
Abstract
The hydrogen bond has justifiably been termed the ‘master key of molecular recognition’. It is an interaction that is weaker than the covalent bond and stronger than the van der Waals interaction. The ubiquity and flexibility of hydrogen bonds make them the most important physical interaction in systems of biomolecules in aqueous solution. Hydrogen bonding plays a significant role in many chemical and biological processes, including ligand binding and enzyme catalysis. In biological processes, both specificity and reversibility are important. Weaker interactions can be made and broken more easily than stronger interactions. In this context, it is of interest to assess the relative significance of strong and weak interactions in the macromolecular recognition processes. Is protein–ligand binding governed by conventional, that is, electrostatic N–H…O and O–H…O hydrogen bonds, or do weaker interactions with a greater dispersive component such as C–H…O hydrogen bonds also play a role? If so, to what extent are they significant? Most proteins, involving as they do, main chains, side chains, and differently bound forms of water, do not really have a static fixed structure, but rather have a dynamic, breathing nature. This tendency may to some extent be lessened by the ligands which are small molecules, but in the end, it is reasonable to expect that the strong and weak hydrogen bonds inside the protein and also at the protein–ligand interface will also have dynamic character; arguably, the weaker the hydrogen bond, the greater its dynamic character. These are often central to the much debated mechanisms of binding such as conformational selection and induced fit. All protein–ligand interactions must compete with interactions with water; both the protein and the ligand are solvated before complexation and lose their solvation shell on complex formation. Conversely, the entropic cost of trapping highly mobile water molecules in the binding site is large. However, in favorable cases, these losses are suitably compensated by the enthalpic gain resulting from water-mediated hydrogen bonds. In effect, the enthalpy–entropy balance is a fine one, and for a water molecule to be able to contribute to binding affinity, it has to be in a binding site that provides the maximum number of hydrogen-bond partners at the optimum distance and orientation. In summary, hydrogen bonds are crucial to the recognition of ligands by proteins. Integration of knowledge gained from more high-quality protein–ligand structures into theoretical and computational molecular models will be an exciting challenge in the coming years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In this centenary year of the hydrogen bond, it appears that there is no end in sight to the limitless possibilities that are afforded by this very special interaction in molecular recognition processes.1 The term ‘master key of molecular recognition’ has been used to describe the hydrogen bond. This terminology seems to be entirely justified. It is an interaction that is weaker than the covalent bond and stronger than the van der Waals interaction. It is made easily and broken easily at temperatures that are close to ambient and correspond to body temperatures. The reversibility conferred by the intermediate energy of this interaction makes it particularly suitable for the conduct of reactions in living tissues: the hydrogen bond, therefore, has profound implications in biochemistry. In a book co-authored by one of us in 1999, it was mentioned that the twin ability to both associate and dissociate at ambient temperatures “renders the interaction well suited to achieving specificity of recognition within short time spans, a necessary condition for biological reactions that must take place around room temperature”. A further, and exquisite, aspect of the interaction X‒H…A‒Y is that it spans an energy range roughly between 40 and 1 kcal/mol, depending on what X, A, and Y are. This means that within the palette of hydrogen bonding, there is a further discrimination between very strong, strong, and weak hydrogen bonds with the relevant differences in lability and reversibility2 (Table 1). This provides a fine-tuning to the overall molecular recognition phenomenon with its obvious implications to areas as disparate as understanding protein function, drug design, and crystallization mechanisms, even the quality of crystals obtained thereby.
Non-covalent interactions between molecules of all sizes and hues predominate cellular functions. All molecular transactions within and between cells involve these interactions. Many of these intermolecular interactions require them to be specific and/or selective, such as enzyme–substrate/inhibitor, antigen–antibody, and ligand–receptor (drug–receptor) interactions. The specificity of biological processes suggests that the intermolecular interactions involved in the underlying recognition events are also specific, with conserved orientation.3,4,5,–6
1.1 Strong and Weak Hydrogen Bonds
Our studies on strong and weak hydrogen bonding in protein–ligand recognition followed from our earlier work on the use of hydrogen bonds in recognition phenomena during crystallization of small molecules, a process whose understanding is essential in the design of crystal structures of functional solids, also referred to as crystal engineering. The key element in this regard is the supramolecular synthon, a small modular unit that encapsulates critical recognition information between molecules.4 Synthons represent both strength and directionality of recognition, and in this regard, it is assumed that they are also of key importance in biological recognition. The persistent recurrence of synthons mediated by weak forces in crystals indicates that such patterns might still be important in solution for transient processes such as those associated with biomolecular structure and conformation [Fig. 1].7 In short, it was felt that the hydrogen bond, as the most reliable directional interaction in supramolecular construction and crystal engineering, would also make it of great importance in the whole domain of biomolecular recognition.2,3,–4, 8

Synthons extracted from Cambridge structure database. X is any non-hydrogen atom. The numbers indicate corresponding hydrogen-bond distances in Ångstroms (Adapted from Ref. 7).
Hydrogen bonds are instrumental not only in mediating drug–receptor binding, but they also affect physicochemical properties of a molecule, such as solubility, partitioning, distribution, and permeability, which are crucial to drug development.9,10,–11 Another compelling aspect of the hydrogen bond is its composite character. The hydrogen bond, or alternatively the ‘hydrogen bridge’, is viewed as an interaction that has covalent, electrostatic, and van der Waals character, and spans a wide energy range.3, 12 The composite nature of the hydrogen bond means that the relative proportions of covalency, electrostatics, and van der Waals character in the X–H…A‒Y interaction vary smoothly, depending on the nature of X, A and Y [Table 1]. This, in turn, renders the interactions chemically tunable with the corresponding implications for function.
Weak hydrogen bonds have been known since the 1960s through the pioneering work of Sutor.13,14,15,16,–17 Weak hydrogen bonds in biological molecules have been studied since the 1980s, but it is only in recent years, with near-atomic resolutions becoming a reality in macromolecular crystallography, that meaningful conclusions have been possible. With respect to the C–H…O bond, work by Derewenda18 on proteins, Sundaralingam19 on nucleic acids and Steiner20 on water is noteworthy. Every protein contains a very large number of C–H…O hydrogen bonds, and for the larger proteins, they occur in the thousands. There are three main configurations of weak C–H…O bonds in proteins: side chain-to-side chain, main chain-to-side chain, and protein–ligand.21 Most of these interactions are weak-to-very weak and their functions are normally supportive at best.22 The most common of these interactions is C–H…O = C interactions in parallel and anti-parallel β-sheets.23 Other C–H…O=C contacts are found in α-helices, buried polar side chains and buried water molecules. An interesting residue is Pro, which cannot donate N–H…O hydrogen bonds. If inserted in an α-helix, the regular pattern of N(i)–H…O=C(i–4) hydrogen bonds is disrupted, leading to a kink in the helix. Bhattacharyya and Chakrabarti have noted that in this situation, the activated proline CδH2 group is often involved in C–H…O interactions with carbonyl acceptors at positions (i–3), (i–4) or (i–5), depending on the local conformation.24 C–H…O hydrogen bonds from amino acid side chains are even weaker than hydrogen bonds formed by Cα–H groups. Other types of weak hydrogen bonds in proteins are formed with \(\pi\)-acceptors. Examples are known with all strong donor types that are present in proteins: main chain and side chain N–H, side chain O–H, water molecules, and O/N–H groups of substrate molecules. The acceptors are the side chains of Phe, Tyr, Trp, and occasionally His residues. The energy range of O/N–H…\(\pi\) hydrogen bonds is about 2–4 kcal/mol25,26,–27 for uncharged systems; in other words, they are more significant than typical C–H…O interactions.28
1.2 Enthalpy and Entropy
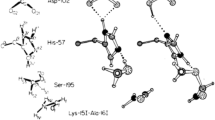
We found that both strong (N–H…O, O–H…O) and weak (C–H…O) hydrogen bonds are involved in ligand binding and that multifurcation is common.7, 29, 30 Therefore, the restrictive geometrical criteria set-up for hydrogen bonds in small-molecule crystal structures may need to be relaxed in macromolecular structures (Fig. 2). For example, there are definite deviations from linearity (θ ~ 180°) for both strong and weak hydrogen bonds. In contrast to small-molecule structures, anti-cooperative geometries are common in biomolecular structures. We found that C–H…O bonds formed by Gly, Phe, and Tyr are noteworthy, and that the numbers of hydrogen-bond donors and acceptors agree with the Lipinski rules that predict drug-like properties.31 Hydrogen bonds formed by water are also seen to be relevant in that ligand C–H…Ow interactions are abundant when compared to N–H…Ow and O–H…Ow. This suggests that ligands prefer to use their stronger hydrogen-bond capabilities for use with the protein residues, leaving the weaker interactions to water. Thus, the interplay between strong and weak interactions in ligand binding leads to a satisfactory enthalpy–entropy balance.

Definitions of the geometrical parameters d, and D for a C–H…O hydrogen bond. The H-atom position should be neutron normalized for systematic analysis (Adapted from ref. 29).
The importance of C–H…O hydrogen bonds in protein–ligand binding has been demonstrated by Pierce et al., in a study of 200 liganded kinase structures.32 The evidence is most convincing for activated C–H groups such as those found adjacent to heteroatoms in kinase ligands (heterocycles). While kinase ligands have been optimized for high affinity binding using other criteria, the strong C–H…O hydrogen bonds that result are a serendipitous added value, making use of these bonds is expected to be of considerable utility in protein modeling, ligand design, and structure–activity analysis. A question that arises immediately is, ‘What is the penalty in binding affinity for replacing a traditional protein–ligand hydrogen bond with an aromatic C–H…O hydrogen bond?’ This penalty appears to be surprisingly small and is rationalized on the basis that N–H and O–H groups must pay a larger desolvation price to leave the aqueous environment to form their hydrogen bonds with the protein. Therefore, perhaps, these two effects (hydrogen-bond formation in the protein–ligand complex and desolvation) largely counterbalance each other, resulting in similar binding affinities for conventional hydrogen bonds and their C–H…O analogs.33 Pierce et al. conclude that if N–H…O and C–H…O hydrogen bonds are interchangeable, the impact on ligand design would be tremendous, because N–H-to-C–H donor swaps would allow the design of novel inhibitors with similar binding affinity but potentially improved non-binding-related properties such as cell permeability or metabolic stability. In fact, the chemical and structural equivalence of N–H…O and O–H…O to C–H…O hydrogen bonds has been amply demonstrated.34,35,–36
1.3 Specificity and Reversibility
The binding properties of proteins are the essence of functional genomics. It is necessary to know when a protein is expressed and where it is localized, but to find out what it does, one needs to find out to what it binds, and how. The specificity of biological processes suggests that the intermolecular interactions involved in the underlying recognition events are also specific, with conserved stereochemical orientation. Hydrogen bonds, even the weakest ones, are electrostatic and, therefore, of long-range character; this is what makes them so important in the whole domain of biomolecular recognition. For example, the phosphate and sulfate transport receptors bind to their ligands with exquisite specificity through multiple hydrogen bonds.37
No less important than specificity is reversibility in biological processes. Weaker interactions can be made and broken more easily than stronger interactions. Accordingly, it is of interest to compare the significance of strong and weak interactions in the macromolecular recognition process. Is protein–ligand binding governed by conventional, that is, electrostatic, N–H…O and O–H…O hydrogen bonds, or do weaker interactions with a greater dispersive component like C–H…O also play a role? If so, to what extent are they significant? Noting that several recent studies have identified and validated the presence of C–H…O and other weak hydrogen bonds in macromolecular structures,23, 38, 39 we undertook a database study of 28 selected high-resolution protein–ligand crystal structures, so that we could assess strong and weak hydrogen bonds simultaneously in a category of biological structures that is of importance in drug design.7 In this analysis, we found that both strong and weak hydrogen bonds are involved in ligand binding. The stronger N‒H…O and O‒H…O interactions show slight but definite deviations from linearity. Multifurcation of strong with weak hydrogen bonds is common in the structures in this study. The propensity of occurrence of acceptor furcated bonds, or anti-cooperative interactions, justifies the need to consider a more liberal distance cut-off criterion for these interactions. The formation of C‒H…O hydrogen bonds is influenced by the activation of the C‒H atoms and by the flexibility of the side chain atoms. C‒H…O bonds formed by Gly, Phe, and Tyr residues are noteworthy. Hydrogen bonds formed by water are also seen to be relevant in ligand binding, as discussed in the section above.
We obtained similar results in a subsequent expanded analysis of 251 protein–ligand complexes using an in-house computer program (HBAT).30 Strong hydrogen bonds retain good geometries up to a resolution of 2.3 Å, whereas for weak bonds, the limit is 2.0 Å. Residues like Gly and Ala, which are smaller in size and have greater flexibility, participate well in both strong and weak hydrogen bonds. Other weak interactions involving halogen atoms (both as electrophiles and nucleophiles), π-acceptors, and S-atom acceptors are also important in the protein–ligand interface. We conclude that the results of our previous study of 28 structures are largely applicable to a set of structures that is nearly ten times as large. An encouraging aspect of this study is that macromolecular crystal structures with resolutions up to 2.0 Å may be used to analyze hydrogen-bond geometry provided a reliable way is found to fix H-atom positions.
1.4 Equivalence of Strong and Weak Hydrogen Bonds
Examples of the interchangeable nature of these hydrogen bonds is provided in our studies where we carried out virtual screening (VS) of (1) 128 EGFR kinase inhibitors based on the 4-anilinoquinazoline fragment40 and (2) a database of ~ 500,000 molecules by a composite docking-pharmacophore screening model to identify new leads for Mycobacterium tuberculosis deoxythymidine monophosphate kinase (TMPKmt) inhibitors.41 We chose these systems because of the known importance of strong and weak (C–H…O) hydrogen bonding.32 VS is a sequence of computational techniques that allows selection and ranking of possible leads from a library of compounds and is of significance in the current drug design scenario, wherein high-throughput screening is proving to be increasingly expensive and perhaps even unreliable.42,43,–44
The docking of ligands for the VS was done in the active site as obtained in the experimental crystal structure of the erlotinib–EGFR complex.45 Erlotinib is an anti-cancer drug from Genentech, belonging to the 4-anilinoquinazoline class. The 128 ligands were docked in the active site and the respective scores were obtained. The obtained poses, which represent positional and orientational information of the ligands, were classified into one of three categories: close, shifted, and misoriented. We identified three key hydrogen bonds (N–H…N, O‒Hw…N, and C–H…O), of comparable stabilization energy, as responsible for anchoring the ligand in the active site (Fig. 3), and a ligand in the close category is docked with all three hydrogen bonds appearing correctly. A shifted ligand has one or more of the hydrogen bonds in place, but the metrics are incorrect. A ligand with a misoriented pose is in a completely wrong orientation and/or position. While the N–H…N bond between Met769 and N(1) (d, 1.81 Å) and the Ow–H…N between water10 and N(3) (d, 2.01 Å).are of moderate strength, the C–H…O to Gln767 is very short (d, 2.19 Å) and involves a highly activated donor; indeed, it is the best conserved interaction in the group. In the currently available docking software, the C–H…O bonds are not modeled explicitly; they fortuitously appear correctly for the close category ligands. The shifted and misoriented ligands could well be false negatives. We argue, accordingly, that if weak hydrogen bonds and other interactions are explicitly incorporated into the software, the efficiency of VS would increase greatly. VS is supposed to rapidly screen large chemical libraries and to cherry pick and rank the few active ones, from the very large number of moderately active and inactive compounds—the so-called needle-in-the-hay-stack problem.

Binding of erlotinib in the EGFR kinase active site. Note the C–H…O bond formed by the activated heterocyclic donor (Adapted from ref. 40).
In the second example, viz., TMPKmt, a detailed docking analysis and pharmacophore modeling was carried out using TMPKmt inhibitors. Docking confirmed the role of weak interactions in promoting enzyme selectivity toward deoxyribonucleotides. It also highlighted the importance of water-mediated cooperative networks and weak hydrogen bonds to ligand-binding affinities.3, 7 Another interesting finding of this study is the role of halogen bonding46 and its stabilization by the water-mediated cooperative hydrogen-bond network. With an appreciation of the functionalities involved in molecular recognition acquired from the above-mentioned methods, a composite pharmacophore model was developed and validated with a database containing known TMPKmt inhibitors. This composite model was used as a 3D query for the successful VS of a database of about 500,000 compounds to find new antitubercular leads. Till today, VS approaches have concentrated on speed and automation. We suggest that future software should explicitly seek out hydrogen-bond forming ability of a ligand or, in other words, address chemical issues directly, so that structure-based VS becomes increasingly accurate and reliable.
The C–H…O hydrogen bond was first invoked in the 1930s, but it is only during the last 25 years or so that C–H…O and other weak interactions have been studied intensively and documented properly. Today, the question is not so much whether this interaction exists, or whether it is important in crystal packing as a structure determinant—these questions have long since been answered in the affirmative—but more about how it may be used and applied. In this regard, possibilities in the biological world appear to be very promising. Future work will show to what extent this promise is realized.
1.5 The Role of Water
We have already seen that the structure and function of biological molecules is to a large degree determined by hydrogen bonding. Water, an essential constituent of protein structure, is in this structure–function context, a crucial agent because of its excellent hydrogen-bonding ability. Indeed, its entire molecular surface is composed of groups that are able to either donate or accept hydrogen bonds. The molecule is of very small size, and as such, it can be accommodated in many locations and environments. Significantly, and in relation to this review, water is known to accept and donate weaker hydrogen bonds. First, even the so-called ‘strong’ O‒Hw…Ow hydrogen bond between water molecules themselves is at the weaker end for this particular hydrogen-bond type, being around 5 kcal/mol. Second, water is freely able to donate and accept weak hydrogen bonds.
Interactions such as C‒H…Ow and to a lesser extent O‒Hw…Ph are ubiquitous in protein structures because of the profusion of aliphatic and aromatic residues. Finally, the regions of donor and acceptor abilities are somewhat smeared out in the water molecule. All this means that water molecules in biological structures are fully coordinated with a range of hydrogen bonds of varying strengths, directionalities, and flexibilities. Therefore, they are intimately and implicitly connected to function.
The structural variety of hydrogen bonding mediated by water in biological structures is truly astounding. The functional aspects of water arise, because the molecule is chameleon-like in its hydrogen-bonding ability: because of its small size and flexibility of placement, it can change its function from a hydrogen-bond donor to a hydrogen-bond acceptor. Such orientational flexibility can alter the hydrogen-bond topology considerably, but the changes in energy are relatively minor; this renders the water molecule to be unique in its structural and functional capabilities. Water almost always donates two hydrogen bonds. In its acceptor capacity, it may accept one or two hydrogen bonds. As a multifurcated acceptor, this number may even go up. Sometimes, it could accept one strong hydrogen bond and one weak hydrogen bond, approximating a tetrahedral environment. Additionally, water forms hydrogen bonds to itself, forming infinite or discrete clusters. All this means that possible water coordination geometries exist in vast number and variety.
In the context of equivalence of strong and weak hydrogen bonds, C–H donors often participate in the coordination of water molecules similar to O–H and N–H. Certainly, water molecules prefer to accept strong hydrogen bonds. However, if these are not available in sufficient numbers and in suitable enough configurations in a given local environment, a water molecule will resort to accepting the weaker C–H···OW hydrogen bonds rather than leaving its acceptor potential unsatisfied. In these arrangements, O/N–H···OW and C–H···OW hydrogen bonds have the same functions, with differences only in the strengths.
The case of OW–H···\(\pi\) hydrogen bonds is also interesting. In small-molecule crystal structures, there are relatively few examples of water molecules donating hydrogen bonds to \(\pi\)-acceptors. More systematic studies have been performed on OW–H···Ph hydrogen bonds in small hydrated peptides. The geometries are very variable, with some of the water O atoms residing almost exactly over the aromatic centroids of Phe or Tyr, whereas others are more off-centered. These water interactions do not represent generally favorable configurations. They are far less common than regular O–H…\(\pi\) hydrogen bonds in biological structures from stronger donors like Tyr, but show that water molecules are able to find stable positions in ‘unfriendly’ environments.
Several of these hydrogen-bonding possibilities are revealed in a study of the active site structure of erlotinib–EGFR complex discussed in the previous section. We found that failure to include the hydrogen-bonded water molecule that forms the Ow‒H…N bond leads to incorrect results. Curiously, we also found that of the three interactions, the C‒H…O formed by an activated C‒H group is the best conserved rather than the supposedly stronger N‒H…N. In the VS context, all three interactions need to be modeled correctly, so that correct poses and affinities are obtained for potential leads. Initially the water molecules were not considered, and so, these crucial hydrogen bonds were not included. Therefore, docking is not effective, and there was little-to-no correlation between the scores and the observed activities. Accordingly, a conscious decision was made to include this water molecule. After including this molecule and four others in the inflexible part of the protein, an improvement was observed.
Though the experimental material is limited at the present, there is little doubt that non-conventional hydrogen bonds occur frequently in protein–solvent interactions. C–H…OW hydrogen bonds typically function in satisfying water acceptor potentials in partly or mainly hydrophobic surroundings. This allows water molecules to find stable positions even in sites that lack conventional hydrogen-bond donors. Specific roles in protein functions can be readily conceived and their elucidation is a promising field of structural research for the future.
1.6 Structure and Function
The connection between structure and function in biomolecules is sometimes subtle. Structure is often correlated with function, but, sometimes, these connections may be hidden or show non-linearities. The energy range of hydrogen bonds offer some interesting possibilities with regard to function, and on occasion, weak hydrogen bonds might facilitate a function that stronger ones do not. Which is more effective, a few strong hydrogen bonds or many weak ones? In this context, it is worthwhile to mention the interesting use of C‒H…O contacts introduced by reductive methylation of nine surface lysine residues to help crystallize a protein that had previously resisted crystallization despite extensive purification and crystallization space screening.47 The C‒H…O hydrogen bonds most likely add to supramolecular coherence of the system and stiffen the protein sufficiently, so that acceptable diffraction data may be collected.
Sometimes, the presence or absence of a single hydrogen bond can determine the pharmacological properties of ligands when bound to their receptors. Misra et al. reported that PAT5A, a chemically distinct unsaturated thiazolidinedione, activates peroxisome proliferator-activated receptor γ (PPARγ) submaximally in vitro with a binding affinity ~ ten times less than that of rosiglitazone, a highly potent thiazolidinedione. PAT5A binds to the same pocket as rosiglitazone, but misses a hydrogen bond with the hydroxyl of tyrosine 473, leading to differential co-activator recruitment and gene activation and, therefore, behaves as a partial agonist of PPARγ, yet only weakly adipogenic, which makes it a better molecule than rosiglitazone.48
Connelly et al. successfully investigated whether potency and insolubility share a common origin, and examined the structural and thermodynamic properties of telaprevir, a sparingly soluble inhibitor of hepatitis C virus protease.10 They compared the hydrogen-bond patterns in crystalline telaprevir with those present in the protease–telaprevir complex and found striking similarities (see Fig. 1b and c from Ref. 10). Also, they reckoned that the thermodynamics of telaprevir dissolution closely resembles that of protein–ligand dissociation. Their findings pointed to a common origin of potency and insolubility rooted in certain amide–amide hydrogen-bond patterns. The insolubility of telaprevir is shown by computational analysis to be caused by interactions in the crystal, rather than unfavorable hydrophobic hydration. Accordingly, they competed out the particular amide–amide hydrogen-bond motifs in crystalline telaprevir with 4-hydroxybenzoic acid that yielded a co-crystalline solid with greater aqueous solubility and oral absorption (Fig. 3a from Ref. 10). Connelly et al. found similar results with the non-nucleoside HIV reverse transcriptase inhibitor, efavirenz (Fig. 3f in Ref. 10).
2 Conclusions
The essence of the hydrogen bond X‒H…A‒Y is that it is both a complex and a composite interaction. It is complex, because it is made up of all the atoms within the interacting system, namely X, A, Y, and the all-important H. In multifurcated arrangements, more atoms are within the hydrogen-bond system. It is of composite character, because it is made up of three main ingredients: electrostatics, covalency, and van der Waals. Because these ingredients can be of varying importance, hydrogen bonds exist across a wide energy range and they are somewhat arbitrarily, but mostly for purposes of convenience, differentiated as very strong, strong, and weak. These different types of hydrogen bonds have broadly similar if graded effects in the building up of all types of crystals from molecules. As far as structure is concerned, more of the weaker interactions are needed for any particular effect to be clearly manifested. Accordingly, while there are geometrical, energetic, and spectroscopic criteria to assess an interaction as a hydrogen bond, there are no hard and fast cut-offs in these criteria. In a recent IUPAC definition49 of the hydrogen bond, it is merely mentioned that “the evidence for hydrogen bond formation may be experimental or theoretical, or ideally, a combination of both. Some criteria useful as evidence and some typical characteristics for hydrogen bonding, not necessarily exclusive, are listed below [..] The greater the number of criteria satisfied, the more reliable is the characterization as a hydrogen bond.” It is exactly for this reason that we have progressed easily from Pauling’s statement that “under certain conditions an atom of hydrogen is attracted by rather strong forces to two atoms instead of only one so that it may be considered to be acting as a bond between them” to “the hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation”.49
Equilibrium crystal structures of small molecules are analyzed and predicted in crystal engineering on the basis of interplay between strong and weak hydrogen bonds. In such crystals, strong and weak hydrogen bonds interact with one another to produce low-energy minima that we call polymorphs. However, in the final equilibrium structures, the atoms are largely fixed with of course the thermally governed oscillations about mean positions. The crystal is in the end, static.
It is in the domain of biomolecular crystals that the functional aspects of hydrogen bonds come into their own. Strong and weak hydrogen bonds X‒H…A‒Y show a broad similarity at the gross level and dissimilarity at the fine level. Because biomolecules are large, and because of the presence of a large amount of water in the crystal and because the molecules are held together with hydrogen bond and other interactions of widely differing energies, different portions of these crystals are static or dynamic to differing extents. Strength is contraposed by weakness and, consequently, directionality with flexibility. Affinity is accompanied by hydrophobicity and specificity with reversibility. There is a tendency to understand and rationalize biomolecular function in terms of just the strong hydrogen bonds present in the system. Nothing could be more dangerous or misleading. The examples which we have presented show that neglecting the weaker hydrogen bonds would in many cases lead to complete non-comprehension of the static structure, and that consideration of strong and weak hydrogen bonds together is the only way to obtaining an understanding of biological function.
References
Desiraju GR (2011) A bond by any other name. Angew Chem Int Ed Engl 35:52–59. https://doi.org/10.1002/anie.201002960
Desiraju GR, Steiner T (1999) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford. https://doi.org/10.1093/acprof:oso/9780198509707.001.0001
Jeffrey GA, Saenger W (1991) Hydrogen bonding in biological structures. Springer, Berlin. https://doi.org/10.1007/978-3-642-85135-3
Desiraju GR (1995) Supramolecular synthons in crystal engineering—a new organic-synthesis. Angew Chem Int Ed 34:2311–2327. https://doi.org/10.1002/anie.199523111
Glusker JP (1998) Directional aspects of intermolecular interactions. In: Weber E (ed) Topics in current chemistry: design of organic solids. Springer, Berlin, pp 1–56. https://doi.org/10.1007/3-540-69178-2_1
Williams MA, Ladbury JE (2003) Hydrogen bonds in protein-ligand complexes. In: Böhm H-J, Schneider G (eds) Protein-ligand interactions: from molecular recognition to drug design. Wiley, Hoboken, pp 137–161. https://doi.org/10.1002/9783527610754.pl02
Sarkhel S, Desiraju GR (2004) N–H…O, O–H…O, and C–H…O Hydrogen bonds in protein–ligand complexes: strong and weak interactions in molecular recognition. Proteins 54:247–259. https://doi.org/10.1002/prot.10567
Williams DH, Stephens E, O’Brien DP, Zhou M (2004) Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and, enzymes. Angew Chem Int Ed 43:6596–6616. https://doi.org/10.1002/anie.200300644
Gandhi R, Pillai O, Thilagavathi R, Gopalakrishnan B, Kaul CL, Panchagnula R (2002) Characterisation of azithromycin hydrates. Eur J Pharm Sci 16:175–184. https://doi.org/10.1016/S0928-0987(02)00087-8
Connelly PR, Snyder PW, Zhang Y, McClain B, Quinn BP, Johnston S, Medek A, Tanoury J, Griffith J, Walters WP, Dokou E, Knezic D, Bransford P (2015) The potency–insolubility conundrum in pharmaceuticals: mechanism and solution for hepatitis C protease inhibitors. Biophys Chem 196:100–108. https://doi.org/10.1016/j.bpc.2014.08.008
Sanphui P, Rajput L, Gopi SP, Desiraju GR (2016) New multi-component solid forms of an anti-cancer drug erlotinib: role of auxiliary interactions in determining a preferred conformation. Acta Crystallogr Sect B 72:291–300. https://doi.org/10.1107/s2052520616003607
Desiraju GR (2002) Hydrogen bridges in crystal engineering: interactions without borders. Acc Chem Res 35:565–573. https://doi.org/10.1021/ar950135n
Sutor DJ (1962) The C-H…O hydrogen bond in crystals. Nature 195:68–69
Sutor DJ (1963) Evidence for the existence of C–H…O hydrogen bonds in crystals. J Chem Soc 1105–1110. https://doi.org/10.1039/jr9630001105
Taylor R, Kennard O (1982) Crystallographic evidence for the existence of C–H…O, C–H…N and C–H…Cl hydrogen bonds. J Am Chem Soc 104:5063–5070. https://doi.org/10.1021/ja00383a012
Extance A (2019) The forgotten female crystallographer who discovered C–H…O bonds. Chem World. https://www.chemistryworld.com/features/the-forgotten-female-crystallographer-who-discovered-cho-bonds/3010324.article
Horowitz S, Trievel R, Scheiner S, Schwalbe C (2019) Do you know about C–H…O? Chem World. https://www.chemistryworld.com/opinion/do-you-know-about-cho/3010705.article
Derewenda ZS, Lee L, Derewenda U (1995) The occurrence of C–H…O hydrogen bonds in proteins. J Mol Biol 252:248–262. https://doi.org/10.1006/jmbi.1995.0492
Wahl MC, Sundaralingam M (1997) C–H…O hydrogen bonding in biology. Trends Biochem Sci 22:97–102. https://doi.org/10.1016/s0968-0004(97)01004-9
Steiner T (1995) Water molecules which apparently accept no hydrogen bonds are systematically involved in C-H…O interactions. Acta Crystallogr Sect D 51:93–97. https://doi.org/10.1107/S0907444994007614
Yesselman JD, Horowitz S, Brooks CL III, Trievel RC (2015) Frequent side chain methyl carbon-oxygen hydrogen bonding in proteins revealed by computational and stereochemical analysis of neutron structures. Proteins 83:403–410. https://doi.org/10.1002/prot.24724
Castellano RK (2004) Progress toward understanding the nature and function of C-H…O interactions. Curr Org Chem 8:845–865. https://doi.org/10.2174/1385272043370384
Fabiola GF, Krishnaswamy S, Nagarajan V, Pattabhi V (1997) C–H…O hydrogen bonds in β-sheets. Acta Crystallogr D 53:316–320. https://doi.org/10.1107/S0907444997000383
Bhattacharyya R, Chakrabarti P (2003) Stereospecific interactions of proline residues in protein structures and complexes. J Mol Biol 331:925. https://doi.org/10.1016/s0022-2836(03)00759-9
Desiraju GR, Sharma CVKM (1991) C–H…O hydrogen-bonding and topochemistry in crystalline 3,5-dinitrocinnamic acid and its 1–1 donor-acceptor complex with 2,5-dimethoxycinnamic acid. J Chem Soc Chem Commun. https://doi.org/10.1039/c39910001239
Viswamitra MA, Radhakrishnan R, Bandekar J, Desiraju GR (1993) Evidence for O–H…C and N–H…C hydrogen-bonding in crystalline alkynes, alkenes, and aromatics. J Am Chem Soc 115:4868–4869. https://doi.org/10.1021/ja00064a055
Thallapally PK, Katz AK, Carrell HL, Desiraju GR (2002) Unusually long cooperative chain of seven hydrogen bonds. An alternative packing type for symmetrical phenols. Chem Commun. https://doi.org/10.1039/b110036j
Steiner T, Koellner G (2001) Hydrogen bonds with π-acceptors in proteins: frequencies and role in stabilizing local 3D structures. J Mol Biol 305:535–557. https://doi.org/10.1006/jmbi.2000.4301
Desiraju GR (2005) C-H…O and other weak hydrogen bonds. From crystal engineering to virtual screening. Chem Commun. https://doi.org/10.1039/b504372g
Panigrahi SK, Desiraju GR (2007) Strong and weak hydrogen bonds in the protein–ligand interface. Proteins 67:128–141. https://doi.org/10.1002/prot.21253
Lipinski CA, Lombardo F, Dominy BW, Feeney PL (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev 23:3–25. https://doi.org/10.1016/S0169-409X(96),00423-1
Pierce AC, Sandretto KL, Bemis GW (2002) Kinase inhibitors and the case for C–H…O hydrogen bonds in protein–ligand binding. Proteins 49:567–576. https://doi.org/10.1002/prot.10259
Adrian JC, Wilcox CS (1992) General effects of binding-site water exclusion on hydrogen-bond based molecular recognition systems—a closed binding-site is less affected by environmental-changes than an open site. J Am Chem Soc 114:1398. https://doi.org/10.1021/ja00030a040
Berkovitch-Yellin Z, Leiserowitz L (1984) The role played by C-H…O and C–H…N interactions in determining molecular packing and conformation. Acta Crystallogr Sect B 40:159–165. https://doi.org/10.1107/s0108768194012061
Anthony A, Jaskolski M, Nangia A, Desiraju GR (1998) Isostructurality in crystalline oxa-androgens: a case of C-H…O and C–H…O interaction mimicry and solid solution formation. Chem Commun. https://doi.org/10.1039/a806607h
Thakur TS, Azim Y, Srinu T, Desiraju GR (2010) N–H…O and C–H…O interaction mimicry in the 1:1 molecular complexes of 5,5-diethylbarbituric acid with urea and acetamide. Curr Sci 98:793–802
Quiocho FA (1996) Atomic basis of the exquisite specificity of phosphate and sulfate transport receptors. Kidney Int 49:943–946
Klaholz BP, Moras D (2002) C–H…O hydrogen bonds in the nuclear receptor RAR—a potential tool for drug selectivity. Structure 10:1197–1204. https://doi.org/10.1016/S0969-2126(02),00828-6
Jiang L, Lai L (2002) C–H…O hydrogen bonds at the protein–protein interfaces. J Biol Chem 277:37732–37740. https://doi.org/10.1074/jbc.M204514200
Aparna V, Rambabu G, Panigrahi SK, Sarma JARP, Desiraju GR (2005) Virtual screening of 4-anilinoquinazoline analogues as egfr kinase inhibitors: importance of hydrogen bonds in the evaluation of poses and scoring functions. J Chem Inf Model 45:725–738. https://doi.org/10.1021/ci049676u
Gopalakrishnan B, Aparna V, Jeevan J, Ravi M, Desiraju GR (2005) A virtual screening approach for thymidine monophosphate kinase inhibitors as antitubercular agents based on docking and pharmacophore models. J Chem Inf Model 45:1101–1108. https://doi.org/10.1021/ci050064z
Gruenberg S, Stubbs MT, Klebe G (2002) Successful virtual screening for novel inhibitors of human carbonic anhydrase: strategy and experimental confirmation. J Med Chem 45:3588–3602. https://doi.org/10.1021/jm011112j
Lyne PD (2002) Structure-based virtual screening: an overview. Drug Discov Today 7:1047–1055. https://doi.org/10.1016/s1359-6446(02)02483-2
Bleicher KH, Bohm H-J, Muller K, Alanine AI (2003) Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discovery 2:369–378. https://doi.org/10.1038/nrd1086
Stamos J, Silwkowski MX, Eigenbrot C (2002) Structure of the epidermal growth factor receptor kinase domain alone and in the complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 48:46265–46272. https://doi.org/10.1074/jbc.M207135200
Auffinger P, Hays FA, Westhof E, Ho PS (2004) Halogen bonds in biological molecules. Proc Natl Acad Sci USA 101:16789–16794. https://doi.org/10.1073/pnas.0407607101
Shaw N, Cheng C, Tempel W, Chang J, Ng J, Wang X-Y, Perrett S, Rose J, Rao Z, Wang B-C, Liu Z-J (2007) (NZ)CH…O Contacts assist crystallization of a ParB-like nuclease. BMC Struct Biol 7:46. https://doi.org/10.1002/pro.420
Misra P, Chakrabarti R, Vikramadithyan RK, Gopalakrishnan B, Suresh J, Jagadheshan H, Cynthia G, Rajjak A, Kashireddy P, Yu S, Surapureddi S, Qi C, Zhu Y-J, Rao MS, Reddy JK, Rajagopalan R (2003) PAT5A: a partial agonist of peroxisome proliferator-activated receptor is a potent antidiabetic thiazolidinedione yet weakly adipogenic. J Pharm Exp Ther 306:763–771. https://doi.org/10.1124/jpet.103.049791
Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B, Nesbitt DJ (2011) Defining the hydrogen bond: an account (IUPAC technical report). Pure Appl Chem 83:1619–1636. https://doi.org/10.1351/PAC-REP-10-01-01
Acknowledgements
GRD thanks the DST for the award of a J. C. Bose fellowship (SR/S2/JCB-18/2006).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bulusu, G., Desiraju, G.R. Strong and Weak Hydrogen Bonds in Protein–Ligand Recognition. J Indian Inst Sci 100, 31–41 (2020). https://doi.org/10.1007/s41745-019-00141-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41745-019-00141-9