
Abstract
Vanadium dioxide (B-VO2) nanowires were successfully prepared under soft conditions by hydrothermal treatment. Structural analysis and phase identification have been carried out by means of X-ray diffractraction (XRD). The morphological properties of the samples have been examined by scanning electron microscopy (SEM). The XRD results revealed single phase oxide B-VO2 with considerable differences concerning to morphology and cristallinity as observed by SEM and XRD. Moreover, VO2(B) prepared for 96 h exhibited different morphology concerning to grain size and shape as well as dissimilar preference in crystal orientation, as a result of the processing conditions. The possible growth mechanism steps leading to nano-VO2(B) wires-like morphology was also proposed. The optical properties of the samples were evaluated by UV–visible spectroscopy and photoluminescence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One-dimensional metal oxides have experienced tremendous growth due to their potential application in various fields such as optical and electronics devices [1,2,3,4]. Among them, nanostructured vanadium oxides have been widely studied due to their particular electronic, chemical and optical properties [5,6,7,8,9,10,11].
The most abundant oxidation states of vanadium are known to be V2+, V3+, V4+ and V5+ characteristic of the oxides VO, V2O3, VO2 and V2O5, respectively. Nevertheless, intermediate phases like V3O5, V4O7, V5O9 and V6O11 can be produced [12, 13]. In the family of vanadium oxides, vanadium dioxide (VO2) is a representative binary compound with different polymorphs, including VO2(M), VO2(R), VO2(B), VO2(A), VO2(C), VO2(D), etc. Among the VO2 polymorphs, VO2(B), VO2(M/R) and VO2(A) have been paid much attention in the past decades [14,15,16,17]. Among these, only the rutile-type VO2 (R/M) undergoes a fully reversible metal–semiconductor phase transition (MST) at about 68°C [18]. These features make VO2(M) and its related compounds to be suitable for various applications [19,20,21,22], including smart window coatings, in optical switches, storage medium, temperature-sensing devices, laser protection, etc. Along with the phase transition, VO2’s optical and electrical properties simultaneously change as well [23]. Thus, the optical constants undergo a large change during the phase transition, causing a sharp drop in transmission. This change is most evident for the near infrared (NIR) region up to 2500 nm [24], thus allowing VO2 to be incorporated into smart window designs [25].
Recently rutile-type VO2 (M/R) nanomaterials have been grown by chemical vapor deposition [26], ion beam sputtering and so on [27, 28]. In another way, the rutile VO2 (M/R) can be synthesized by transforming from VO2 (B) to VO2 (M/R) by heat treatment [29]. Vanadium dioxide B-phase (B-VO2) has attracted great interest as a promising cathode material for Li-ion batteries due to its tunnel structure, through which the Li-ions can perform intercalation and deintercalation in reversible Li-ion battery [30,31,32]. It is also characterized by its reversible metal–insulator transition at 68 °C, which is accompanied by significant changes in optical properties with a transition from a transparent state at low temperature to a more opaque state at high temperature [33]. This material has been extensively studied since its discovery by Morin [34], both for understanding the mechanisms involved during the transition and for its innovative applications meeting energy needs. From a crystallographic point of view, the transition involves a transformation of two crystalline phases: the monoclinic (M) and the rutile (R) [35,36,37,38]. Chemically produced vanadium oxide can lead to the formation of metastable phases. This is the case of the two phases B-VO2 and A-VO2 which are obtained by hydrothermal treatment at low temperature. F. Theobald et al. [14] obtained the B-VO2 phase by hydrothermal treatment of a mixture of V2O3 and V2O5 at a temperature between 180 and 220 °C. Above 220 °C, B-VO2 transforms into A-VO2, then into R-VO2 from 350 °C. Since the observation of its phase transition by Morin [34], many studies have been carried out to integrate the VO2 oxide into electronic or optical devices. For example, smart windows [39,40,41], satellite thermal controllers [42, 43], gas detectors [44], electrodes for lithium batteries [45], solar collectors [46]. These various applications make VO2 a material with high potential, which is why it has been synthesized in various forms: nanoparticles [47], nanowires [48], nanorods [49], nanoplatelets [50], etc.
The physico-chemical properties depend not only on the structural but also on the morphology of the material as well as the size of the crystallites. In this context, several studies have been devoted to the synthesis of nanostructured B-VO2 phase and the improvement of their electrochemical activity [51,52,53,54,55,56]. So far, the synthesis of B-VO2 oxide is carried out from hydrothermal treatment or reduction of vanadium oxide precursors under different atmospheres. In fact, the hydrothermal process has many advantages by offering several experimental parameters such as: reaction time, temperature, autoclave filling rate, nature of the reducing and structuring agent. These parameters are often very useful to obtain materials with desired morphologies and to explore new phases.
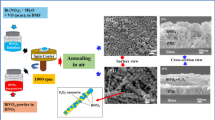
B-VO2 phase has attracted great interest as a promising cathode material for Li-ion batteries. The present study provides a rapid way to obtain B-VO2 under economical and environmentally friendly conditions. In fact, B-VO2 nanowires were successfully synthesized by one-step hydrothermal process at 180 °C for 96 h using V2O5 as vanadium source and 3-amino-1-propanol as reducing and structure-directing agent for the first time. The obtained samples were characterized by several techniques such as X-ray powder diffraction, scanning electron microscopy, Fourier transform infrared and Raman spectroscopy. In addition, the optical properties have been studied by UV–visible spectroscopy and photoluminescence.
Experimental
Hydrothermal synthesis
All the chemical reagents were procured from Acros Organic. Vanadium (V) oxide was used as vanadium source. The organic reagent 3-amino-1-propanol has been used as reducing and structure-directing agent for the first time. Nano-B-VO2 wires-like morphology was prepared by hydrothermal treatment of a mixture of V2O5, 3-amino-1-propanol and distilled water, in a molar ratio 2:1: 350. After a few hours of stirring, the mixture was heated under hydrothermal conditions for different reaction times. A mixture of a supernatant solution and a black precipitate was obtained. This was recovered, washed with distilled water and ethanol and then dried in air at 60°C then characterized by multiple physico-chemical techniques. The black coloring of the precipitate implies the existence of V4+ ions in the structure resulting from the reduction of V5+ ions by the oxidation of the organic compound [57].
In order to study the effect of the reaction time on the structural, morphological and optical properties several preparations were carried out.
Characterization techniques
Powder X-ray diffraction analysis was performed on a Panalytical X`Pert Pro diffractometer, with a continuous scan of CuKα radiation (λ = 1.5406 Å) and graphite monochromator in the 6—70° range. The morphology of the samples was examined using Stereoscan 120 scanning electron microscope. Infrared spectra were recorded from 4000 to 400 cm−1 on a Nicolet 380 spectrometer on pellets of the samples dispersed in KBr. A Raman Jobin Yvon T 64000 spectrometer was used to analyze chemical bonding between various elements. The optical properties of the samples were evaluated from UV–visible spectroscopy (Shimadzu-3101PC UV–vis spectrophotometer) and photoluminescence measurements (JobinYvon luminescence instrument).
Results and discussion
XRD analysis: structural study
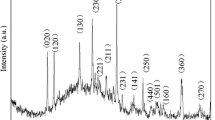
In order to determine certain experimental factors which can influence the structural properties, a study of the effect of the reaction time on the crystallinity was carried out. The crystallinity and the identification of the crystalline phases of the obtained powders after hydrothermal treatment were examined by X-ray diffraction. To do this, preparations were made for reaction times equal to 24, 48, 72 and 96 h. The X-ray diffractograms of the obtained samples are shown in Fig. 1a. It is clear that the reaction time plays an important role on the nature and the crystallinity of the material. Indeed, when the synthesis was carried out for 24 h, the powder obtained after hydrothermal treatment presents a diffractogram characteristic of an amorphous phase in the presence of a few diffraction peaks. Note that the increase in reaction time leads to the appearance of characteristic peaks of VO2(B). Indeed, when the hydrothermal treatment was carried out for 48 or 72 h, all the diffraction peaks have been identified at the monoclinic B-VO2 phase with the lattice parameters a = 12,030 Å; b = 3,693 Å; c = 6,420 Å according to JCPDS 31–1438, thus indicating the reduction of V5+ ions to V4+ ions by the organic precursor (reducing agent). However, when the hydrothermal treatment was carried out for 96 h, the diffractogram shows the presence of high intensity diffraction peaks which have been perfectly indexed to the crystalline B-VO2 phase (JCPDS 31–1438). It should be noted that no other diffraction peak related to the presence of other phases or impurities was detected, which indicates the purity of the crystalline B-VO2 phase produced under these conditions. This study shows that the reduction rate of vanadium ions is proportional to the reaction time. Thus, the characteristic peaks observed at 2θ values of 14.435°, 15.375°, 25.406°, 29.093°, 30.182, 33.933°, 44.116°, 45.149°, 53.884° and 59.164° are associated to (001), (200), (110), (002), (\(\overline{4 }\) 01), (\(\overline{3 }\) 01), (003) (\(\overline{5 }\) 01), (312), (601) and (\(\overline{7 }\) 01) plans of VO2(B) (JCPDS # 71–2248). According to F. Theobald [14], VO2(B) structure is formed by the packing of edge-sharing octahedra that are only linked at corners in the (001) plane, as shown inset in Fig. 2. There are two types of distorted oxygen octahedra in the structure, with vanadium atoms being shifted from the center [58, 59]. The compound VO2(B) crystallizes in space group C2/m and exhibits a three-dimensional framework of VO6 octahedra. This structure is formed by two layers of identical atoms along the [010] direction. Moreover, there are stacks of octahedral structure sharing the edges which are bound only by the corners in the (010) planes.

Powder X-ray diffraction patterns of the resulting products synthesized for different reaction times: 24 h, 48 h, 72 h and 96 h

FTIR spectra of the as-synthesized samples for 72 h and 96 h
Furthermore, the average crystallite size of the samples prepared for 24, 48, 72 and 96 h was determined using the Debye Scherrer formula below:
The results relating to the most intense peaks showed that the crystallites size decreases when the reaction time increases. In fact, this size goes from 94 nm for a sample prepared for 24 h to 43 nm when the synthesis was carried out for 96 h (Table 1). Thus, this study allowed us to observe that the size of the crystallites evolves exponentially with the reaction time.
FTIR spectroscopy
To investigate the chemical bonding between vanadium and oxygen ions and to confirm the phase purity, we performed FTIR spectroscopy. The infrared spectra of the as-synthesized samples at 180°C for different reaction time are shown in Fig. 2. Analysis of these reveals that all spectra are superimposed and show the same absorption bands. The attribution of the vibration bands of the vanadium–oxygen bonds is carried out from the structure of VO2(B) which comprises two types of octahedra [14]. The main vibrational bands observed from the FTIR spectra are at 530, 757, 934 and 997 cm−1 can be attributed to the various vibrational bands of vanadium–oxygen bonds. The absorption peaks among 1100 and 400 cm−1 are assigned to V–O vibration band in the infrared spectra [60, 61]. Indeed, the 995–1000 cm−1 band observed for B phase VO2 is present in many vanadium oxide compounds with intermediate oxidation state between V5+ and V4+. It is attributed to the symmetric stretching vibration of short V4+ = O bonds [59], while that observed around 757 cm−1 is attributed to the stretching vibration of the vibrator O–V–O connection [62] and that at 934 cm−1 is ascribed to V–O bonds according to J.-C. Valmalette [59]. However, the located band at around 530 cm−1 is attributed to the elongation vibration of the V–O–V octahedra bending modes [62,63,64]. Therefore, the vibration modes detected by infrared absorption spectroscopy are characteristic of vanadium oxide B-VO2 which is in agreement with the results of X-ray diffraction.
Raman spectroscopy
Typical Raman spectra, recorded at room temperature, of the samples synthesized for 72 and 96 h are shown in Fig. 3. In fact, the spectra exhibit a series of Raman bands in the range of 100 to 1000 cm−1 centered at 283, 405, 512, 688, 836, 870 and 990 cm−1, characteristics of the O–V–O and V–O–V bending modes and the V–O stretching modes [65,66,67]. Indeed, the detected bands at 280 and 405 cm−1 are attributed to the bending vibration of the O3–V = O bonds and to that of the V–O3–V bonds, respectively [67]. However, the observed band at 512 cm−1 is attributed to the triply coordinated oxygen stretching mode (V3–O) [67, 68]. Nevertheless, the band at 688 cm−1 corresponds to the stretching vibration of doubly coordinated oxygen (V2–O) [67,68,69,70,71]. However, the high frequency Raman band at 990 cm−1 is attributed to the terminal stretching mode of oxygen (V = O) which arises from unshared oxygen [67, 68]. Nevertheless, the spectrum of the sample prepared for 96 h reveals the presence of two narrow, well resolved and well defined bands at 836 and 873 cm−1 compared to that prepared for 72 h. These bands are attributed to the bending vibration of V–O-V groups involving doubly coordinated oxygen’s. In addition, no other bands related to the presence of other phases or impurities were detected, indicating the purity of the B-VO2 crystalline phase produced under these conditions, which synchronizes well with the XRD and EDX results. The narrowness and well-defined character of the bands indicate that the sample prepared for 96 h has a high crystallinity compared to that prepared for 72 h.

Raman spectra of the as-synthesized samples for 72 h and 96 h
SEM analysis: morphological study.
The evolution of the morphological properties of the prepared samples was carried out by scanning electron microscopy. Figure 4 illustrates the micrographs of the samples for different reaction time. It is clear that observation by scanning electron microscopy reveals an evolution of the morphology as a function of the reaction time. Indeed, when the synthesis was carried out for 24 h, the sample is formed of a heterogeneous phase consisting of a majority amorphous phase decorated on its surface by a few crystallized particles (Fig. 4a). By increasing the reaction time to 48 h, it is observed that the number of particles dispersed on the surface increases with the reaction time. Indeed, the analysis of the micrograph reveals that the material has two aspects, thus showing the existence of two different phases. In fact, it is a majority phase consisting of a few crystallized particles in the presence of an amorphous minority phase (Fig. 4b). When the synthesis is carried out for 72 h, the analysis of the SEM micrograph reveals a notable change in morphology and the particles evolve into wires (Fig. 4c). Thus the material obtained becomes more homogeneous than that prepared for 48 h. It can be said that the increase in reaction time in the hydrothermal bomb causes this noticeable change in morphology. This is confirmed by the fact that after 96 h of hydrothermal treatment (Fig. 4d), the obtained material consists of a homogeneous phase, formed by an assembly of wires, thus forming turns of nanowires whose average wire width is of the order of 20 µm and 50 nm thick (Fig. 5). These results confirm that the reaction time plays an important role with respect to the dispersion, the homogeneity and the shape of the material.

SEM micrographs of the resulting samples synthesized for different reaction times: 24 h (a), 48 h (b), 72 h (c) and 96 h (d)

SEM micrograph of B-VO2 phase synthesized for 96 h
To determine the chemical composition of the nanowires, we resorted to microanalysis using energy dispersive X-ray (EDX) analysis (Fig. 6). Thereby, the EDX spectrum reveals the presence of the elements oxygen and vanadium with a molar ratio very close to 2, which confirms the results obtained by X-ray diffraction. However, the weak signal corresponds to the carbon atom coming from the scanning electron microscope grating.

EDX spectrum of B-VO2 nanowires
Proposal mechanism formation of nano-VO2(B) wires-like morphology
In the formation process, 3-amino-1-propanol as a template was used in the reaction system and V2O5 was introduced as a vanadium source. Based on the layered structure of vanadium pentoxide and the experimental results, the 3-amino-1-propanol intercalating and splitting process was proposed to explain the formation of the 1D nanostructure. Then, as shown in Fig. 7, the interlayer spaces in the layered structure of V2O5 are occupied by molecules of 3-amino-1-propanol giving rise to an intermediate precursor of lamellar structure whose stability can be explained by the presence of 3-amino-1-propanol molecules inserted into the vanadium oxide interlayer space. Then, during the hydrothermal process and under the thermal effect, the intercalated molecules progressively deintercalate in a statistical way and the inorganic layers become destabilized. This allowed us to think about the breaking of bonds and the formation of others. Thus, the interactions between the layers are weakened and after heat treatment, the layered structure gradually splits to form VO2(B) nanowires.

Schematic representation of possible mechanism steps leading to nanowires of B-VO2
Uv–visible and photoluminescence study: optical properties
The optical properties of the samples prepared for 72 and 96 h were estimated by UV–visible spectroscopy. Thus from the UV–visible spectra it is possible to determine the value of the width of the forbidden band (the gap). Generally, the absorption bands observed in the 200–1000 nm range correspond to oxygen—vanadium Vn+ (O→Vn+) charge transfers or d-d type transitions, when the vanadium oxide exhibits a mixed valence [70,71,72,73,74]. The absorption spectra of the two samples are represented in Fig. 8 which shows a practically total superposition of the two spectra. Indeed, we note the presence of a band towards low wavelengths attributed to a charge transfer O→V5+ or V4+ where the vanadium ion is in an octahedral environment [70, 74,75,76].

UV–visible absorption spectra of the samples synthesized for 72 h and 96 h
Generally, vanadium oxides are direct gap semiconductors and the value of the gap (Eg) can thus be determined from the following equation [76,77,78]:
Figure 9 represents (αhv)2 as a function of hv of the samples prepared for 72 and 96 h. This study shows that the graph of (αhv)2 as a function of hv gives a straight line, which allows us to conclude that the optical transition is direct with a gap energy Eg = 3.511 eV and 3.614 eV for samples prepared for 72 h and 96 h, respectively. Indeed, due to the confinement of electrons and holes, the band gap energy increases between the valence band and the conduction band with decreasing particle size. This energy can be attributed to a direct allowed transition from the occupied O2p orbital of oxygen to the unoccupied 3d orbital of vanadium in B-VO2 oxide.

Variation of (αhν)2 with photon energy of the samples synthesized for 72 h and 96 h
The optical properties of the B-VO2 nanowires were also evaluated by photoluminescence (PL). Indeed, this technique makes it possible to explain the nature of the intrinsic defect in the structure of the B-VO2 phase. Figure 10a-b shows the photoluminescence spectra of samples prepared for 72 and 96 h. The results obtained reveal that the spectra obtained are similar with the presence of a series of bands around 363, 435, 457, 485 and 530 nm. Indeed, the emission band in the UV localized at 363 nm is attributed to free excitation emission [79]. Although the emission band at 435 nm is related to charge transfer from O2− to V5+ [72, 73]. However, the emission band observed at 455 nm is attributed to electric charge transfer and corresponds to the low energy of the V = O double bond [64]. However, the bands located around 485 nm and 530 nm seem to be attributed to energy defects within the material [79, 80].

The photoluminescence spectra of the B-VO2 synthesized for 72 h and 96 h
Conclusion
In summary, nano-VO2(B) wires-like morphology has been selectively synthesized via controlled and eco-friendly hydrothermally route using V2O5 as vanadium source and 3-amino-1-propanol as reducing and structure-directing agent for the first time. In this study, we found that the reaction time played an important role in the structural, the morphological and optical properties of the material. SEM micrographs revealed that the particles have a wire-like morphology with a uniform size distribution whose average wire width is of the order of 20 µm and 50 nm thick. The optical properties of the as-synthesized samples were investigated by UV–visible and photoluminescence spectroscopy. Optical band gap was found to vary with reaction time and crystallites size. Thus, when the hydrothermal treatment was carried out for 72 h, the gap is equal to 3.628 eV. However, it becomes equal to 3.774 eV when the synthesis has been performed for 96 h. Furthermore, we have used photoluminescence spectroscopy in order to explain the nature of intrinsic defects in the structure of the B-VO2 phase. In fact, this study allowed us to highlight the presence of the emissions bands relating to the emission of free excitation, to the charge transfer from O2− to V5+ as well as to the energy defects within the material. The formation mechanism of B-VO2 nanowires was also proposed and allowed us to understand the processes leading to the observed nanowires-like morphology.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated or analyzed during this study are included in this published article.
References
Zhou, D., Liu, S., Wang, H., Yan, Gu.: Na2V6O16·0.14H2O nanowires as a novel anode material for aqueous rechargeable lithium battery with good cycling performance. J. Power Sources 227, 111–117 (2013). https://doi.org/10.1016/j.jpowsour.2012.11.022
Salmaoui, S., Sediri, F., Gharbi, N.: Christian, Perruchot, Mohamed Jouin, Hexagonal hydrated tungsten oxide nanomaterials: Hydrothermal synthesis and electrochemical properties. Electrochim. Acta 108, 634–643 (2013). https://doi.org/10.1016/j.electacta.2013.07.086
Cui, C.-J., Wu, G.-M., Shen, J., Zhou, B., Zhang, Z.-H., Yang, H.-Y., She, S.-F.: Synthesis and electrochemical performance of lithium vanadium oxide nanotubes as cathodes for rechargeable lithium-ion batteries. Electrochim. Acta 55, 2536–2541 (2010). https://doi.org/10.1016/j.electacta.2009.12.022
Wang, Y., Zhang, Z.: Synthesis and field emission property of VO2 nanorods with a body-centered-cubic structure. Physica E 41, 548–551 (2009). https://doi.org/10.1016/j.physe.2008.10.006
Hong, W.G., Kim, J.M., Kim, H.J., Kim, B.H.: Exceptional electrochemical performance of two-year aged V2O5 nanowires for lithium storage. Curr. Appl. Phys. 15, 1488–1491 (2015). https://doi.org/10.1016/j.cap.2015.08.018
Tien, L.C., Chen, Y.J.: Influence of growth ambient on the surface and structural properties of vanadium oxide nanorods. Appl. Surf. Sci. 274, 64–70 (2013). https://doi.org/10.1016/j.apsusc.2013.02.092
Sharma, R.K., Kumar, P., Reddy, G.B.: Synthesis of vanadium pentoxide (V2O5) nanobelts with high coverage using plasma assisted PVD approach. J. Alloys Compds 638, 289–297 (2015). https://doi.org/10.1016/j.jallcom.2015.02.178
Ragupathy, P., Shivakumara, S., Vasan, H.N., Munichandraiah, N.: Preparation of Nanostrip V2O5 by the Polyol Method and Its Electrochemical Characterization as Cathode Material for Rechargeable Lithium Batteries. J. Phys. Chem. C 112, 16700–16707 (2008). https://doi.org/10.1021/jp804182z
Fei, H., Liu, X., Lin, Y., Wei, M.: Facile synthesis of ammonium vanadium oxide nanorods for Na-ion battery cathodes. J. Colloid. Interf. Sci. 428, 73–77 (2014). https://doi.org/10.1016/j.jcis.2014.04.029
Kim YK, Park SJ, Koo JP, Kim GT, Hong SH, and Ha JS (2007) Control of adsorption and alignment of V2O5 nanowires via chemically functionalized patterns, Nanotechnol. 18:015304-. https://doi.org/10.1088/0957‐4484/18/1/015304.
Yan, B., Liao, L., You, Y., Xu, X., Zheng, Z., Shen, Z., Ma, J., Tong, L., Yu, T.: Single-Crystalline V2O5 Ultralong Nanoribbon Waveguides. Adv. Mater. 21, 2436–2440 (2009). https://doi.org/10.1002/adma.200803684
Wriedt, H.A.: The O-V (oxygen-Vanadium) system. Bull. Alloy Phase Diagrams 10(3), 271–277 (1989). https://doi.org/10.1007/BF02877512
Ji, Y.D., Pan, T.S., Bi, Z., Liang, W.Z., Zhang, Y., Zeng, H.Z., Wen, Q.Y., Zhang, H.W., Chen, C.L., Jia, Q.X., Lin, Y.: Epitaxial growth and metal-insulator transition of vanadium oxide thin films with controllable phases. Appl. Phys. Lett. 101, 71902 (2012). https://doi.org/10.1063/1.4745843
Théobald, F., Cabala, R., Bernard, J.: Essai sur la Structure de VO2(B). J. Solid State Chem. 17, 431–438 (1976). https://doi.org/10.1016/S0022-4596(76)80013-8
Andersson, G.: Studies on Vanadium Oxides. II. The Crystal Structure of Vanadium Dioxide. Acta Chem. Scand. 10, 623–628 (1956). https://doi.org/10.3891/acta.chem.scand.10-0623
Westman, S.: Note on a Phase Transition in VO2. Acta Chem. Scand. 15, 217 (1961). https://doi.org/10.3891/acta.chem.scand.15-0217
Oka, Y., Sato, S., Yao, T., Yamamoto, N.: Crystal Structures and Transition Mechanism of VO2(A). J. Solid State Chem. 141, 594–598 (1998). https://doi.org/10.1006/jssc.1998.8025
Cao, C.X., Gao, Y.F., Luo, H.J.: Pure Single-Crystal Rutile Vanadium Dioxide Powders: Synthesis, Mechanism and Phase-Transformation Property. J. Phys. Chem. C 112, 18810–18814 (2008). https://doi.org/10.1021/jp8073688
Zhang, Y., Zhang, J., Zhang, X., Mo, S., Wu, W., Niu, F., Zhong, Y., Liu, X., Huang, C., Liu, X.: Direct preparation and formation mechanism of belt-like doped VO2(M) with rectangular cross sections by one-step hydrothermal route and their phase transition and optical switching properties. J. Alloys Compd. 570, 104–113 (2013). https://doi.org/10.1016/j.jallcom.2013.03.053
Zhang, Y., Zhang, X., Huang, Y., Huang, C., Niu, F., Meng, C., Tan, X.: One-step hydrothermal conversion of VO2(B) into W-doped VO2(M) and its phase transition and optical switching properties. Solid State Commun. 180, 24–27 (2014). https://doi.org/10.1016/j.ssc.2013.10.028
Gonçalves, A., Resende, J., Marques, A.C., Pinto, J.V., Nunes, D., Marie, A., Goncalves, R., Pereira, L., Martins, R., Fortunato, E.: Smart optically active VO2 nanostructured layers applied in roof-type ceramics tiles for energy efficiency, Solar Energy Mater. Solar Cells 150, 1–9 (2016). https://doi.org/10.1016/j.solmat.2016.02.001
Chen, X., Lv, Q., Yi, X.: Smart window coating based on nanostructured VO2 thin film. Optic 123, 1187–1189 (2012). https://doi.org/10.1016/j.ijleo.2011.07.048
Tao, Z., Han, T.-R.T., Mahanti, S.D., Duxbury, P.M., Yuan, F., Ruan, C.-Y., Wang, K., Wu, J.: Decoupling of Structural and Electronic Phase Transitions in VO2. Phys. Rev. Lett. 109, 166406 (2012). https://doi.org/10.1103/PhysRevLett.109.166406
Barker, A.S., Verleur, H.W., Guggenheim, H.J.: Infrared optical properties of vanadium dioxide above and below the transition temperature. Phys. Rev. Lett. 36, 2137 (1976). https://doi.org/10.1103/PhysRevLett.17.1286
Cui, Y., Ke, Y., Liu, C., Chen, Z., Wang, N., Zhang, L., Zhou, Y., Wang, S., Gao, Y., Long, Y.: Thermochromic VO2 for Energy-Efficient Smart Windows. Joule 2, 707–1746 (2018). https://doi.org/10.1016/j.joule.2018.06.018
Sahana, M.B., Dharmaprakash, M.S., Shivashankar, S.A.: Microstructure and properties of VO2 thin films deposited by MOCVD from vanadyl acetylacetonate. J. Mater. Chem. 12, 333–338 (2002). https://doi.org/10.1039/B106563G
Yi, X., Changhong, C., Luqin, L., Yingrui, W., Bifeng, X., Hongchen, W., Sihai, C.: A new fabrication method for vanadium dioxide thin films deposited by ion beam sputtering,. Infrared Phys Technol. 44, 137–141 (2003). https://doi.org/10.1016/S1350-4495(02)00187-1
Schlag, H.J., Scherber, W.: New sputter process for VO2 thin films and examination with MIS-elements and C-V-measurements. Thin Solid Films 366, 28–31 (2000). https://doi.org/10.1016/S0040-6090(00)00711-2
Tsang, C., Manthiram, A.: Synthesis of Nanocrystalline VO 2 and Its Electrochemical Behavior in Lithium Batteries. J. Electrochem. Soc. 144, 520 (1997). https://doi.org/10.1149/1.1837442
Wu, C.Z., Xie, Y.: Promising vanadium oxide and hydroxidenanostructures: from energy storage to energy saving. Energy Environ Sci 3, 1191–1206 (2010). https://doi.org/10.1039/C0EE00026D
Ni, J.A., Jiang, W.T., Yu, K., Gao, Y.F., Zhu, Z.Q.: Hydrothermal synthesis of VO2(B) nanostructures and application in aqueous Li-ion battery. Electrochim. Acta 56, 2122–2126 (2011). https://doi.org/10.1016/j.electacta.2010.11.093
Zhang, Y., Chen, C., Wu, W., Niu, F., Liu, X., Zhong, Y., Cao, Y., Liu, X., Huang, C.: Facile hydrothermal synthesis of vanadium oxides nanobelts by ethanol reduction of peroxovanadium complexes. Ceram. Int. 39, 129–141 (2013). https://doi.org/10.1016/j.ceramint.2012.06.001
Zylbersztejn, A., Mott, N.F.: Metal-insulator transition in vanadium dioxide. Phys. Rev. B 11, 4383–4395 (1975). https://doi.org/10.1103/PhysRevB.11.4383
Morin, F.J.: Oxides which show a metal-to-insulator transition at the Neel tem-perature. Phys. Rev. Lett. 34–36 (1959) https://doi.org/10.1103/PhysRevLett.3.34.
Zhang, S.D., Shang, B., Yang, J.L., Yan, W.S., Wei, S.Q., Xie, Y.: From VO2(B) to VO2(A) nanobelts: first hydrothermal transformation, spectroscopic study and first principles calculation. Phys. Chem. Chem. Phys. 13, 15873–15881 (2011). https://doi.org/10.1039/C1CP20838A
Liu, W.T., Cao, J., Fan, W., Hao, Z., Martin, M.C., Shen, Y.R., Wu, J., Wang, F.: Intrinsic optical properties of vanadium dioxide near the insulator-metal transition. Nano Lett. 11, 466–470 (2011). https://doi.org/10.1021/nl1032205
Qu, B.Y., Liu, L., Xie, Y., Pan, B.C.: Theoretical study of the new compound VO2(D). Phys. Lett. A 375, 3474–3477 (2011). https://doi.org/10.1016/j.physleta.2011.08.004
Sun, Y., Jiang, S., Bi, W., Long, R., Tan, X., Wu, C., Wei, S., Xie, Y.: New aspects of size-dependent metal-insulator transition in synthetic single-domain monoclinic vanadium dioxide nanocrystals. Nanoscale 3, 4394–4401 (2011). https://doi.org/10.1039/C1NR10976F
Chen, Z., Gao, Y., Kang, L., Du, J., Zhang, Z., Luo, H., Miao, H., Tan, G.: VO2-based double-layered films for smart windows: Optical design, all-solution preparation and improved properties. Sol. Energy Mater. Sol. Cells 95, 2677–2684 (2011). https://doi.org/10.1016/j.solmat.2011.05.041
Wang, C., Zhao, L., Liang, Z., Dong, B., Wan, L., Wang, S.: New intelligent multifunctional SiO2/VO2 composite films with enhanced infrared light regulation performance, solar modulation capability, and superhydrophobicity. Phys. Rev. B 18, 563–573 (2017). https://doi.org/10.1080/14686996.2017.1360752
Batista, C., Ribeiro, R.M., Teixeira, V.: Synthesis and characterization of VO2-based thermochromic thin films for energy-efficient windows. Phys. Rev. B 6, 301 (2011). https://doi.org/10.1186/1556-276X-6-301
Benkahoul, M., Chaker, M., Margot, J., Haddad, E., Kruzelecky, R., Wongc, B., Jamroz, W., Poinas, P.: Thermochromic VO2 film deposited on Al with tunable thermal emissivity for space applications. Sol. Energy Mater. Sol. Cells 95, 3504–3508 (2011). https://doi.org/10.1016/j.solmat.2011.08.014
Hendaoui, A., Émond, N., Dorval, S., Chaker, M., Haddad, E.: VO2-based smart coatings with improved emittance-switching properties for an energy-efficient near room-temperature thermal control of spacecrafts. Sol. Energy Mater. Sol. Cells 117, 494–498 (2013). https://doi.org/10.1016/j.solmat.2013.07.023
Strelcov, E., Lilach, Y., Kolmakov, A.: Gas Sensor Based on Metal-Insulator Transition in VO2 Nanowire Thermistor. Nano Lett. 9, 2322–2326 (2009). https://doi.org/10.1021/nl900676n
Luo, J.-Y., Cui, W.-J., He, P., Xia, Y.-Y.: Raising the cycling stability of aqueous lithium-ion batteries by eliminating oxygen in the electrolyte. Nat. Chem. 2, 760–765 (2010). https://doi.org/10.1038/nchem.763
Mercs, D., Didelot, A., Capon, F., Pierson, J.-F., Hafner, B., Pazidis, A., Föste, S., Reineke-Koch, R.: Innovative Smart Selective Coating to Avoid Overheating in Highly Efficient Thermal Solar Collectors. Energy Procedia 91, 84–93 (2016). https://doi.org/10.1016/j.egypro.2016.06.177
Lopez, R., Feldman, L.C., Haglund, R.F., Jr.: Size-Dependent Optical Properties of VO2 Nanoparticle Arrays. Phys. Rev. Lett. 93, 177403 (2004). https://doi.org/10.1103/PhysRevLett.93.177403
Guiton, B.S., Gu, Q., Prieto, A.L., Gudiksen, M.S., Park, H.: Single-Crystalline Vanadium Dioxide Nanowires with Rectangular Cross Sections. J. Am. Chem. Soc. 127, 498–499 (2005). https://doi.org/10.1021/ja045976g
Ji, S., Zhao, Y., Zhang, F., Jin, P.: Direct formation of single crystal VO2(R) nanorods by one-step hydrothermal treatment. J. Cryst. Growth 312, 282–286 (2010). https://doi.org/10.1016/J.JCRYSGRO.2009.10.026
Strelcov, E., Davydov, A.V., Lanke, U., Watts, C., Kolmakov, A.: In Situ Monitoring of the Growth, Intermediate Phase Transformations and Templating of Single Crystal VO2 Nanowires and Nanoplatelets. ACS Nano 5, 3373–3384 (2011). https://doi.org/10.1021/nn2007089
Sediri, F., Gharbi, N.: From crystalline V2O5 to nanostructured vanadium oxides using aromatic amines as templates. J. Phys. Chem. Solids 68, 1821–1829 (2007). https://doi.org/10.1016/j.jpcs.2007.06.012
Li, X., Chen, X., Han, C., Shi, C.: Hydrothermal synthesis and characterization of VO2(B) nanorods array. J. Cryst. Growth 309, 43–47 (2007). https://doi.org/10.1016/j.jcrysgro.2007.09.003
Armstrong, G., Canales, J., Armstrong, A.R., Bruce, P.: The synthesis and lithium intercalation electrochemistry of VO2(B) ultra-thin nanowires. J. Power Sources 178, 723–728 (2008). https://doi.org/10.1016/j.jpowsour.2007.11.028
Kong, L.F., Liu, Z.P., Shao, M.W., Xie, Q., Yu, W.C., Qian, Y.T.: Controlled synthesis of single-crystal VOx·nH2O nanoribbons via a hydrothermal reduction method. J. Solid State Chem. 177, 690–695 (2004). https://doi.org/10.1016/j.jssc.2003.08.035
Liu, J., Li, Q., Wang, T., Yu, D., Li, Y.: Metastable Vanadium Dioxide Nanobelts: Hydrothermal Synthesis, Electrical Transport, and Magnetic Properties. Angew Chem Int. Ed. 43, 5048–5052 (2004). https://doi.org/10.1002/anie.200460104
Sediri, F., Gharbi, N.: Nanorod B phase VO2 obtained by using benzylamine as a reducing agent. Mater. Sci. Eng. B 139, 114–117 (2007). https://doi.org/10.1016/j.mseb.2006.12.011
Bouhedja, L., Stenou, N., Maquet, J., Livage, J.: Synthesis of Polyoxovanadates from Aqueous Solutions. J. Solid State Chem. 162, 315–321 (2001). https://doi.org/10.1006/jssc.2001.9325
Leroux, Ch., Nihoul, G., Van Tendeloo, G.: From VO2(B) to VO2(R): Theoretical structures of VO2 polymorphs and in situ electron microscopy. Phys. Rev. B 57, 5111–5121 (1998). https://doi.org/10.1103/PhysRevB.57.5111
Valmalette, J.C., Gavarri, J.R.: High efficiency thermochromic VO2(R) resulting from the irreversible transformation of VO2(B). Mater. Sci. Eng. B 26, 168–173 (1998). https://doi.org/10.1016/S0921-5107(98)00148-2
Kong, D.B., Li, X.G., Zhang, W.B., Hai, X., Wang, B., Qiu, X.Y., Song, Q., Yang, Q.H., Zhi, L.J.: Encapsulating V2O5 into carbon nanotubes enables the synthesis of flexible high-performance lithium ion batteries. Energy Environ. Sci. 9, 906–911 (2016). https://doi.org/10.1039/C5EE03345D
Gilson, T.R., Bizri, O.F., Cheetham, N: ingle-crystal Raman and infrared spectra of vanadium (V) oxide. J. Chem. Soc. Dalton Trans. 291–294 (1973). https://doi.org/10.1039/DT9730000291.
Sediri, F., Gharbi, N.: Controlled hydrothermal synthesis of VO2(B) nanobelts. Mater. Lett. 63, 15–18 (2009). https://doi.org/10.1016/j.matlet.2008.08.022
Sediri, F., Touati, F., Gharbi, N.: From V2O5 foam to VO2(B) nanoneedles. Mater. Sci. Eng. B 129, 251–255 (2006)
Ponzio, E.A., Benedetti, T.M., Torresi, R.M.: Electrochemical and morphological stabilization of V2O5 nanofibers by the addition of polyaniline. Electrochim. Acta 52, 4419–4427 (2007). https://doi.org/10.1016/j.electacta.2006.12.023
Li, R.: Liu Ch-Y, VO2(B) nanospheres: hydrothermal synthesis and electrochemical properties. Mater. Res. Bull. 45, 688–692 (2010). https://doi.org/10.1016/j.materresbull.2010.02.021
Mjejri, I., Etteyeb, N., Sediri, F.: H2V3O8 nanobelts as a novel stable electrode material with good reversible redox performance. J. Alloys Compd. 611, 372–380 (2014). https://doi.org/10.1016/j.jallcom.2014.05.151
Faggio, G., Modafferi, V., Panzera, G., Alfieri, D., Santangelo, S.: Micro Raman and photoluminescence analysis of composite vanadium oxide/poly-vinyl acetate fibres synthesised by electro-spinning. J. Raman Spectrosc. 43, 761–768 (2012). https://doi.org/10.1002/jrs.3089
Baddour-Hadjean, R., Golabkan, V., Pereira-Ramos, J.P., Mantoux, A., Lincot, D.: A Raman study of the lithium insertion process in vanadium pentoxide thin films deposited by atomic layer deposition. J. Raman Spectrosc. 33, 631–638 (2002). https://doi.org/10.1002/jrs.893
Baddour-Hadjean, R., Marzouk, A., Pereira-Ramos, J.P.: Structural modifications of LixV2O5 in a composite cathode (0≤x≤2) investigated by Raman microspectrometry. J. Raman Spectrosc. 43, 153–160 (2012). https://doi.org/10.1002/jrs.2984
Lee, S.-H., Cheong, H.M., Seong, M.J., Liu, P., Tracy, C.E., Mascarenhas, A., Pitts, J.R., Deb, S.K.: Raman spectroscopic studies of amorphous vanadium oxide thin films. Solid State Ionics 165, 111–116 (2003). https://doi.org/10.1016/j.ssi.2003.08.022
Rama, N., Rao, M.S.R.: Synthesis and study of electrical and magnetic properties of vanadium oxide micro and nanosized rods grown using pulsed laser deposition technique. Solid State Commun. 150, 1041–1044 (2010). https://doi.org/10.1016/j.ssc.2010.01.049
Blasco, T., Concepción, P., Nieto, J.M.L.: Oxidative dyhydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A 157, 117–142 (1995). https://doi.org/10.1016/S0926-860X(97)00029-X
Larrubia, M.A., Busca, G.: An ultraviolet–visible–near infrared study of the electronic structure of oxide-supported vanadia–tungsta and vanadia–molybdena. Mater. Chem. Phys. 72, 337–346 (2001). https://doi.org/10.1016/S0254-0584(01)00329-7
Čapek, L., Adam, J., Grygar, T., Bulánek, R., Vradman, L., Kučrová, G.K., Čičmanec, P., Knotek, P.: Oxidative dehydrogenation of ethane over vanadium supported on mesoporous materials of M41S family. Appl. Catal. A 342, 99–106 (2008). https://doi.org/10.1016/j.apcata.2008.03.003
Naydenov, V., Tosheva, L., Sterte, J.: Spherical silica macrostructures containing vanadium and tungsten oxides assembled by the resin templating method. Microporous Mesoporous Mater. 55, 253–263 (2002). https://doi.org/10.1016/S1387-1811(02)00427-4
Enache, D.I., Richard, E.B., Ensuque, A., Verduraz, F.B.: Vanadium oxide catalysts supported on zirconia and titania: I. Preparation and characterization. Appl. Catal. A 278, 93–102 (2004). https://doi.org/10.1016/j.apcata.2004.09.032
Gontier, S., Tuel, A.: Characterization of vanadium-containing mesoporous silicas, Microporous. Mater. 5, 161–171 (1995). https://doi.org/10.1016/0927-6513(95)00057-G
Tauc, J., Grigorovici, R., Vancu, A.: Optical Properties and Electronic Structure of Amorphous Germanium. Phys. Status Solidi B 15, 627–637 (1966). https://doi.org/10.1002/pssb.19660150224
Anpo, M., Tanahashi, I., Kubokawa, Y.: Photoluminescence and photoreduction of vanadium pentoxide supported on porous Vycor glass. J. Phys. Chem. 84, 3440–3443 (1980). https://doi.org/10.1021/j100462a026
Anpo, M., Yamashita, H., Matsuoka, M., Park, D.R., Shul, Y.G., Park, S.E.: Design and development of titanium and vanadium oxide photocatalysts incorporated within zeolite cavities and their photocatalytic reactivities, J. Industrial. Eng. Chem. 6, 59–71 (2000)
Author information
Authors and Affiliations
Contributions
W. Ben Youssef: Conceptualization, Methodology, Data curation Original draft preparation. H. Nefzi: Visualization, Investigation, Supervision. S. Somrani: Supervision. F. Sediri: Validation, Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Youssef, W.B., Nefzi, H., Somrani, S. et al. B-VO2 nanowires: controlled hydrothermal synthesis and optical properties. J Aust Ceram Soc 60, 195–206 (2024). https://doi.org/10.1007/s41779-023-00940-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41779-023-00940-5