
Abstract
Carbonated hydroxyapatite (CHAp) was prepared from snail shells calcined at 850 °C (calcined powder) and a diammonium hydrogen phosphate (DHP) solution for bone material. The titration route of the preparation was varied (route A and route B) to have two products (CHApA and CHApB). In route A, DHP solution was titrated against calcined powder (CP) solution to produce CHApA, whereas in route B, CP solution was titrated against DHP solution to produce CHApB. Thermogravimetric analysis-differential scanning calorimetry (TGA–DSC) showed different thermal profiles for the CHAps, which resulted in different morphologies that influenced mineralization. The mineralization studies of the CHAps were performed in simulated body fluid (SBF) for 7, 14, and 21 days. The X-ray fluorescence (XRF) data of the SBF solution before and after CHAp immersion showed that CHApB has higher dissolution of Ca and P minerals on day 14 than CHApA. Similarly, on day 21, the mineral uptake by the CHAp was higher in CHApB than in CHApA. Scanning electron microscopy (SEM) images showed spheroids and apatite-like bundles in CHApB but were absent in CHApA. In addition, X-ray diffractometry (XRD) and Fourier transform infrared (FTIR) spectroscopy confirmed the growth of carbonated apatite that started from day 14 to day 21. The findings indicate that the titration routes have a great influence on the formation of a highly resorbable material for bone implant coatings and augmentation applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mineralization is a process of converting a material partly or wholly into inorganic material to improve its biological functions. Literature reports that mineralized biomaterials improve bone regeneration when compared to non-mineralized counterparts [1]. Normally in bone tissue engineering, growth factors including bone morphogenetic proteins (BMPs) and fibroblast growth factors (FGFs) are added to non-mineralized materials to illicit cellular activity but with the emergence of mineralized materials have reduced or eliminated the dosage of growth factors [2, 3]. Mineralized materials outperform non-mineralized materials, particularly in surface and mechanical characteristics that improve osteoconductivity and osteoinductivity [4]. The osteoconductive material stimulates the growth of apatite film on the surface of the material which is a crucial process for bone healing [5]. Moreover, the osteoconductive efficiency is focused on the reaction of the material with the physiological fluids through the exchange of ions such as Ca, Si, P, and Na for bond formation [6, 7]. Thus, the bond formation results in a bone-like apatite film which aids in fixation of bone tissue with the material surface [7,8,9,10,11].
Mineralization occurs naturally in a biological environment, but research has proven that synthetic approaches can be used to produce a material that can facilitate the process. The synthetic approach uses organic fillers or template matrices to induce crystallization of a crystalline inorganic phase that mimics the biological process [12]. Literature reports on the incorporation of hydroxyapatite, HAp, and HAp-based materials (mineralized material) into biomaterial scaffolds (e.g., collagen) to improve bone regeneration. Also, naturally derived and synthetic or composite scaffolds have been developed and modified through mineralization processes for bone regeneration. As a result, various efforts have been made to mimic biological mineralization in order to design novel synthetic materials with improved properties. For instance, iron and manganese have been incorporated into HAp to develop intrafibillar mineralized collagen scaffolds with an enhanced osteoinductivity [13]. The mineralization process is mainly done by simulated body fluid (SBF), sequential mineralization, mineralization by cells, or mineralization by direct incorporation [4, 14]. In SBF and sequential mineralization processes, the scaffold is incubated in SBF and mineralization (Ca2+ and PO3−) solutions, respectively. For the mineralization by cells, cells are cultured on the scaffold in vitro to form a mineral sheet. However, the mineralization by direct incorporation approach involves the treatment of the scaffold with a mineral (microscale and nanoscale) solution. Although all the mineralization strategies are used, SBF is commonly used because it has inorganic ion levels close to that of human blood plasma which can easily replicate the apatite on bioactive materials [15,16,17]. However, the mineralization depends greatly on the surface characteristics (geometry and texture) of the material produced by the synthetic procedure by varying preparation routes, temperature treatment, and dopant concentrations [18,19,20]. Literature reports on the preparation of several carbonated hydroxyapatite (which is a mineralized HAp-based material) using wet chemical precipitation method, but the variation of the titration routes on the material surface characteristics has not been studied extensively on mineralization.
Herein, the authors have synthesized a reactive carbonated hydroxyapatite (CHAp) material using calcined powder (CP: prepared from Achatina achatina snail shells) and diammonium hydrogen phosphate (DHP) solution as a bone material. Carbonated hydroxyapatite (CHAp) is typically prepared by combining three chemicals: calcium (Ca) precursor from calcium nitrate tetrahydrate [Ca(NO3)2·4H2O], phosphate (PO4) precursor from diammonium hydrogen phosphate [(NH4)2·HPO4], and carbonate (CO3) precursor from ammonium hydrogen carbonate [NH4·HCO3], resulting in a complicated procedure. Our CHAp preparation, on the other hand, is a simple procedure because we obtained calcium and carbonate precursors from the CP, which contains CaCO3 and Ca(OH)2. We also varied the titration route of the CHAp preparation to study the morphological effects on mineralization. Before mineralization, the CHAp thermal behaviour was studied using thermogravimetric analysis—differential scanning calorimetry (TGA–DSC), after which in vitro mineralization performed in SBF solution was studied using X-ray fluorescence (XRF). In addition, infrared (IR) spectroscopy, X-ray diffractometry (XRD), and scanning electron microscopy (SEM) were used to affirm the mineralization data.
Materials and methods
Materials
Shells of Achatina achatina (AA) were obtained from the Madina market in Ghana, and diammonium hydrogen phosphate [DHP: (NH4)2HPO4, 132.06 g/mol, 98.5%] was obtained from Kosdag Listed.
Calcination of AA shells
AA shells were fully cleaned with water and then dried in the sun for 6 h. The dried shells were ground into powder and calcined for 2 h at 850 °C to produce a calcined powder (CP) of a mixed phase of calcite and calcium hydroxide powder.
CHAp preparation
The carbonated hydroxyapatite (CHAp) was synthesized using calcined powder (CP) solution and diammonium hydrogen phosphate (DHP) solution by varying the titration route. For CP solution, a quantity of 5.7 g of CP was mixed with 150 ml of distilled water and stirred using a hot plate magnetic stirrer for 1 h to produce a CP solution. For DHP solution, a 5.9 g of DHP was mixed with 150 ml of distilled water to produce a DHP solution. The CHAp preparation route was varied (route A and route B) as illustrated in Fig. 1 to produce two CHAp materials (CHApA and CHApB). For route A, the DHP solution was added to the CP solution dropwise, and the resulting solution was agitated for 1 h at 40 °C using a hot plate magnetic stirrer. The resultant solution was then allowed to settle for 24 h to grow apatite crystals. After that, the solution was filtered, and the precipitate was dried in an oven for 2 h at 100 °C to obtain CHApA. For route B, CP solution was kept as the based material, whereas DHP solution was titrated against it, after which the same preparation conditions for route A were adopted to produce CHApB.

A schematic diagram showing CHAp preparation route. a RouteA producing CHApA. b route B producing CHApB
In vitro mineralization studies
An in vitro evaluation of the material was conducted in simulated body fluid (SBF) prepared using the ISO 23317 protocol, but HEPES was used to replace TRIS. SBF prepared with HEPES is more stable and has an ideal concentration of ions for mineralization studies [21]. SBF (pH 7.0) in a volume of 20 mL was poured into two separate falcon tubes. Under static conditions, 40 mg of the prepared materials (CHApA and CHApB) was immersed in 20 mL of SBF at 37 °C for 7, 14, and 21 days. The mineral levels in the SBF before and after CHApA and CHApB immersions were then determined using X-ray fluorescence (XRF) spectroscopy. The spectrometer uses a helium gas (flow rate 0.660 mL/min) to perform the analysis. The materials were dried in an oven for 1 h, and the apatite-like bundle identified using a scanning electron microscope (SEM; Thermo Sci., Phenom ProX). The images were taken at a working distance of 200 µm and an accelerating voltage of 15 kV.
Characterization techniques
X-ray diffractometry (PANalytical, Empyrean) equipped with CuKα radiation (~ 1.5406) was used to identify the phase composition and to determine the crystallite size using Scherrer’s equation. Raman spectroscopy (micro-Raman-532TEC-Ci spectrometer, Technospex) coupled with µ-soft 2.0 analytical software was used to affirm the phases of the uncalcined and calcined powders. The analysis was performed on the powders using a single-mode frequency-stabilized cobalt excitation laser (wavelength 532 nm and power 50 mW) at a spectral range of 100–3400 cm−1 with 7 cm−1 resolution. The CHApA and CHApB TGA–DSC profiles were obtained using the Q600 SDT thermal analyser (TA instrument). The thermal profiles were obtained at a temperature range of 22 to 850 °C at a heating rate of 10 °C/min under nitrogen gas at a flow rate of 100 cm3/min. The functional groups in CHApA and CHApB before and after immersion in SBF were identified using the Fourier transform infrared spectrometer (FTIR; Bruker, Tensor 27). The spectra were acquired over a wavenumber range of 400–4000 cm−1.
Statistical analysis
The relation between the mineral concentration of calcium and phosphorus groups at day 0, day 7, day 14, and day 21 of HApA and HApB in SBF was determined using the one-way analysis of variance (ANOVA) and Tukey’s post hoc test. At a confidence level of p > 0.05, statistical significance was determined. The data used were the means, and the errors were reported as the standard deviations of the means.
Results
XRD and Raman spectroscopy of uncalcined and calcined powders
Figure 2a and b show the X-ray diffractometry (XRD) patterns and Raman spectra of uncalcined and calcined AA powders, respectively. The uncalcined powders in Fig. 2a show phases of aragonite structure, whereas the calcined powder shows mixed phases of calcite and calcium hydroxide [22, 23]. In Fig. 2b, the Raman shifts occurring at 154, 207, 702, and 1086 cm−1 affirm the aragonite structure [24], whereas the shifts at 154, 283, 1086, and 356 cm−1 confirm calcite [24] and calcium hydroxide [25], respectively. The other Raman shifts occurring at 1305, 1754, and 2433 cm−1 are carbonate bonds [26, 27] in both uncalcined and calcined powders.

a, b XRD pattern and Raman spectra of raw uncalcined AA powder (dark) and calcined AA powder at 850 °C (red)
TGA/DSC characterization of CHApA and CHApB
Figure 3a and b show the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) profile of the carbonated hydroxyapatite prepared through titration route A (CHApA) and titration route B (CHApB), respectively. For CHApA, heat loss occurred from 6.95 to − 19.69 mW at 22.3 to 40.9 °C, and indicate an exothermic reaction. Heat was then gained gradually to − 1.70 mW at temperature range of 40.9–160.9 °C and remained fairly constant to a temperature of about 197 °C. The weight loss remained fairly constant to a temperature of about 400 °C, where weight loss occurs, and this is confirmed by the derivative weight loss. A slight upward peak in weight loss is observed at a temperature of about 500 °C, with a downward peak in the derivative weight also observed. Further weight loss of about 20.41% occurred from a temperature range of about 620 to 843.4 °C.The CHApA gradually losses energy of − 19.8 mW at a temperature of 701 °C. Furthermore, a rapid endothermal reaction occurs from 701 to 726 °C and followed by an exothermic reaction which occurs to 844.4 °C. For CHApB, Fig. 3b shows an increase in the heat flow from − 58.87 to 0.67 mW at 35.3 to 97.2 °C indicating an endothermic reaction. The heat flow remained fairly constant until 320 °C (1.01 mW) followed by exothermic reaction to − 20.92 mW at 709.0 °C. Another endothermic reaction occurred starting from 709.0 to 732.8 °C and followed by another exothermic reaction occurring at 844.4 °C. The DSC profile of CHApA and CHApB indicates three step transitions occurring at different temperature range with its respective weight loss as shown in Table 1. The variation in the heat flow of CHApA resulted in 79.54% total weight remaining whereas that of CHApB resulted in 81.06%.

TGA/DSC profile of a CHApA demonstrating exothermic reaction and b CHApB demonstrating endothermic reaction. The symbols I, II, and III indicate the step transitions in the reaction
XRF characterization of CHApA and CHApB
Figure 4 shows the X-ray fluorescence (XRF) data of mineral levels in SBF for before and after immersion for CHApA and CHApB. For CHApA in Fig. 4a, there was an uptake of mainly Na, S, Cl, and Ca for day 0 to day 7, and dissolute rapidly in day 14 and day 21. Ca and P are two key minerals for bone mineralization and thus were the further focus of the XRF analysis. Mainly calcium (Ca) levels increased insignificantly (p > 0.05) by 8.9% in day 7 and decreased significantly (p > 0.05) by 29.1% in day 14 and insignificantly (p > 0.05) by 24.5% in day 21 from day 14. Phosphorous (P) levels decrease insignificantly (p > 0.05) in the SBF by 5.8%, 11.3%, and 11.7% for day 0 to day 7, day 7 to day 14, and day 14 to day 21, respectively. For CHApB in Fig. 4b, the mineral levels show the same trend as that of CHApA. Ca levels increase insignificantly for day 0 to day 7 but increase significantly (p > 0.05) and decrease significantly (p > 0.05) by 57.5 and 55.3% from day 7 to day 14 and day 14 to day 21, respectively. Phosphorous (P) levels decrease significantly (p > 0.05) by 29.41%, increased significantly (p > 0.05) by 241.7%, and decrease significantly (p > 0.05) by 62.36% for day 0 to day 7, day 7 to day 14, and day 14 to day 21, respectively.

Mineral variation in SBF before and after immersion of a CHApA, b CHApB, and c Ca and P concentrations in a and b
FTIR and XRD characterization of CHApA and CHApB
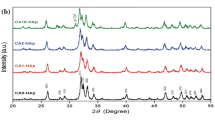
Figures 5 and 6 show the Fourier transform infrared (FTIR) spectra and X-ray diffraction patterns, respectively, for CHApA and CHApB before and after immersion in SBF. For FTIR spectra, CHApA before immersion (CHApA_Day0) shows functional groups of (PO4)3− \(\upsilon 1\) and \(\upsilon 3\) asymmetric stretching mode occurring at 959 and 1018 cm−1 whereas (PO4)3− \(\upsilon 4\) asymmetric bending mode occurs at 559 and 604 cm−1. There is (CO3)2− \(\upsilon 2\) asymmetric bending and (CO3)2− \(\upsilon 3\) stretching mode appearing at 873 and 1456 cm−1, respectively. Furthermore, bands at 1235 and 1742 cm−1 wavenumber likely correspond to \(\mathrm{C}-\mathrm{O}\) group and \(\mathrm{C}=\mathrm{O}\) of carbonate groups, respectively [28, 29]. Finally, hydrogen bonded to H2O molecules’ \(\upsilon 1\) and \(\upsilon 3\) stretching mode and structural hydroxy (OH−) groups occurs at 3405 and 3752 cm−1, respectively. Similar to the FTIR spectra of CHApA, the functional groups of CHApB occur at the same wavenumbers, but the (CO3)2− \(\upsilon 3\) stretching mode appearing at 1456 cm−1 for HApB_Day14 and CHApB_Day21 rises up than that of CHApA. For the XRD patterns, CHApA and CHApB in Fig. 6a and b show two phases, hydroxyapatite and calcite, and after 7 days of immersion, CHApB_Day7 show an additional phase, apatite, which continuous to grow to day 21 (see CHApB_Day14 and CHApB_Day21). Unlike CHApB, the apatite phase started after 14 days of CHApA immersion (CHApA_Day14). Other differences between the XRD patterns of CHApA and CHApB are the crystallite sizes as presented in Table 2. CHApA produces crystallite size larger than crystallite size produced by CHApB.

FTIR of CHAp before and after immersion in SBF. a CHApA. b CHApB. After immersion in day 7, the CO3)2− \(\upsilon 3\) stretching mode appearing at 1414–1456 cm.−1 begins to rise and becomes more pronounced in b than in a

XRD patterns of CHAp before and after immersion in SBF. a CHApA. b CHApB. The patterns indicate phases of calcite (o) and hydroxyapatite (•). After immersion in day 7, the peaks appearing at 31.9 and 45.5 2θ/° grow to become taller in a and b
SEM surface characterization of CHApA and CHApB
Figures 7 and 8 show the surface morphology of CHApA and CHApB before and after immersion in SBF for 7, 14, and 21 days. For CHApA, before immersion (Fig. 7a), the CHApA surface shows rod-like structures of diameter (5–10 µm), chunks of particles of random sizes and aggregates with voids. After immersion, in day 7 (Fig. 7b), the rod-like structures and the aggregates become more pronounce until day 14 (Fig. 7c), where the diameter of the rod-like structures becomes larger between 10 and 15 µm. In day 21 (Fig. 7d), the surface of the CHApA is more brighter with uniform chunks and aggregates to form clusters as magnified in Fig. 7d′. For CHApB, the surface of the CHApB before immersion in Fig. 8a is plane with microcracks and after immersion; in day 7 (Fig. 8b), the microcracks become dominant with bright chunks. Also, the surface shows microwells of bundles as magnified in Fig. 8b′. In day 14 (Fig. 8c), the CHApB surface shows massive chunks separated by voids with brighter spheroid aggregate to form spherical cluster as shown in Fig. 8c′. In day 21 (Fig. 8d), the surface is bright with few chunks and more bundles of random sizes magnified in Fig. 8d′.

SEM image of CHApA before and after immersion in SBF: (a) day 0, (b) day 7, (c) day 14, and (d) day 21. d′ is a magnification of d which shows a cluster of bright chuncks indicating apatite-like nucleation

SEM image of CHApB before and after immersion in SBF: (a) day 0, (b) day 7, (c) day 14, and (d) day 21. b′, c′, and d′ are magnifications of b, c, and d, respectively. b′ shows microwells, c′ shows aggregates of spheroids, and d′ shows bright chunks of apatite-like bundles
Discussion
The XRD revealed calcite and calcium hydroxide in the calcined powder (CP). However, when the CP water solution was titrated against the diammonium hydrogen phosphate (DHP) water solution, the calcium hydroxide reacted with the DHP to produce hydroxyapatite (HAp) and ammonium liquid. The reaction pathway is in Eq. (1). The calcite phase in the CP solution also dissociated into calcium and carbonate during stirring. The calcium from the calcite serves as a calcium precursor that contributes to the calcium from calcium hydroxide for the HAp formation. After filtration, the HAp is produced in association with carbonate. Further drying at 100 °C for 2 h substitutes carbonate into the HAp structure to replace phosphate, which yields carbonated hydroxyapatite (CHAp) material.
Surprisingly, when the CP solution is kept as the base and the DHP solution titrated against it (route A), the TGA–DSC profile of the material reveals different thermal properties as compared to when the DHP solution is kept as the base and the CP solution titrated against it (route B). Both route A and route B produce two CHAp materials, CHApA and CHApB, respectively. In a temperature range of 22.1 to 100 °C, the DSC for CHApA and CHApB produced exothermic and endothermic reactions, which were caused by how the surfaces adsorbed water. According to the literature, asperity (unevenness) on the surfaces of materials promotes water adsorption. [30,31,32]. The SEM images of CHApA show more surface asperity than CHApB, indicating that CHApA adsorbed more water into the surface than CHApB. Thus, the adsorbed water layer thickness was sufficient to form a possible ordered water structure via strong hydrogen bond interactions with the CHApA. However, the exothermic reaction at temperatures ranging from 22.3 to 41.6 °C is caused by ordered adsorbed water forming bonds with free anions and cations at the CHApA surface. XRF analysis confirmed significant levels of ions including chlorine, phosphorus, calcium, and sodium in the CHAp materials during dissolution in SBF. Thus, phosphorus oxychloride (POCl3), calcium chloride (CaCl2), calcium hydroxide (CaOH2), and sodium oxide (Na2O) are likely exothermic reactions that could occur on the CHApA surface. Following the formation of the bonds, the endothermic reaction at 41.6–100 °C indicates the evaporation of excess surface water. Also, the weight loss at a temperature of about 400 °C is attributed to the release of tightly bound water molecules (structural absorbed water) [33] in the CHAp structure. The peak at a temperature of about 500 °C in the TGA and derivative weight of CHApA are associated with the reactions such as oxidation [34] that take place in CHApA. The weight loss that occurred in the temperature range of 620 to 843.4 °C was due to the loss of some structural water and decomposition of residue [35] in both CHApA and CHApB. In CHApB, however, the adsorbed water layer thickness is thought to be lower, preventing interactions with surface ions. As a result, the CHApB was heated from 34.1 to 100 °C to evaporate the surface adsorbed water. The different thermal pathways resulted in weight loss that was inversely related to the crystallite sizes of the CHAp, which affects mineralization. The CHApA with the smallest weight loss (2.78%) and the largest crystallite size (28.13 nm) was unable to exchange phosphorus and calcium minerals in the SBF. The poor mineral exchange is caused by the larger crystallite size, which is available to the small volume of minerals in the SBF, as well as the exothermic reaction caused by bond formation, which consumes the majority of the surface minerals. Conversely, the CHApB with the greatest weight loss (3.88%) and smallest crystallite size (18.76 nm) allowed for phosphorous and calcium mineral exchange. The high dissolution of Ca and P in the SBF on day 14 indicates the formation of Ca-rich and P-rich minerals, whereas the CHApB surface becomes Ca-poor and P-poor. As shown in the SEM images, the poor CHAp surface interacted with the mineral-rich SBF and initiated the nucleation of the apatite (Fig. 8c). On day 21, the Ca and P levels in SBF drop significantly, indicating that the CHApB is consuming more minerals. The CHApB’s rapid uptake resulted in the formation of apatite-like bundles (see Fig. 8d). According to the literature, the majority of the carbonate in bone is B-type [36, 37]. However, the presence of carbonate functional groups at 1210, 1469, and 1742 cm−1 and the structural OH peak at 3752 cm−1 revealed by the IR spectra indicate a resemblance of the CHApA and CHApB to the B-type carbonate bone, with CHApB being more pronounced. Furthermore, the XRD shows phases of calcite (CaCO3) and apatite peaking at the 29.4 and 45.5 2θ/°, respectively, on days 7 to 21 in CHApB, supporting the IR’s claim that carbonate apatite-like bundle formation begins earlier in CHApB than in CHApA. This research work is in support of an earlier work where CHAp was annealed and, due to preferred orientation, produced smaller crystallite sizes that promote apatite bundle formation than unannealed CHAp [38]. Furthermore, literature indicates that biogenic apatite has crystallite sizes ranging from 10 to 35 nm [39]. The report confirms that the smaller crystallite size of biogenic apatite will have a higher surface area, allowing for greater adsorption of ions and molecules [39] before crystallites begin to enlarge in size [40], as in line with our results in Table 2. The findings show that the crystallite size of CHApB is smaller than CHApA and has higher mineral adsorption and promotes the apatite-like film formation than CHApA.
Conclusion
Carbonated hydroxyapatite was produced using calcined powder (CP) and diammonium hydrogen phosphate (DHP) solution. The titration route of the preparation was varied; in route A, CP solution was used as the base material and DHP solution was titrated against it to produce CHApA material, whereas in route B, DHP solution was used as the base material and CP solution was titrated against it to produce CHApB material. Thermal profiling revealed that the routes follow different reaction pathways and produce crystallites with varying sizes and morphologies, which affect mineralization. The CHApA and CHApB begin with exothermic and endothermic reactions, resulting in crystallite sizes of 28.13 and 18.76 nm, respectively. In vitro mineralization studies in simulated body fluid for 7, 14, and 21 days revealed that calcium and phosphorus dissolution was higher in CHApB than in CHApA on day 14. The lower dissolution in CHApA was attributed to the fact that some of the surface minerals were used for bond formation during the exothermic process. On day 21, the CHApB morphology revealed bright spheroids and apatite-like bundles of varying sizes, which were confirmed by X-ray diffractometry and infrared spectroscopy to be B-type carbonate apatite bundles. Day 21’s morphology, on the other hand, suggests apatite nucleation for CHApA. The routes produce a bone-like material, with CHApB being a fast-resorbing one that should be considered for bone implant coating and augmentation.
Data availability
Not applicable.
Code availability
Not applicable.
References
Patel, A., Zaky, S.H., Schoedel, K., Li, H., Sant, V., Beniash, E., Sfeir, C., Stolz, D.B., Sant, S.: Design and evaluation of collagen-inspired mineral-hydrogel nanocomposites for bone regeneration. Acta Biomater. 112, 262–273 (2020). https://doi.org/10.1016/j.actbio.2020.05.034
Carvalho, M.S., Cabral, J., da Silva, C.L., Vashishth, D.: Bone matrix non-collagenous proteins in tissue engineering: creating new bone by mimicking the extracellular matrix. Polymers (Basel). 13, 1095 (2021)
Rosowski, J.: Model for tooth development in vitro. Technische Universitaet Berlin (Germany) (2019)
Wu, X., Walsh, K., Hoff, B.L., Camci-Unal, G.: Mineralization of biomaterials for bone tissue engineering. Bioengineering 7, 132 (2020)
Liu, C., Ren, Z., Xu, Y., Pang, S., Zhao, X., Zhao, Y.: Biodegradable magnesium alloys developed as bone repair materials: a review. Scanning. 2018, (2018)
Valerio, P., Pereira, M.M., Goes, A.M., Leite, M.F.: Effects of extracellular calcium concentration on the glutamate release by bioactive glass (BG60S) preincubated osteoblasts. Biomed. Mater. 4, 45011 (2009)
Srinath, P., Abdul Azeem, P., Venugopal Reddy, K.: Review on calcium silicate-based bioceramics in bone tissue engineering. Int. J. Appl. Ceram. Technol. 17, 2450–2464 (2020). https://doi.org/10.1111/ijac.13577
Abinaya, B., Prasith, T.P., Ashwin, B., Viji Chandran, S., Selvamurugan, N.: Chitosan in surface modification for bone tissue engineering applications. Biotechnol. J. 14, 1900171 (2019)
Fattah-alhosseini, A., Chaharmahali, R., Keshavarz, M.K., Babaei, K.: Surface characterization of bioceramic coatings on Zr and its alloys using plasma electrolytic oxidation (PEO): a review. Surfaces and Interfaces. 25, 101283 (2021)
O’Sullivan, C., Kennedy, G., O’Neill, L., Crean, A.M., Ryan, K.B.: Inorganic biomaterials to support the formation and repair of bone tissue. Biomed. Appl. Inorg. Mater. 10, 242 (2021)
Kim, D., Cho, H.H., Thangavelu, M., Song, C., Kim, H.S., Choi, M.J., Song, J.E., Khang, G.: Osteochondral and bone tissue engineering scaffold prepared from Gallus var domesticus derived demineralized bone powder combined with gellan gum for medical application. Int. J. Biol. Macromol. 149, 381–394 (2020)
De Yoreo, J.: Research methods in biomineralization science. Elsevier (2013)
Yu, L., Rowe, D.W., Perera, I.P., Zhang, J., Suib, S.L., Xin, X., Wei, M.: Intrafibrillar mineralized collagen-hydroxyapatite-based scaffolds for bone regeneration. ACS Appl. Mater. Interfaces. 12, 18235–18249 (2020). https://doi.org/10.1021/acsami.0c00275
Wang, Q., Wu, M., Xu, X., Ding, C., Luo, J., Li, J.: Direct current stimulation for improved osteogenesis of MC3T3 cells using mineralized conductive polyaniline. ACS Biomater. Sci. Eng. 7, 852–861 (2020)
Sergi, R., Bellucci, D., Candidato, R.T., Jr., Lusvarghi, L., Bolelli, G., Pawlowski, L., Candiani, G., Altomare, L., De Nardo, L., Cannillo, V.: Bioactive Zn-doped hydroxyapatite coatings and their antibacterial efficacy against Escherichia coli and Staphylococcus aureus. Surf. Coatings Technol. 352, 84–91 (2018)
Solanke, S., Gaval, V., Sanghavi, S.: In vitro tribological investigation and osseointegration assessment for metallic orthopedic bioimplant materials. Mater. Today Proc. 44, 4173–4178 (2021)
Zamin, H., Yabutsuka, T., Takai, S., Sakaguchi, H.: A comparitive in vitro bioactivy evaluation of polyvinylidene fluoride and polycaprolactone incorporated with amorphous calcium phosphate particles. Phosphorus Res. Bull. 36, 15–22 (2020)
Baino, F., Yamaguchi, S.: The use of simulated body fluid (SBF) for assessing materials bioactivity in the context of tissue engineering: review and challenges. Biomimetics. 5, 57 (2020)
Shah, R., Gashi, B., Hoque, S., Marian, M., Rosenkranz, A.: Enhancing mechanical and biomedical properties of protheses—surface and material design. Surfaces and Interfaces. 27, 101498 (2021)
Dziaduszewska, M., Zieliński, A.: Structural and material determinants influencing the behavior of porous Ti and its alloys made by additive manufacturing techniques for biomedical applications. Materials (Basel). 14, 712 (2021)
Oyane, A., Kim, H., Furuya, T., Kokubo, T., Miyazaki, T., Nakamura, T.: Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. Part A An Off. J. Soc. Biomater. Japanese Soc. Biomater Aust. Soc. Biomater. Korean Soc. Biomater. 65, 188–195 (2003)
Asimeng, B.O., Afeke, D.W., Tiburu, E.K.: Biomaterial for bone and dental implants: synthesis of B-type carbonated hydroxyapatite from biogenic source. In: Biomaterials. IntechOpen (2020)
Liang, Z., Wang, Q., Dong, B., Jiang, B., Xing, F.: Ion-triggered calcium hydroxide microcapsules for enhanced corrosion resistance of steel bars. RSC Adv. 8, 39536–39544 (2018)
Borromeo, L., Egeland, N., Wetrhus Minde, M., Zimmermann, U., Andò, S., Madland, M.V., Korsnes, R.I.: Quick, easy, and economic mineralogical studies of flooded chalk for EOR experiments using Raman spectroscopy. Minerals. 8, 221 (2018)
Schmid, T., Dariz, P.: Shedding light onto the spectra of lime: Raman and luminescence bands of CaO, Ca (OH) 2 and CaCO3. J. Raman Spectrosc. 46, 141–146 (2015)
Asimeng, B.O., Amenyaglo, E.K., Dodoo-Arhin, D., Efavi, J.K., Kwakye-Awuah, B., Tiburu, E.K., Foster, E.J., Czernuska, J.: Snail based carbonated-hydroxyapatite material as adsorbents for water iron (II). Materials (Basel). 15, 3253 (2022)
Peccati, F., Bernocco, C., Ugliengo, P., Corno, M.: Properties and reactivity toward water of A type carbonated apatite and hydroxyapatite surfaces. J. Phys. Chem. C. 122, 3934–3944 (2018). https://doi.org/10.1021/acs.jpcc.7b12738
Jofre-Reche, J.A., García-Pacios, V., Costa, V., Colera, M., Martín-Martínez, J.M.: Role of the interactions between carbonate groups on the phase separation and properties of waterborne polyurethane dispersions prepared with copolymers of polycarbonate diol. Prog. Org. Coatings. 88, 199–211 (2015). https://doi.org/10.1016/j.porgcoat.2015.06.029
Mekhemer, G.A.H., Bongard, H., Shahin, A.A.B., Zaki, M.I.: FTIR and electron microscopy observed consequences of HCl and CO2 interfacial interactions with synthetic and biological apatites: influence of hydroxyapatite maturity. Mater. Chem. Phys. 221, 332–341 (2019). https://doi.org/10.1016/j.matchemphys.2018.09.007
Xiao, C., Shi, P., Yan, W., Chen, L., Qian, L., Kim, S.H.: Thickness and structure of adsorbed water layer and effects on adhesion and friction at nanoasperity contact. Colloids and Interfaces. 3, 55 (2019)
Tang, B., Tang, C., Chen, L., Xiao, C., Rosenkranz, A., Qian, L.: Nanoscopic humidity-dependent adhesion behaviors of 2D materials. Appl. Surf. Sci. 572, 151394 (2022)
Tong, C.Y., Derek, C.J.C.: Membrane surface roughness promotes rapid initial cell adhesion and long term microalgal biofilm stability. Environ. Res. 206, 112602 (2022)
Degli Esposti, L., Adamiano, A., Siliqi, D., Giannini, C., Iafisco, M.: The effect of chemical structure of carboxylate molecules on hydroxyapatite nanoparticles. A structural and morphological study. Bioact. Mater. 6, 2360–2371 (2021). https://doi.org/10.1016/j.bioactmat.2021.01.010
Zhang, S., Schoenitz, M., Dreizin, E.L.: Nearly pure aluminum powders with modified protective surface. Combust. Sci. Technol. 185, 1360–1377 (2013)
Lazić, S., Zec, S., Miljević, N., Milonjić, S.: The effect of temperature on the properties of hydroxyapatite precipitated from calcium hydroxide and phosphoric acid. Thermochim. Acta. 374, 13–22 (2001)
Yoder, C.H., Bollmeyer, M.M., Stepien, K.R., Dudrick, R.N.: The effect of incorporated carbonate and sodium on the IR spectra of A-and AB-type carbonated apatites. Am. Mineral. J. Earth Planet. Mater. 104, 869–877 (2019)
Lakrat, M., Elansari, L.L., Mejdoubi, E.: Synthesis of B-type carbonated hydroxyapatite by a new dissolution-precipitation method. Mater. Today Proc. 31, S83–S88 (2020)
Asimeng, B.O., Tiburu, E.K., Kan-Dapaah, K., Efavi, J.K., Asiamah, R., Afeke, D.W.: Influence of preferred orientation on the bioactivity of hydroxyapatite: a potential tooth repair and implant surface coating material. Cerâmica 66, 340–346 (2020)
Palmer, L.C., Newcomb, C.J., Kaltz, S.R., Spoerke, E.D., Stupp, S.I.: Biomimetic systems for hydroxyapatite mineralization inspired by bone and enamel. Chem. Rev. 108, 4754–4783 (2008). https://doi.org/10.1021/cr8004422
Kaya, Y., Jodati, H., Evis, Z.: Effects of biomimetic synthesis route and sintering temperature on physicochemical, microstructural, and mechanical properties of hydroxyapatite. J. Aust. Ceram. Soc. 57, 1117–1129 (2021)
Acknowledgements
The authors thank Mr. Solomon Kingsley Katu and Mr. Selassie Wonder King Hatekah of the University of Ghana’s Department of Biomedical Engineering for their laboratory assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Asimeng, B.O., Agyen, E.O., Asiamah, R. et al. Titration route affects biomimetic mineralization of carbonated hydroxyapatite bone material. J Aust Ceram Soc 58, 1321–1331 (2022). https://doi.org/10.1007/s41779-022-00772-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41779-022-00772-9