotoxicity. With increments of CT content up to 6%wt, cells were elongated and attached with long filopodia (Fig. 18). At CT contents from 8 to 20%wt, cells proliferated and spread with extended lamellipodia and filopodia. Cells could attach and form a cellular network on the composite surfaces, which indicated the biocompatibility of the composites and the promotion of tissue formation and growth. Although both HAp and the CT-HAp composites exhibited cell biocompatibility, the CT-HAp composite was more biocompatible than pure HAp since cellular proliferation and expansion were greater. The positive ion content of the composite surface and the surface morphology were factors that improved biocompatibilty [6, 63]. The presence of highly conductive CT particles in the HAp matrix activated bioaffinity and the rougher and more irregular surface favored cell attachment.

SEM images of MC3T3-E1 osteoblast cells attached to hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents after three days of culture
The surfaces of HAp and CT-HAp samples soaked in CGM for seven days were observed by SEM. When compared to the original surfaces (Fig. 7), certain changes were observed. The surfaces of HAp and CT-HAp composites were covered with rough layers that grew perpendicularly to the surfaces and these layers were deposited with increasing thickness as CT content increased (Fig. 19). EDX analysis indicated that the Ca/P ratio of sample surfaces increased from 2.50 for HAp to 3.63 for 20%wtCT-HAp. A previous report suggested [65] that calcium and phosphate ions obtained from culture medium and/or from the vicinity of degraded ceramics were possibly precipitated as new calcium-phosphate onto the material surface.

SEM images-EDX profiles show the surface morphologies and chemical compositions of calcium titanate-hydroxyapatite composites with different calcium titanate contents. The images and analyses were obtained after osteogenic induction in osteogenic induction medium for 24 h followed by soaking in complete growth medium for seven days
ALP, a common osteogenic marker, was used to evaluate the growth of MC3T3-E1 osteoblast cells cultured in CGM for seven days after osteogenic induction (Fig. 20). Since the original blue color of the stain, labeled with NBT/BCIP, was unchanged in ODM alone, no secretion of ALP occurred from the cells to ODM. Purple ALP staining indicated the presence of MC3T3-E1 cells on all HAp and CT-HAp samples. However, the adherence of the cells varied at different contents of CT. The visual evidence was consistent with the response to osteogenic induction by ALP activity (Fig. 21). Zhang et al. [66] reported that CT enhanced the attachment and induced the spread and proliferation of MC3T3-E1 cells, and promoted ALP activity. Extracellular Ca2+ ions from CaTiO3 increased intracellular Ca2+ ions, which induced the proliferation and differentiation of osteoblast cells [67, 68].

Alkaline phosphatase activity in response to osteogenic induction in MC3T3-E1 osteoblast cells on hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents was indicated by NBT/BCIP staining

Alkaline phosphatase (ALP) activity on hydroxyapatite and calcium titanate-hydroxyapatite composites at different calcium titanate contents and its variations in response to osteogenic induction in MC3T3-E1 osteoblast cells was assessed under a stereomicroscope. *denotes a statistically significant difference with respect to control (0%wt of calcium titanate)
Conclusions
Calcium titanate-hydroxyapatite (CT-HAp) bioceramic composites were synthesized with various CT contents (0–20%wt) and sintered conventionally at 1250 °C for 2 h in air. The phase formation, morphology, physical, mechanical and biological properties of the fabricated composites were examined. Based on XRD analysis, the composites formed a stable phase of calcium titanate and hydroxyapatite without any phase transformation. Microstructural observations revealed that increasing the CT content of the composite promoted the agglomeration of CT. Differences in the densification and porosity of composites with different CT contents could be explained by the presence of a non-densifying phase (CT). In addition, the grain size of the composites was slightly affected by CT content. However, the results showed that a CT-HAp composite containing 8%wt of CT exhibited the best mechanical properties (flexural strength, hardness and fracture toughness). The very low solubility of all composites after 28 days incubation indicated that the proposed composite degraded very slowly. In-vitro biological studies showed that osteoblast cells were viable, adhered, spread, proliferated and expressed alkaline phosphatase on the CT-HAp composites. Moreover, the cells covered a greater surface area of the composite with increments of CT. Thus, the mechanical properties, biocompatibility and cellular osteoinduction of the proposed CT-HAp composite are suitable properties for application in bone tissue engineering.
References
Inthong, S., Kamnoy, M., Intatha, U., Intawin, P., Pengpat, K., Tunkasiri, T., Eitssayeam, S.: Phase, mechanical and bioactivity properties of hydroxyapatite-calcium titanate composite. Mater. Res. Express. 6(2), 1–6 (2018). https://doi.org/10.1088/2053-1591/aaeec2
Hou, X., Zhang, L., Zhou, Z., Luo, X., Wang, T., Zhao, X., Lu, B., Chen, F., Zheng, L.: Calcium phosphate-based biomaterials for bone repair. Funct. Biomater. 13(187), 1–39 (2022). https://doi.org/10.3390/jfb13040187
Dubey, A.K., Tripathi, G., Basu, B.: Characterization of hydroxyapatite-perovskite (CaTiO3) composites: Phase evaluation and cellular response. J. Biomed. Mater. Res. B. Appl. Biomater. 95(2), 321–329 (2010). https://doi.org/10.1002/jbm.b.31716
Dubey, A.K., Mallik, P.K., Kundu, S., Basu, B.: Dielectric and electrical conductivity properties of multi-stage spark plasma sintered HA-CaTiO3 composites and comparison with conventionally sintered materials. J. Eur. Ceram. 33, 3445–3453 (2013). https://doi.org/10.1016/j.jeurceramsoc.2013.07.012
Mallik, P.K., Basu, B.: Better early osteogenesis of electroconductive hydroxyapatite-calcium titanate composites in a rabbit animal model. J. Biomed. Mater. Res. A 102A(3), 842–851 (2014). https://doi.org/10.1002/jbm.a.34752
Balinh, N.T., Mondal, D., Lee, B.T.: In vitro study of CaTiO3-hydroxyapatite composites for bone tissue engineering. ASAIO J. 60(6), 722–729 (2014). https://doi.org/10.1097/MAT.0000000000000126
Ravikumar, K., Mallik, P.K., Basu, B.: Twinning induced enhancement of fracture toughness in ultrafine grained hydroxyapatite-calcium titanate composites. J. Eur. Ceram. 36(3), 805–815 (2016). https://doi.org/10.1016/j.jeurceramsoc.2015.10.044
Ozturk, S., Yetmez, M.: Studies on characterization of bovine hydroxyapatite/CaTiO3 biocomposites. Adv. Mater. Sci. Eng. 2016, 1–7 (2016). https://doi.org/10.1155/2016/6987218
Inthong, S., Sweatman, D.R., Eitssayeam, S., Tunkasiri, T.: Influence of sintering temperature on physical and mechanical properties of hydroxyapatite-calcium titanate composite. Key Eng. Mater. 805, 65–70 (2019). https://doi.org/10.4028/www.scientific.net/KEM.805.65
Choy, M.T., Tang, C.Y., Chen, L., Law, W.C., Tsui, C.P., Lu, W.W.: Microwave assisted-in situ synthesis of porous titanium/calcium phosphate composites and their in vitro apatite-forming capability. Compos. B Eng. 83, 50–57 (2015). https://doi.org/10.1016/j.compositesb.2015.08.046
Trzaskowska, M., Vivcharenko, V., Przekora, A.: The impact of hydroxyapatite sintering temperature on its microstructural, mechanical, and biological properties. Int. J. Mol. Sci. 24(5083), 1–21 (2023). https://doi.org/10.3390/ijms24065083
Pires, L.A., de Meira, C.R., Tokuhara, C.K., de Oliveira, F.A., Dainezi, V.B., Zardin Graeff, M.S., Fortulan, C.A., de Oliveira, R.C., Puppin-Rontani, R.M., Borges, A.F.S.: Wettability and pre-osteoblastic behavior evaluations of a dense bovine hydroxyapatite ceramics. J. Oral Sci. 62(3), 259–264 (2020). https://doi.org/10.2334/josnusd.19-0007
Wu, S., Tu, B., Lin, J., Wang, Z., Wang, X., Shen, M., Hu, R.: Evaluation of the biocompatibility of a hydroxyapatite-CaTiO3 coating in vivo. Biocybern Biomed Eng. 35(4), 296–303 (2015). https://doi.org/10.1016/j.bbe.2015.05.001
Rodríguez, A.P., Sánchez, M.A., Felice, B., Zamora, M.L., Tsujigiwa, H., Takabatake, K., Kawai, H., Nakano, K., Nagatsuka, H.: In vitro efficacy of CaCO3 content in CaTiO3- CaCO3 composites for bone growth. J. Hard Tissue Biol. 27(3), 250–256 (2018). https://doi.org/10.2485/jhtb.27.250
Morris, J.P., Backeljau, T., Chapelle, G.: Shells from aquaculture: a valuable biomaterial, not a nuisance waste product. Rev. Aquac. 11(1), 42–57 (2019). https://doi.org/10.1111/raq.12225
Islam, K.N., Bakar, M.Z.B.A., Noordin, M.M., Hussein, M.Z.B., Abd Rahman, N.S.B., Ali, M.E.: Characterisation of calcium carbonate and its polymorphs from cockle shells (Anadara granosa). Powder Technol. 213(1–3), 188–191 (2011). https://doi.org/10.1016/j.powtec.2011.07.031
Mohamed, M., Yusup, S., Maitra, S.: Decomposition study of calcium carbonate in cockle shell. J. Eng. Sci. Technol. 7(1), 1–10 (2012). https://doi.org/10.1016/j.jclepro.2012.07.050
Hamester, M.R.R., Balzer, P.S., Becker, D.: Characterization of calcium carbonate obtained from oyster and mussel shells and incorporation in polypropylene. Mater. Res. 15(2), 204–208 (2012). https://doi.org/10.1590/S1516-14392012005000014
Seesanong, S., Boonchom, B., Chaiseeda, K., Boonmee, W., Laohavisuti, N.: Conversion of bivalve shells to monocalcium and tricalcium phosphates: An approach to recycle seafood wastes. Materials. 14(4395), 1–15 (2021). https://doi.org/10.3390/ma14164395
Azis, Y., Jamarun, N., Arief, S., Nur, H.: Facile synthesis of hydroxyapatite particles from cockle shells (Anadaragranosa) by hydrothermal method. Orient. J. Chem. 31(2), 1–7 (2015). https://doi.org/10.13005/ojc/310261
Azis, Y., Jamarun, N., Zultiniar, Arief, S., Nur, H.: Synthesis of hydroxyapatite by hydrothermal method from cockle shell (Anadara granosa). J. Chem. Pharm. Res. 7(5), 798–804 (2015)
Khiri, M.Z.A., Matori, K.A., Zainuddin, N., Abdullah, C.A.C., Alassan, Z.N., Baharuddin, N.F., Zaid, M.H.M.: The usability of ark clam shell (Anadara granosa) as calcium precursor to produce hydroxyapatite nanoparticle via wet chemical precipitate method in various sintering temperature. Springerplus. 5(1206), 1–15 (2016). https://doi.org/10.1186/s40064-016-2824-y
Ferdynanto, R.A., Dharmayanti, P.E.S., Dewi, P.T.K., Prananingrum, W.: The effect of various concentrations of HA-TCP derived from cockle shell synthesis on scaffold porosity. Dent. J. 51(3), 114–118 (2018). https://doi.org/10.20473/j.djmkg.v51.i3.p114-118
Saharudin, S.H., Shariffuddin, J.H., Nordin, N.I.A.A., Ismail, A.: Effect of aging time in the synthesis of biogenic hydroxyapatite derived from cockle shell. Mater. Today: Proc. 19(4), 1208–1215 (2019). https://doi.org/10.1016/j.matpr.2019.11.124
Fatimah, I., Fahrani, D., Harmawantika, T., Sahroni, I., Kamari, A., Rahmatillah, C.S., Nurillahi, R.: Functionalization of hydroxyapatite derived from cockle (Anadara granosa) shells into hydroxyapatite-nano TiO2 for photocatalytic degradation of methyl violet. Sustain. Environ. Res. 29(40), 1–14 (2019)
Mohd Puad, N.A.S., Te Chuan, L., Salman, N.S., Selimin, M.A., Abdul Latif, A.F., Muhamad, M.S., Abdullah, H.Z., Idris, M.I., Mustapha, S.N.H.: Synthesis of bioactive calcium phosphate from cockle shell for biomedical applications. Biointerface Res. Appl. Chem. 10(4), 5787–5791 (2020). https://doi.org/10.33263/BRIAC104.787791
Sinurat, E., Dewi, F.R., Fransiska, D., Nurbayasari, R.: Synthesis and characterization of hydroxyapatite of cockle shells (Anadara granosa) originated from Indonesia through precipitation method. IOP Conf. Ser.: Earth Environ. 1118(012035), 1–12 (2022). https://doi.org/10.1088/1755-1315/1118/1/012035
Mtavangu, S.G., Mahene, W., Machunda, R.L., Van der Bruggen, B., Njau, K.N.: Cockle (Anadara granosa) shells-based hydroxyapatite and its potential for defluoridation of drinking water. Results Eng. 13(100379), 1–16 (2022). https://doi.org/10.1016/j.rineng.2022.100379
Fatimah, I., Nur Ilahi, R., Pratami, R.: Low cost CaTiO3 perovskite synthesized from scallop (Anadara granosa) shell as antibacterial ceramic material. IOP Conf. Ser.: Mater. Sci. Eng. 299(012034), 1–6 (2022). https://doi.org/10.1088/1757-899X/299/1/012034
Ozawa, M., Hattori, M., Satake, K.: Waste management and application of fish bone hydroxyapatite for waste water treatment. Proc. of International Symposium on EcoTopia Science, (2007), p. 957–958
Cherdchom, S., Rattanaphan, T., Chanadee, T.: Calcium titanate from food waste: combustion synthesis, sintering, characterization, and properties. Adv. Mater. Sci. Eng. 2019, 1–10 (2019). https://doi.org/10.1155/2019/9639016
Maitreekeaw, K., Chanadee, T.: Calcium titanate ceramics obtained by combustion synthesis and two-step sintering. Sci. Sinter. 52, 491–502 (2020). https://doi.org/10.2298/SOS2004491M
Sri-o-sot, S., Vepulanont, K., Kamkit, C., Srichumpong, T., Chanadee, T.: Fabrication, characterization, and properties of hydroxyapatite ceramics derived from cockle shell. J. Aust. Ceram. Soc. 58, 1081–1093 (2022). https://doi.org/10.1007/s41779-022-00757-8
Ćorić, D., Ćurković, L., Majić Renjo, M.: Statistical analysis of vickers indentation fracture toughness of Y-TZP ceramics. Trans. Famena. 41(2), 1–16 (2017). https://doi.org/10.21278/TOF.41201
Callister, W.D., Rethwisch, D.G.: materials science and engineering: an introduction. Wiley, USA (2018)
Kokubo, T., Takadama, H.: How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27, 2907–2915 (2006). https://doi.org/10.1016/j.biomaterials.2006.01.017
Liu, X., Sun, Y., Chen, B., Li, Y., Zhu, P., Wang, P., Yan, S., Li, Y., Yang, F., Gu, N.: Novel magnetic silk fibroin scaffolds with delayed degradation for potential long-distance vascular repair. Bioact. Mater. 7, 126–143 (2022). https://doi.org/10.1016/j.bioactmat.2021.04.036
Louis, K.S., Siegel, A.C.: Cell viability analysis using trypan blue: manual and automated methods. In: Stoddart, M. (ed.) Mammalian cell viability. Methods in molecular biology, vol. 740, pp. 7–12. Humana Press, New York (2011). https://doi.org/10.1007/978-1-61779-108-6_2
Dogan, S.C., Polat, Z.A., Deren, S., Yayci, S.F.: Cetin. Effects of neuromuscular blocking drugs on viability of human umbilical vein endothelial cells (HUVECs). Int. J. Med. Res. 48(6), 1–7 (2020). https://doi.org/10.1177/0300060520910888
Chatree, K., Sriboonaied, P., Phetkong, C., Wattananit, W., Chanchao, C., Charoenpanich, A.: Distinctions in bone matrix nanostructure, composition, and formation between osteoblast-like cells, MG-63, and human mesenchymal stem cells, UE7T-13. Heliyon. 9, 1–16 (2023). https://doi.org/10.1016/j.heliyon.2023.e15556
Suchanek, W., Yoshimura, M.: Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J. Mater. Res. 13(1), 94–117 (1998). https://doi.org/10.1557/JMR.1998.0015
Xie, J., Ye, Q., Zhou, J., Liao, Y., Qian, G.: The photocatalytic activity of CaTiO3 derived from the microwave-melting heating process of blast furnace slag. Nanomaterials. 13(1412), 1–16 (2023). https://doi.org/10.3390/nano13081412
Bulina, N.V., Makarova, S.V., Baev, S.G., Matvienko, A.A., Gerasimov, K.B., Logutenko, O.A., Bystrov, V.S.: A Study of thermal stability of hydroxyapatite. Minerals. 11(1310), 1–15 (2021). https://doi.org/10.3390/min11121310
Gralik, G., Thomsen, A.E., Moraes, C.A., Raupp-Pereira, F., Hotza, D.: Processing and characterization of CaTiO3 perovskite ceramics. Process. Appl. Ceram. 8(2), 53–57 (2014). https://doi.org/10.2298/PAC1402053G
Ramírez, M.A., Parra, R., Reboredo, M.M., Varela, J.A., Castro, M.S., Ramajo, L.: Elastic modulus and hardness of CaTiO3, CaCu3Ti4O12 and CaTiO3/CaCu3Ti4O12 mixture. Mater. Lett. 64, 1226–1228 (2010). https://doi.org/10.1016/j.matlet.2010.02.058
Aswad, M.A.: Measurement of the fracture toughness and mechanical properties of hydroxyapatite using vickers indentation technique. Nanosistemi, Nanomateriali, Nanotehnologii. 19(3), 571–584 (2021)
Hervas, I., Montagne, A., Van Gorp, A., Bentoumi, M., Thuault, A.A.: lost. Fracture toughness of glasses and hydroxyapatite: A comparative study of 7 methods by using vickers indenter. Ceram. Inter. 42(11), 12740–12750 (2016). https://doi.org/10.1016/j.ceramint.2016.05.030
Quinn, J.B., Quinn, G.D.: On the hardness and brittleness of ceramics. Key Eng. Mater. 132–136, 460–463 (1997). https://doi.org/10.1002/9780470314821.ch7
Miranda-Hernandez, J.G., Vargas-Hernandez, J., Casarrubias-Vargas, H., Gonzalez-Moran, C.O., Refugio-García, E., de Jesús, J., Flores Cuautle, A.: Synthesis and effect of CaTiO3 formation in CaO.Al2O3 by solid-state reaction from CaCO3.Al2O3 and Ti. Mater. Chem. Phys. 232, 57–64 (2019). https://doi.org/10.1016/j.matchemphys.2019.04.050
Czikó, M., Bogya, E.S., Barabás, R., Bizo, L., Stefan, R.: In vitro biological activity comparison of some hydroxyapatite-based composite materials using simulated body fluid. Cent. Eur. J. Chem. 11(10), 1583–1598 (2013). https://doi.org/10.2478/s11532-013-0293-5
So, K., Fujibayashi, S., Neo, M., Anan, Y., Ogawa, T., Kokubo, T., Nakamura, T.: Accelerated degradation and improved bone-bonding ability of hydroxyapatite ceramics by the addition of glass. Biomaterials 27, 4738–4744 (2006). https://doi.org/10.1016/j.biomaterials.2006.05.020
Shuai, C., Yang, W., Feng, P., Peng, S., Pan, H.: Accelerated degradation of HAP/PLLA bone scaffold by PGA blending facilitates bioactivity and osteoconductivity. Bioact. Mater. 6, 490–502 (2021). https://doi.org/10.1016/j.bioactmat.2020.09.001
Vlădescu, A., Pârâu, A., Pană, I., Cotrut, C.M., Constantin, L.R., Vlădescu, A., Pârâu, A., Pană, I., Cotrut, C.M., Constantin, L.R.: In vitro activity assays of sputtered HAp coatings with SiC addition in various simulated biological fluids. Coatings 9(383), 1–17 (2019). https://doi.org/10.3390/coatings9060389
Jeong, J., Kim, J.H., Shim, J.H., Hwang, N.S., Heo, C.Y.: Bioactive calcium phosphate materials and applications in bone regeneration. Biomater. Res. 23(4), 1–11 (2019). https://doi.org/10.1186/s40824-018-0149-3
Hsu, P.Y., Kuo, H.C., Tuan, W.H., Shih, S.J., Naito, M., Lai, P.L.: Manipulation of the degradation behavior of calcium sulfate by the addition of bioglass. Prog. Biomater. 8, 115–125 (2019). https://doi.org/10.1007/s40204-019-0116-7
Sanchez, Mate, de Val, J.E., Calvo-Guirado, J.L., Gomez-Moreno, G., Perez-Albacete Martınez, C., Mazon, P., De Aza, P.N.: Influence of hydroxyapatite granule size, porosity, and crystallinity on tissue reaction in vivo. Part A: synthesis, characterization of the materials, and SEM analysis. Clin. Oral. Impl. Res. 27(11), 1331–1338 (2016). https://doi.org/10.1111/clr.12722
Pattanayak, D.K., Prasad, R.C., Rao, B.T., Rama Mohan, T.R.: Apatite wollastonite-titanium biocomposites: Synthesis and in vitro evaluation. J. Am. Ceram. Soc. 89(7), 2172–2176 (2006). https://doi.org/10.1111/j.1551-2916.2006.01068.x
Jamaludin, S.B., Shima Adzali, N.M., Nazree Derman, M.: Microstructure and in-vitro test bioactivity behavior of Co-CrMo (F-75)/hydroxyapatite in phosphate buffered saline solution. Acta Metall. Slovaca. 20(1), 82–88 (2014). https://doi.org/10.12776/ams.v20i1.192
Wan, Y.Z., Hong, L., Jia, S.R., Huang, Y., Zhu, Y., Wang, Y.L., Jiang, H.J.: Synthesis and characterization of hydroxyapatite-bacterial cellulose nanocomposites. Compos. Sci. Technol. 66, 1825–1832 (2006). https://doi.org/10.1016/j.compscitech.2005.11.027
Yang, Z., Xia, L., Li, W., Han, J.: Effect of Ca/P ratio on bioactivity of PEO coatings. Adv. Biomed. Eng. 2, 13–19 (2015). https://doi.org/10.15379/2409-3394.2015.02.01.2
Hassan Ibrahim, A.M., Takacova, M., Jelenska, L., Csaderova, L., Balog, M., Kopacek, J., Svastova, E., Krizik, P.: The effect of surface modification of TiMg composite on the in-vitro degradation response, cell survival, adhesion, and proliferation. Mater. Sci. Eng. C 127, 1–11 (2021). https://doi.org/10.1016/j.msec.2021.112259
Dziadek, M., Zagrajczuk, B., Menaszek, E., Cholewa-Kowalska, K.: A new insight into in vitro behaviour of poly(ε-caprolactone)/bioactive glass composites, in biologically related fluids. J. Mater. Sci. 53, 3939–3958 (2018). https://doi.org/10.1007/s10853-017-1839-2
Thrivikraman, G., Mallik, P.K., Basu, B.: Substrate conductivity dependent modulation of cell proliferation and differentiation in vitro. Biomaterials 34, 7073–7085 (2013). https://doi.org/10.1016/j.biomaterials.2013.05.076
Deligianni, D.D., Katsala, N.D., Koutsoukos, P.G., Missirlis, Y.F.: Effect of surface roughness of hydroxyapatite on human bone marrow cell adhesion, proliferation, differentiation and detachment strength. Biomaterials 22, 87–96 (2001). https://doi.org/10.1016/s0142-9612(00)00174-5
Zhang, J., Luo, X., Barbieri, D., Barradas, A.M.C., de Bruijn, J.D., van Blitterswijk, C.A., Yuan, H.: The size of surface microstructures as an osteogenic factor in calcium phosphate ceramics. Acta Biomater. 10(7), 3254–3263 (2014). https://doi.org/10.1016/j.actbio.2014.03.021
Zhang, Y., Wang, K., Dong, K., Cui, N., Lu, T., Han, Y.: Enhanced osteogenic activity of poly ether ether ketone using calcium plasma immersion ion implantation. Colloids Surf. B 187, 1–10 (2020). https://doi.org/10.1016/j.colsurfb.2016.02.056
Zayzafoon, M.: Calcium/calmodulin signaling controls osteoblast growth and differentiation. J. Cell. Biochem. 97, 56–70 (2006). https://doi.org/10.1002/jcb.20675
Hu, J., Zhou, Y., Huang, L., Liu, J., Lu, H.: Effect of nano-hydroxyapatite coating on the, osteoinductivity of porous biphasic calcium phosphate ceramics. BMC Musculoskelet. Disord. 15(114), 1–13 (2014). https://doi.org/10.1186/1471-2474-15-114
Acknowledgements
This work was financially supported by a Basic Research Grant from Prince of Songkla University under contract no. SCI6302056S and the Faculty of Science Research Fund, Prince of Songkla University, under contract no.1-2564-02-005. Sincere thanks and appreciation are extended to Mr. Thomas Duncan Coyne for editing and proofreading this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sri-o-Sot, S., Vepulanont, K., Pitakpornpreecha, T. et al. CaTiO3-hydroxyapatite bioceramic composite: Synthesis of reactant powders from waste cockle shell, sintering, characterization and investigation of physical, mechanical and in-vitro biological properties. J Aust Ceram Soc 60, 65–87 (2024). https://doi.org/10.1007/s41779-023-00987-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41779-023-00987-4