
Abstract
In this study, mesoporous strontium substituted hydroxyapatite (SrHAP) powders with individual sub-micrometric particle morphologies of irregular particles (SrHAP-IP) and nanostructured spherical particles (SrHAP-SP) were tested for vancomycin loading and releasing abilities. Initially, structural and physicochemical properties of synthesized products were comprehensively investigated. XRD analysis identified HAP as a chief phase in both SrHAP-IP and SrHAP-SP powders. Rietveld refinement suggested that HAP nanocrystals had appropriate lattice parameters and subjected to tensile strain. FESEM and HRTEM evaluations suggested that the particles of SrHAP-IP and SrHAP-SP powders were irregular (0.14 ± 0.04 μm) and nanostructured assembled burr-like spheres (0.88 ± 0.23 μm) in shape, respectively. SAED and lattice fringes corroborated the polycrystalline structure of both powders. EDX corroborated the formation of strontium substituted apatite in both powders. Isotherms suggested the mesoporous structure having specific surface area of 74.666 and 32.450 m2 g−1 for SrHAP-IP and SrHAP-SP powders, respectively. SrHAP-SP powder was 2.3 times more porous than SrHAP-IP powder. Accordingly, SrHAP-SP powder exhibited 44% higher vancomycin loading efficiency than SrHAP-IP powder. Although, both powders exhibited sustained drug release during longer incubation, but drug releasing rate of SrHAP-SP powder was relatively more than SrHAP-IP powder. Thus, this study made it apparent that difference in structural and physicochemical properties of SrHAP powders influenced their drug loading and releasing efficacies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among various bioceramic materials like calcium phosphate cements (CPC), hydroxyapatite [HAP, Ca10(PO4)6(OH)2], and β-tricalcium phosphate [β-TCP, β-Ca3(PO4)2] etc., HAP has been widely acclaimed for dental and orthopedic applications owing to excellent biocompatibility, osteoconductivity, and resemblance to components of human bone [1]. On the other hand, products composed of monolithic HAP phase are deficient in stimulating the development of new bone and that limits its clinical applications [2]. This shortcoming of monolithic HAP powders can be mitigated by employing several techniques such as incorporation of growth factors [1, 3], tailoring grain size and particle morphological designs [1, 4], and substitution of functional trace elements [1, 5] and so forth. A recently developed osteoporosis healing drug, i.e., strontium (Sr) ranelate has exhibited high potential in terms of stimulation of differentiation of osteoblast cells and inhibition of bone resorption functions, which significantly lessens the frequency of bone fractures in osteoporotic patients [1, 6]. Furthermore, the replacement of Ca atoms by Sr atoms significantly improves the biological abilities of apatitic powders [1, 7].
Over the past few years, efforts have been focused on the development of efficient carriers for loading and releasing applications of various molecules like anti-cancer drugs, antibiotics, growth factors, and so forth. The development of such controlled drug delivery systems is believed to offer multi-advantages such as gradual and long-term release of drugs, consequent improved therapeutic index, and causes minimum harmful effects to adjoining tissue(s) [8]. The carriers in drug delivery systems play a significant function as these store the drug and release it gradually with time. Among different drug delivery systems consisted of silica, graphene, and polymeric materials, mesoporous HAP bioceramic particles have been recognized as a novel and potential drug carrier material.
It has been well proposed that mesoporous structures of drug carriers significantly improve its drug loading capacity (DLC) as well as in vitro drug releasing abilities. Salient properties like considerable specific surface area of particles and their inherent pore volume significantly increase the DLC of such carriers. In addition, nanostructured mesoporous conduits in drug carriers act as transfer channels and enhance their drug loading and releasing abilities [8]. Such nanostructured mesoporous materials are superior to other particles [9]. Besides the mesoporous structure, particle morphology of drug carriers also critically influences the drug administration capacity and other intended biological functions [10]. For example, microspherical particles exhibited high loading and controlled release of vancomycin drug [8], nanorods were suggested as a novel contrast agent for both molecular imaging and photothermal cancer therapy [10], and superparamagnetic iron oxide-based nanoworms were studied for tumor-targeting applications [11] and so forth. On the other hand, owing to the complex crystal structure of HAP phase, tailoring the particle morphology of mesoporous HAP powders using customary synthesis routes is usually an intricate process [8, 12]. Consequently, synthesis of nanostructured HAP powders having specific particle morphologies using appropriate and profitable methods has also become a promising issue. It is believed that mesoporous strontium doped hydroxyapatite (SrHAP) powders having different particle morphologies are believed to exhibit better biological properties to be advantageous for futuristic drug delivery applications. Moreover, synthesis of mesoporous SrHAP powders with nanostructured hierarchical particles for drug delivery applications has been rarely studied.
Taking these aspects into consideration, the present study was designed to synthesize mesoporous SrHAP powders having individual particle morphologies of irregular (SrHAP-IP) and spherical (SrHAP-SP) shapes and thereby tested for their influence on drug loading and releasing efficiencies. The doping percentage of Sr was fixed at 5% by weight in both powders. Prior to drug loading and in vitro release testings, structural and physicochemical constitutions of synthesized products were comprehensively examined using different material characterization techniques. Rietveld refinement was employed to obtain the crystallographic information of products. Thus, this study conferred important insights involving relationships among structural, physicochemical, and drug loading and releasing abilities of mesoporous SrHAP powders.
Materials and method
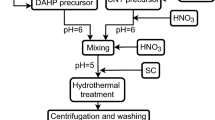
All chemicals were analytical reagents and used in as-received form. Experiments were conducted in ambient conditions. Calcium nitrate tetrahydrate (CNT, Merck, 99%), diammonium hydrogen orthophosphate (DAHP, Merck, 99%), and strontium nitrate (SN, Merck, 98.5%) were employed as Ca, P, and Sr ion sources, respectively. Disodium ethylene diamine tetra acetic (EDTA, Merck, 98%) and trisodium citrate (TSC, Merck, 99%) were used as complex agents. Ammonium hydroxide (NH4OH, Merck, 25%) and nitric acid (HNO3) were used to adjust the pH of solutions. Figure 1 summarizes the adopted synthesis protocols of SrHAP-IP and SrHAP-SP powders.

Synthesis protocols of a SrHAP-IP and b SrHAP-SP powders
Briefly, for the synthesis of SrHAP-IP powder, aqueous solutions of CNT (0.304 M) and EDTA (0.304 M) were gradually mixed together and formed solution A. Later, aqueous SN solution (0.02 M) followed by DAHP solution (0.1 M) was slowly added into solution A and formed SrHAP solution. The pH of this SrHAP solution was adjusted to nine using NH4OH. After vigorous stirring for 10 min, the resultant milky solution was irradiated at 800 W for 30 min in household MW (IFB, 2.45 GHz). After annealing, the solution was centrifuged at 5000 rpm for 5 min, washed repeatedly, and dried at 100 ± 2 °C for 24 h in an air oven. Prepared products were crushed using mortar and pestle.
For the synthesis of SrHAP-SP powder, 0.1 M DAHP solution of six pH (adjusted with HNO3) was rapidly added into a mixed solution of CNT (0.162 M) and SN (0.01 M) under vigorous stirring and formed SrHAP solution. The pH of this SrHAP solution was further adjusted to five using HNO3. A TSC solution (0.405 M) was dropwise added into SrHAP solution, and the mixture was vigorously stirred for 10 min. Later, this solution was stored into a 150-ml Teflon bottle, sealed in an autoclave, heated at 180 °C for 2 h, and annealed in an electric furnace. The precipitates were centrifuged at 5000 rpm for 5 min, washed with DDW, and dried at 100 ± 2 °C for 24 h in an air oven.
Characterization
The crystal structure of powders was studied using XRD (Bruker D8) equipped with CuKα radiation (λ = 154 pm, scan range = 10°–80°, step size 0.02°). MAUD 2.7 software was employed for the Rietveld refinement of diffraction patterns. The XRD patterns were modeled using polynomial function of fifth order and Pseudo-Voigt algorithm was adopted to study the shape of the diffracted peaks. The experimental and calculated profiles were refined until the profiles fitted closely. The JCPDS database for HAP (09-0432), β-TCP (09-0169), calcium nitrate tetrahydrate (CNT, CaH8N2O10, 01-070-0394), strontium calcium phosphate (SCP, Ca8.542Sr1.458(PO4)6(OH)2, 034-0483), and sodium chlorate dihydrate (SCD, NaClH2O5, 001-0603) were used for simulation purposes.
Debye Scherrer formula (Eq. 1) was used to calculate the crystallite size (Xs in nanometer) of HAP:
Where λ is the wavelength of X-rays, β (in radians) is the width at half maxima (FWHM), and θ (in degree) is Bragg’s angle. The diffraction peaks, i.e., (1 0 2), (3 1 1), (1 1 3), (2 1 3), and (0 0 4) and (1 0 1), (2 0 0), (1 1 1), (3 0 0), and (2 1 2) of HAP crystals were considered to calculate the crystal size of SrHAP-IP and SrHAP-SP powders, respectively. Crystal size, lattice strain, and Young’s modulus of HAP were calculated using Williamson-Hall-ISM model (Eq. 2) and Williamson-Hall-ASM model (Eq. 3).
Where ε is lattice strain and E is Young’s modulus of HAP crystals. The particle shape and elemental composition were studied using FESEM (S-4800, Hitachi) equipped with EDX (XFlash 4010, Bruker). HRTEM examination was conducted on FEI Tecnai G2 S-Twin transmission electron microscope. The particle size was calculated using ImageJ software [10]. The specific surface area and porosity of powders was evaluated by N2 adsorption-desorption (Autosorb-1-C, Quantachrome) using Branauer-Emmett-Teller (BET) method.
Drug loading test
Vancomycin was employed as a model drug. The 100 mg of SrHAP-IP and SrHAP-SP powders was separately dispersed into 5 ml of ethanol having drug concentration of 20 mg ml−1. The suspensions were shaken in a sealed vessel at 180 rpm incubated at 37 °C for 24 h. Thereafter, solutions were centrifuged for 5 min at 5000 rpm, and precipitate and supernatant were separated. The drug concentration in the supernatant was tested using UV-VIS at 230 nm wavelength. The drug loading efficiency (η) of powders was calculated using Eq. (4).
Where a and b denote the initial and final drug concentrations of the drug solution, respectively [13].
In vitro vancomycin release test
The 100 mg of drug-loaded powder was suspended in 50 ml of simulated body fluid (SBF) medium in separate glass bottles and incubated at 37 °C under sterile condition. After selected time intervals, 5 ml solution was taken out and centrifuged at 5000 rpm. The remaining solution in glass bottles was immediately refreshed with extra 5 ml of fresh SBF medium. Drug concentration in the supernatant was tested spectrophotometrically at a wavelength of 230 nm.
Results and discussion
Particle development mechanisms
The formation of irregular-shaped particles in SrHAP-IP powder can be described by nucleation-aggregation-growth mechanism theory as suggested by Sanosh et al. (2010) [14] and is schematically shown in Fig. 2a. This mechanism suggested that particle formation transformed through various steps including (a) nucleation of nanocrystallites; (b) aggregation of nanocrystals by molecular attractions; and (c) crystal growth and formation of agglomerates. The particle size increased by aggregation of agglomerated particles and formed the secondary particles.

Schematic diagrams showing particle formation mechanisms of a SrHAP-IP and b SrHAP-SP powders
For the synthesis of microspherical particles in SrHAP-SP powder, development of high temperature and high pressure in hydrothermal cell improved the nucleation-growth of SrHAP crystals [16]. Following nucleation, the adsorbed citrate ions on the surface of SrHAP crystals would change the surface energy of crystal facets and consequently influenced the growth of SrHAP crystals along the directions of adsorbed citrate ions [16, 17]. Furthermore, as the COO− groups in citrate ions matched that of Ca2+ ions oriented along c-axis in SrHAP crystals, the citrate ions caused the preferred orientated led growth of SrHAP along the c-axis [18]. This oriented growth of crystals led to the formation of long needle-like SrHAP particles as illustrated in Fig. 6d. Thereafter, the adsorbed citrate molecules self-assembled these SrHAP needle-like particles together by intermolecular hydrogen bonding which caused the orderly aggregation of particles and ultimately led to the formation of spherical particles [16]. Schematic representation showing development of SrHAP-SP powder is shown in Fig. 2b [19]. More details pertaining to the development of irregular and microspherical morphologies of HAP powders have been discussed elsewhere [14,14,15,16,17,18,19].
Phase structure
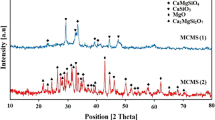
XRD patterns of SrHAP-IP and SrHAP-SP powders are shown in Fig. 3. Rietveld refinement suggested that both SrHAP-IP and SrHAP-SP powders were triphasic in composition. SrHAP-IP powder was composed of HAP, SCP, and β-TCP phases, whereas SrHAP-SP powder was consisted of HAP, CNT, and SCD phases. Narrow and high intensity diffraction peaks indicated good crystallinity of both powders [20]. Characteristic peaks of SCP phase were observed in SrHAP-IP powder only as shown in Fig. 3a. Presence of β-TCP phase indicated the calcium-deficient HAP (CDHAP) structure of SrHAP-IP powder. This calcium deficiency indicated that Ca2+ ions in HAP lattices were substituted by Sr2+ ions [21, 22]. On the other hand, surplus presence of CNT and SCD impurities in SrHAP-SP powder indicated that these could not be removed during centrifugation and washing process. Furthermore, disparity (in terms of different positions of the same peaks) between experimental and stoichiometric XRD patterns of HAP phase suggested that the Sr2+ ions replaced the Ca2+ ions in HAP crystals [1, 5]. Substitution of ions caused strain (Table 2) in lattices and deformation of lattice parameters, and hence was responsible for the peak-shift phenomena in the XRD pattern of SrHAP-SP powder. The speculated chemical reactions involved in the synthesis of SrHAP-IP and SrHAP-SP powders are proposed in Eq. (5) and Eq. (6), respectively.

XRD patterns of a SrHAP-IP and b SrHAP-SP powders
Crystallographic structure
The experimental, calculated, and difference profiles of XRD patterns of SrHAP-IP and SrHAP-SP powders are shown in Fig. 4. Table 1 enumerates the values of goodness of fit parameters (σ, Rwp, Rexp) pertaining to each powder. The values of σ parameter were less than four, and were acceptable as per the fundamental principle of the goodness of fit [23].

Experimental and calculated XRD profiles of a SrHAP-IP and b SrHAP-SP powders together with their difference profiles
The quantitative phase analysis of both powders suggested that HAP was a major phase, i.e., 82% in SrHAP-IP powder and 56% in SrHAP-SP powder. The weight% of β-TCP and SCP phases were 16 and 2%, respectively, in SrHAP-IP powder, whereas weight% of CNT and SCD phases were approximated to be 46 and 4%, respectively, in SrHAP-SP powder. Disparity between calculated (a = 10.203 Å and c = 6.956 Å) and stoichiometric (a = 9.418 Å and c = 6.884 Å) lattice parameters of HAP phase was observed in case of SrHAP-IP powder, whereas, in case of SrHAP-SP powder, HAP lattice parameters were relatively close to the stoichiometric lattice parameters (Table 2). The elongated dimensions of HAP lattices in SrHAP-IP powder were attributed to the substitution of smaller ionic radii Ca2+ ions by bigger ionic radii Sr2+ ions [1]. The ionic radius of Ca2+ is about 0.99 Å, while the ionic radius of Sr2+ ions is about 1.13 Å. Although, different models (Rietveld, Scherrer, WH-ISM, and WH-ASM) suggested different sizes of HAP crystals, but confirmed their nanodimensional size in both SrHAP-IP and SrHAP-SP powders. Both Rietveld and WH-ISM models suggested that HAP crystals in both powders were subjected to tensile strain. The elastic modulus of HAP was calculated to be 120 and 147 GPa for SrHAP-IP and SrHAP-SP powders, respectively.
Morphological and elemental structure
The particle morphologies of SrHAP-IP and SrHAP-SP powders are shown in Fig. 5. Nanodimensional irregular-shaped particles of SrHAP-IP powder were in agglomerated state as shown in Fig. 5a. Agglomeration is an inherent property of nanoparticles owing to their high surface energy. Average length and width of particles were 100 ± 26 and 43 ± 15 nm. On the other hand, particles of SrHAP-SP powder were burr-like spheres in shape consisted due to assembly of nanodimensional particles as shown in Fig. 5b. It has been assumed that primordial elongated particles assembled and formed nanostructured hierarchical spherical particles. Average diameter of these particles was 2 ± 0.4 μm. Assembly of these nanodimensional particles formed mesoporous structure, which was also corroborated by pore size distribution curves as shown in Fig. 8.

FESEM micrographs showing particle morphology of a SrHAP-IP and b SrHAP-SP powders
TEM micrographs confirmed the irregular morphology of particles of SrHAP-IP powder as shown in Fig. 6a. Average width of particles was measured to be 0.14 ± 0.04 μm. The bright spots indexed to (1 0 0), (1 1 1), (2 0 2), and (2 2 2) planes of HAP phase as shown in Fig. 6b corroborated the polycrystalline nature of SrHAP-IP powder. The interplanar distance between adjacent lattice fringes was 0.26 and 0.19 nm attributed to (2 0 2) and (2 2 2) planes of HAP crystals, respectively, as shown in Fig. 6c. Similarly, Fig. 6d shows spherical ended dumbbell-shaped particles consisted of hierarchically assembled long needle-like particles. Average diameter of spheres was 0.88 ± 0.23 μm. SAED pattern (Fig. 6e) confirmed the polycrystalline structure of SrHAP-SP powder, analogous to SrHAP-IP powder. The interplanar distance of 0.388 nm was attributed to (1 1 1) plane of HAP phase in SrHAP-SP powder.

HRTEM micrographs of (a-c) SrHAP-IP and (d-f) SrHAP-SP powders
EDX micrographs showing elemental compositions of SrHAP-IP and SrHAP-SP powders are shown in Fig. 7. Presence of elements such as Ca, P, and O required for apatitic formation were present in both powders. Presence of Sr element confirmed the formation of Sr-doped apatite. Atomic molar ratio and the weight ratio of Ca and P elements were 1.62 and 2.17 for SrHAP-IP powder and 1.57 and 2.35 for SrHAP-SP powder. On the other hand, Suganthi et al. [24] reported that the range of Ca/P molar ratio varied from 1.53 to 1.59 for HAP nanopowders doped with the different concentrations of strontium.

EDX micrographs showing elemental composition and corresponding quantitative analysis of a SrHAP-IP and b SrHAP-SP powders
Mesoporous structure
The adsorption-desorption isotherms, BET, BJH, and Langmuir plots of SrHAP-IP and SrHAP-SP powders are shown in Fig. 8. As per IUPAC standards, isotherms of both powders belonged to type II with H3 loop which suggested the formation of mesoporous product consisted of plate-like elongated particles [25]. Furthermore, high adsorption of gas at p/po 0.8–1.0 indicated the presence of macropores in both powders. These results were consistent with FESEM results (Fig. 5). Results of BET, BJH, and Langmuir analysis are mentioned in Table 3. According to BET results, specific surface area of SrHAP-IP and SrHAP-SP powders was 74.666 and 32.450 m2 g−1, respectively. In addition, SrHAP-SP powder was more porous (~ 2.3 times by volume according to both BET and BJH results) than SrHAP-IP powder. The formation of ultrafine pores having diameters from 2 to 12 nm corroborated the formation of mesoporous structure in both powders. The adsorbent was degassed at 200 °C for 3 h before conducting the test. Langmuir modeled specific surface area for both powders was more than the BET modeled specific surface area and results are given in Table 3. For reference purposes, values of both BET constant (C) and Langmuir constant (B) are also mentioned in Table 3. BET specific surface area of SrHAP-IP powder, i.e., 74.666 m2 g−1, was more than the specific surface area of 70.4 m2 g−1 for SrHAP nanopowder as reported by Zhang et al. [27]. Similarly, Lin et al. [1] reported the specific surface area of 29.71 m2 g−1 for SrHAP nanostructured microspheres that was lesser than the specific surface area of SrHAP-SP powder synthesized in the present study. Furthermore, Park et al. [28] reported that the specific surface area of SrHAP microspherical nanopowders decreased from 133 to 24 m2 g−1 with the increase in the concentration of strontium.

a N2 adsorption-desorption, b BET, c BJH, and d Langmuir plots of SrHAP-IP and SrHAP-SP powders
In vitro vancomycin loading and release behaviors
The vancomycin loading efficiency (η) and drug releasing profiles pertaining to both SrHAP-IP and SrHAP-SP powders are shown in Fig. 9. Vancomycin loading efficiency of SrHAP-SP powder was ~ 44% more than SrHAP-IP powder as shown in Fig. 9a [1]. The higher drug loading efficiency of SrHAP-SP powder was mainly attributed to its 3D hierarchical nanostructured particles having more volume of mesopores. These mesopores in the microspherical particles enhanced the specific surface area of particles, which improved the drug loading efficiency of SrHAP-SP powder [26]. In addition to improved diffusion of drug molecules, macropores also facilitated the space for attracting molecules of vancomycin drug [26]. The interactions between hydrogen bonding of OH− ions present in drug molecules and SrHAP powders increased the attachment of drug molecules to particles [8]. SrHAP-IP powder exhibited relatively low drug loading efficiency of 36 ± 3% than SrHAP-SP powder as shown in Fig. 9a. It was believed to be attributed to lesser specific surface area, lesser volume of porosity, and smaller pore diameters of SrHAP-IP powder. On the other hand, observed drug loading efficiency of 36% for SrHAP-IP powder was more than the ibuprofen loading efficiency of 32.9% as reported by Zhang et al. [27]. But, drug loading efficiency of SrHAP-SP powder was compared to be lower than the results reported by Lin et al. [1]. Park et al. [28] reported that drug loading efficiency decreased from 59 to 32% in hierarchically nanostructured SrHAP nanopowders with the increase in concentration of strontium.

a Drug loading efficiency (η) and b cumulative drug release profiles of vancomycin drug from SrHAP-IP and SrHAP-SP powders
The drug releasing profiles of SrHAP-IP and SrHAP-SP powders are shown in Fig. 9b. Both drug delivery systems exhibited similar drug releasing trends with good drug releasing ability without a burst-release effect. Furthermore, SrHAP-SP powder released more drug relative to SrHAP-IP powder. Cumulative drug releasing amount gradually increased up to 40 h and later exhibited stability in drug releasing rates. Such drug releasing profiles are beneficial for rapid delivery of drug required to provide antibacterial effects at the infected sites followed by sustained release to aid long-term healing [13]. The initial quick release might occured due to the weak adsorption of vancomycin molecules weakly attached to the outer surface of particles, whereas the sustained release of the drug could be due to the strong bonding between molecules of vancomycin and drug carrier [27]. The sites of OH− ions of carrier particles acted as the reaction sites, when vancomycin adsorbed on the surface [27]. During drug releasing, the SBF solvent entered the vancomycin-carrier phase through pores [27]. Therefore, the surface adsorbed drug was slowly dissolved into SBF and gradually diffused from the carrier system [27]. Thus, the slow and sustained releasing of vancomycin drug exhibited by both SrHAP-IP and SrHAP-SP powders demonstrated that both carriers can be suitably used as controlled drug delivery systems.
Conclusion
SrHAP powders having sub-micrometric dimensional irregular and hierarchically nanostructured sphere-like particles were successfully synthesized, comprehensively characterized, and tested for drug delivery applications. Both SrHAP-IP and SrHAP-SP powders were triphasic in composition with HAP as a major phase. Rietveld refinement presented the crystallographic design of both powders. Irregular-shaped particles of SrHAP-IP powder were in agglomerated state, whereas particles of SrHAP-SP powder were hierarchically nanostructured spheres in shape. Average particle size of SrHAP-IP and SrHAP-SP powders was 0.14 ± 0.04 and 0.88 ± 0.23 μm, respectively. SAED and HRTEM results corroborated the polycrystalline structure of both powders. Elemental composition also confirmed the formation of strontium-doped apatite in both powders. BET specific surface area for SrHAP-IP and SrHAP-SP powders was 74.666 and 32.450 m2 g−1, respectively. SrHAP-SP powder was more porous than SrHAP-IP powder. Owing to the mesoporous 3D hierarchical nanostructured morphology of SrHAP-SP powder, its vancomycin loading efficiency was 44% more than SrHAP-IP powder. Both drug delivery systems exhibited similar drug releasing trends and showed good drug releasing ability without a burst-release effect. The slow and sustained release of vancomycin drug demonstrated that both SrHAP-IP and SrHAP-SP powders could be suitably used as controlled drug delivery systems.
References
Lin, K., Liu, P., Wei, L., Zou, Z., Zhang, W., Qian, Y., Shen, Y., Chang, J.: Strontium substituted hydroxyapatite porous microspheres: surfactant-free hydrothermal synthesis, enhanced biological response and sustained drug release. Chem. Eng. J. 222, 49–59 (2013)
Yuan, H., Fernandes, H., Habibovic, P., de Boer, J., Barradas, A.M., de Ruiter, A., Walsh, W.R., van Blitterswijk, C.A., de Bruijn, J.D.: Osteoinductive ceramics as a synthetic alternative to autologous bone grafting. Proc. Natl. Acad. Sci. 107(31), 13614–13619 (2010)
Saranya, N., Saravanan, S., Moorthi, A., Ramyakrishna, B., Selvamurugan, N.: Enhanced osteoblast adhesion on polymeric nano-scaffolds for bone tissue engineering. J. Biomed. Nanotechnol. 7(2), 238–244 (2011)
Lin, K., Chen, L., Chang, J.: Fabrication of dense hydroxyapatite nanobioceramics with enhanced mechanical properties via two-step sintering process. Int. J. Appl. Ceram. Technol. 9(3), 479–485 (2012)
Lin, K., Zhou, Y., Zhou, Y., Qu, H., Chen, F., Zhu, Y., Chang, J.: Biomimetic hydroxyapatite porous microspheres with co-substituted essential trace elements: surfactant-free hydrothermal synthesis, enhanced degradation and drug release. J. Mater. Chem. 21(41), 16558–16565 (2011)
Landi, E., Tampieri, A., Celotti, G., Sprio, S., Sandri, M., Logroscino, G.: Sr-substituted hydroxyapatites for osteoporotic bone replacement. Acta Biomater. 3(6), 961–969 (2007)
Capuccini, C., Torricelli, P., Sima, F., Boanini, E., Ristoscu, C., Bracci, B., Socol, G., Fini, M., Mihailescu, I.N., Bigi, A.: Strontium-substituted hydroxyapatite coatings synthesized by pulsed-laser deposition: in vitro osteoblast and osteoclast response. Acta Biomater. 4(6), 1885–1893 (2008)
Long, T., Guo, Y.P., Liu, Y.Z., Zhu, Z.A.: Hierarchically nanostructured mesoporous carbonated hydroxyapatite microspheres for drug delivery systems with high drug-loading capacity. RSC Adv. 3(46), 24169–24176 (2013)
Chung, H.J., Park, T.G.: Surface engineered and drug releasing pre-fabricated scaffolds for tissue engineering. Adv. Drug Deliv. Rev. 59(4), 249–262 (2007)
Liu, Y., Tan, J., Thomas, A., Ou-Yang, D., Muzykantov, V.R.: The shape of things to come: importance of design in nanotechnology for drug delivery. Ther. Deliv. 3(2), 181–194 (2012)
Park, J.H., von Maltzahn, G., Zhang, L., Derfus, A.M., Simberg, D., Harris, T.J., Ruoslahti, E., Bhatia, S.N., Sailor, M.J.: Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting. Small. 5(6), 694–700 (2009)
Kothapalli, C.R., Wei, M., Legeros, R.Z., Shaw, M.T.: Influence of temperature and aging time on HA synthesized by the hydrothermal method. J. Mater. Sci. Mater. Med. 16(5), 441–446 (2005)
Venkatasubbu, G.D., Ramasamy, S., Ramakrishnan, V., Kumar, J.: Nanocrystalline hydroxyapatite and zinc-doped hydroxyapatite as carrier material for controlled delivery of ciprofloxacin. 3 Biotech. 1(3), 173–186 (2011)
Sanosh, K.P., Min-Cheol Chu, A., Balakrishnan, T.N., Kim, S.J.C.: Sol-gel synthesis of pure nano sized β-tricalcium phosphate crystalline powders. Curr. Appl. Phys. 10, 68–71 (2010)
Liu, J., Li, K., Wang, H., Zhu, M., Yan, H.: Rapid formation of hydroxyapatite nanostructures by microwave irradiation. Chem. Phys. Lett. 396(4), 429–432 (2004)
Yang, H., Hao, L., Du, C., Wang, Y.: A systematic examination of the morphology of hydroxyapatite in the presence of citrate. RSC Adv. 3(45), 23184–23189 (2013)
Zhang, C., Yang, J., Quan, Z., Yang, P., Li, C., Hou, Z., Lin, J.: Hydroxyapatite nano- and microcrystals with multiform morphologies: controllable synthesis and luminescence properties. Cryst. Growth Des. 9(6), 2725–2733 (2009)
Hu, Y.Y., Rawal, A., Schmidt-Rohr, K.: Strongly bound citrate stabilizes the apatite nanocrystals in bone. Proc. Natl. Acad. Sci. 107(52), 22425–22429 (2010)
Martins, M.A., Santos, C., Almeida, M.M., Costa, M.E.V.: Hydroxyapatite micro-and nanoparticles: nucleation and growth mechanisms in the presence of citrate species. J. Colloid Interface Sci. 318(2), 210–216 (2008)
Jiang, F., Wang, D.P., Ye, S., Zhao, X.: Strontium-substituted, luminescent and mesoporous hydroxyapatite microspheres for sustained drug release. J. Mater. Sci. Mater. Med. 25(2), 391–400 (2014)
Abert, J., Bergmann, C., Fischer, H.: Wet chemical synthesis of strontium-substituted hydroxyapatite and its influence on the mechanical and biological properties. Ceram. Int. 40(7), 9195–9203 (2014)
Kaygili, O., Keser, S., Kom, M., Eroksuz, Y., Dorozhkin, S.V., Ates, T., Ozercan, I.H., Tatar, C., Yakuphanoglu, F.: Strontium substituted hydroxyapatites: synthesis and determination of their structural properties, in vitro and in vivo performance. Mater. Sci. Eng. C. 55, 538–546 (2015)
Wu, E., Kisi, E.H., Gray, E.M.A.: Modelling dislocation-induced anisotropic line broadening in Rietveld refinements using a Voigt function. II. Application to neutron powder diffraction data. J. Appl. Crystallogr. 31(3), 363–368 (1998)
Suganthi, R.V., Elayaraja, K., Joshy, M.A., Chandra, V.S., Girija, E.K., Kalkura, S.N.: Fibrous growth of strontium substituted hydroxyapatite and its drug release. Mater. Sci. Eng. C. 31(3), 593–599 (2011)
Mohammadi, M.R., Fray, D.J.: Synthesis of highly pure nanocrystalline and mesoporous CaTiO3 by a particulate sol-gel route at the low temperature. J. Sol-Gel Sci. Technol. 68(2), 324–333 (2013)
Guo, Y.P., Guo, L.H., Yao, Y.B., Ning, C.Q., Guo, Y.J.: Magnetic mesoporous carbonated hydroxyapatite microspheres with hierarchical nanostructure for drug delivery systems. Chem. Commun. 47(44), 12215–12217 (2011)
Zhang, C., Li, C., Huang, S., Hou, Z., Cheng, Z., Yang, P., Peng, C., Lin, J.: Self-activated luminescent and mesoporous strontium hydroxyapatite nanorods for drug delivery. Biomaterials. 31(12), 3374–3383 (2010)
Park, S.Y., Kim, K.I., Park, S.P., Lee, J.H., Jung, H.S.: Aspartic acid-assisted synthesis of multifunctional strontium-substituted hydroxyapatite microspheres. Cryst. Growth Des. 16(8), 4318–4326 (2016)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Singh, R.P., Singh, G. & Singh, H. Sub-micrometric mesoporous strontium substituted hydroxyapatite particles for sustained delivery of vancomycin drug. J Aust Ceram Soc 55, 405–414 (2019). https://doi.org/10.1007/s41779-018-0248-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41779-018-0248-6