
Abstract
A sensitive and reliable analytical method for the simultaneous determination of seven endocrine disrupting compounds (EDCs) in water was developed and validated. This quantification method is based on solid phase extraction (SPE) followed by a derivatization with BSTFA and finally the seven EDCs were analyzed by gas chromatography–mass spectrometry (GC–MS). A 23 factorial design was used to optimize the extraction procedure. Three factors, namely sample volume, elution solvent, and pH of sample were investigated using Doehlert matrix. The optimal conditions of SPE method were: HLB cartridge, 540 mL of water sample with a pH 8 and 10 mL of mixture of ethyl acetate/acetone with a ratio of (55/45, v/v) in the elution step. For validation of the technique, accuracy, precision, detection and quantification limits, linearity, sensibility and selectivity were determined. Extraction recovery of these seven EDCs were above 90% with relative standard deviations (RSD) ≤ 2%. The method limit of detection and limit of quantification were in the range of 0.33–3.33 and 1–10 ng/L, respectively.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Endocrine disrupting compounds (EDCs) cover an important range of natural and synthetic substances which disturb the hormone function (Jiang et al. 2013; Yang et al. 2013; Laurenson et al. 2014), such as alkylphenols, bisphenol A, endogenous and synthetic hormones, among authors (Reinen et al. 2010). Recently, EDCs have become an important issue in water pollution because of their potential risk on human health and their universal distribution (Lee et al. 2010; Sim et al. 2010). These compounds can interfere with the endocrine system by antagonizing the action of naturally produced hormones, or by preventing the action of endogenous hormones, altering the function and synthesis of hormone matrix receptors, or by modifying the metabolism, transport and excretion of hormones (Reinen et al. 2010; Ballesteros-Gómez et al. 2009; Munaretto et al. 2013). The EDCs are presents in wastewater and surface water (Vandenberg et al. 2012; Zoeller et al. 2012; Sun et al. 2014; Net et al. 2015; Rabodonirina et al. 2015). However, the measurement of EDCs residues is a very difficult task due to their low concentration levels in complex matrices. To overcome these difficulties, various methods have been developed. Currently, the most prevailing methodological approach designed to analyze EDCs incorporates a mass-based analysis process. Overall, the methods employing mass spectrometry (MS) such as gas chromatography–mass spectrometry (GC–MS) and liquid chromatography–mass spectrometry (LC–MS) show relatively low detection limits compared to other methods such as liquid chromatography–UV detection (LC–UV) (Huang et al. 2011; Munaretto et al. 2013; Yang et al. 2015). Among these method, GC-MS allows exellent separation because of the long columns of fused silica that have literally hundreds of thousands of theoretical plates, which allow excellent separation of hormones from their isomers or interfering substances (Thurman et al. 2013).
Enrichment separation approaches including solvent extraction, solid phase extraction (SPE) and solid phase microextraction (SPME) have been commonly used to improve the instrumental limits of detection (LODs) (Latrous El Atrache et al. 2013). The SPE is an extraction and pre-concentration technique the most commonly used for liquid samples (Guedes-Alonso et al. 2013). The SPE has an important role in pre-concentration step due to its simplicity, high enrichment factors and environmental friendliness. However, several factors can affect the extraction efficiency and should be optimized. The conventional strategies for developing analytical methods by univariated experiments can lead to a number of disadvantages. They require more experiments and do not offer the information on the interactions between factors (Friedrich et al. 2016). Multivariate techniques have also been used in the recent years to optimize analytical methods, allowing the optimization of many variables simultaneously. Multivariate techniques allow saving time, practicality, economy and reducing number of experiments (Facco et al. 2015; Kemmerich et al. 2015; Hibbert 2012). This mathematical model allows an estimation of the significance of effects on processes as well as of the effects of interactions between factors. Factorial design is one of the available statistical processes for multivariate techniques. It was used in analytical method development (Hibbert 2012). However, to determine the real functionality established among the analytical response and the significant factors, the second order designs are used.
The objective of this study is to develop a rapid and simple procedure of extraction, preconcentration and determination of seven EDCs in water, based on off-line SPE, BSTFA derivatization and GC–MS quantification. A two-level full factorial design was used to evaluate the experimental variables including eluent solvent, sample volume and pH sample. The experiments for the optimization were performed according to Doehlert matrix. The developed method was applied to the identification and quantification of these compounds in wastewater samples obtained from the effluents of two wastewater treatment plants (WWTPs) of Tunis (Tunisia).
Experimental
Chemicals and Materials
EDCs standards were purchased from Sigma-Aldrich (Saint-Louis, USA) and Restek (Bellefonte, USA) with a purity of 99%. Supel-Select HLB, Supelclean ENVI-18 and Supelclean LC-18 SPE cartridges (200 mg/6 mL) were purchased from Sigma-Aldrich (Saint-Louis, USA). HPLC grade ethyl acetate, methanol and acetonitrile were purchased from Dislab (Lens, France). Ultrapure water (Milli-Q) was produced by a Millipore apparatus with 18.2 MΩ/cm resistivity. The derivatization reagent N,O-bis(trimethylsilyl)trifluoroacetamide with 1% of trimethylchlorosilane (BSTFA, 1% TMCS) were obtained from Sigma-Aldrich. Standard stock solutions of 500 mg/L were prepared by weighing and dissolving 12.5 mg of each compound in 25 mL of acetonitrile. These solutions were stored at 4 °C. The analyzed seven EDCs with their chemical structures and other characteristics are shown in Table 1.
Sample Preparation
The samples of water were fortified with the seven EDCs. 1 mL of ECDs standard solution with the concentration of 10 μg/L was added to the sample. SPE cartridges were first conditioned with 5 mL of acetone, 5 mL of ethyl acetate then with 5 mL of deionized water. The pH of samples was adjusted with a solution of HCl 1 M. The solutes were eluted with 10 mL of mixture of ethyl acetate/acetone with a ratio of (5.67/4.33; v/v). The eluent was collected in a graduated tube and concentrated, under stream of nitrogen to 1 mL.
Sample Collection
Wastewater samples were collected from two WWTP located in Tunis (Fig. 1). The first WWTP is Chotrana (S1) located in Chotrana 1 Ariana. Chotrana WWTP used a biological treatment system, while the second WWTP, located in Médina Jadida-Ben Arous (S2), used an activated sludge treatment system combined with chemical treatment. The samples were collected in 2-L amber glass bottles. Samples were filtered with glass fiber filters with 0.45 μm Whatman glass microfiber filters. The filtered waters were stored in the dark at 4 °C.

Studied sites at Tunis, Tunisia. (S1) WWTP of Chotrana 1 Ariana (S2) WWTP of Médina Jadida-Ben Arous
Derivatization Procedure
For compounds that are thermally unstable, are too low in volatility, or yield poor chromatographic separation due to their high polarity, a derivatization step must be included prior to GC/MS analyses (Nielsen 2014). 50 µL of each SPE extract was transferred into micro vials followed by the addition of 50 µL of BSTFA (1% TMCS), then heated in a heating block at 65 °C for 90 min. The heated extracts were cooled to room temperature during 15 min prior to GC–MS analysis.
GC–MS Analysis
The GC–MS analysis were performed using a Varian 3900 GC equipped with a deactivated fused silica guard column (5 m, 0.25 mm i.d.) and a fused silica capillary Phenomenex XLB (60 m length, 0.25 mm i.d., 0.25 μm film thickness) and coupled with a Varian Ion Trap Saturn 2000 MS. The carrier gas was helium, held at a constant flow rate of 1 mL/min. 1 µL of each sample was injected in the splitless mode at 280 °C and the injector was purged with helium after 1 min. The temperature of the GC was programmed as follows: 100 °C, held for 2 min, 5 °C min−1 ramped to 250 °C then 3 °C min−1 ramped to 300 °C and held for 2.33 min. The transfer line and the ion trap were, respectively, held at 280 and 220 °C. Each targeted compound was identified based on the retention time (RT) and the mass spectrum (m/z) from chromatogram of standard solutions acquired in full scan (FS) mode. Quantification was then performed in the single ion storage (SIS) modes for better selectivity. The detailed MS detection parameters for each EDC are presented in Table 1.
Results and Discussion
Effect of Sorbent Type
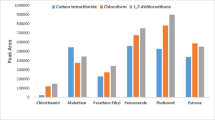
To evaluate the influence of the sorbent type on the extraction recovery of EDCs, three SPE cartridges which were reported to give good efficiency were chosen: Supel-Select HLB (Huang et al. 2011; Grover et al. 2009), Supelclean ENVI-18 (Xu et al. 2014; Gao et al. 2013) and Supelclean LC-18 (Guedes-Alonso et al. 2013; Zhang et al. 2011). The selected cartridges were charged with 500 mL of water sample spiked with 1 µL of EDCs standard solution, then eluted with 5 mL of acetone and 5 mL of ethyl acetate. The experiment was carried out in triplicate. The Fig. 2 presents the recoveries of the seven EDCs obtained from the three types of cartridges. The result showed better recovery for the simultaneous extraction of these seven EDCs obtained with HLB cartridge (Fig. 2).

Influence of the sorbent type on the extraction recovery of studied EDCs
Determination of the Optimal Condition for the Solid Phase Extraction of EDCs
A 23 full factorial design with the variables, sample volume (U 1), eluent solvent (U 2), and pH sample (U 3) was carried out to determine the influence of selected factors and their interactions on the extraction of EDCs. The maximum and minimum values of each factor are listed in Table 2. The choice of the limits of the investigated region is based on data available from the literature (Sun et al. 2014; Huang et al. 2011; Guedes-Alonso et al. 2013; Rice and Hale 2009; Jin et al. 2013; Aydin and Talinli 2013; Labadie and Budzinski 2005; Belhaj et al. 2015).
Doehlert matrix was used to represent the responses of the three factors in all the experimental studied fields. In fact, the Doehlert matrix presents a number of advantages such as a uniform distribution of experimental points in the studied field, the ability to explore the whole of the experimental region, the usefulness of interpolating the response, and the possibility of adding new factors defined on the basis of preliminary result factors without altering the quality of the matrix. To compare the effects of the different factors in the experimental field, concerned coded variables were used. The factors are given in the form of coded variables (Xi) without units to permit comparison of factors of different natures. The transformation of natural variables (U i ) into the corresponding coded variables (X i ) is made on the basis of the Eq. (1) obtained from NEMROWD software,
where \( X_{i} \) is the value taken by the coded variable i; \( U_{i} \) is the value taken by the factor i; \( \overline{{U_{i} }} \) is the value taken by the factor i in the center of the experimental field; ΔU i is the range of variation of the factor I and α is the maximum coded value of \( {\text{X}}_{i} \): X 1 = 1; X 2 = 0866; X 3 = 0,816.
The factorial design is governed by a function described by the experimental variables that can be approximated by a polynomial function, providing a description of the factors and the response obtained:
where Y is the experimental response, X i is the coded variable, b i is the estimation of the principal effect of the factor i for the response Y, and b ij is the estimation of the interaction effect between factors i and j for the response Y.
The number of experiments required (N) is given by N = k 2 + k + 1, where k is the number of variables. In the present case, k = 3, and therefore the matrix was constituted of 13 experiments (Table 3). The levels of the independent variable (effective variables U i ) were calculated according to these following relations:
Replicates at the central level of the variables are performed to validate the model by means of an estimate of experimental variance. The experiment at the center (experiment number 13) was carried out in triplicate (Table 4) to estimate the experimental error. According to these results, the coefficients of the polynomial model were calculated using the NEMROD software (Table 5).
Figure 3 shows typical response surface profiles drawn versus the main factors sample volume, eluent ratio and pH sample and the three-dimensional representation of the same plots using the NEMROD software.

a Contour plots of NP extraction recovery versus eluent ratio and pH at a fixed sample volume (500 mL); b the corresponding three-dimensional plot; c contour plots of PG extraction recovery versus sample volume (mL) and pH at a fixed eluent ratio (4.56); d the corresponding three-dimensional plot; e contour plots of PG extraction recovery versus sample volume (mL) and eluent ratio at a fixed pH (6); f corresponding three-dimensional plot; results obtained from Doehlert matrix (Table 3)
The analysis of the iso-response curves at the chosen experimental field delimited by a circle show that the maximum extraction recovery was obtained when 540.4 mL of water sample with a pH 8 is loaded on HLB cartridge then eluted with 10 mL of mixture of ethyl acetate/acetone with a ratio of (5.67/4.33; v/v).
Validation of the Optimized Method
For the validation of the method, selectivity, linearity, precision, accuracy, and detection and quantification limits (LODs and LOQs) were studied. (1) Selectivity Validation parameters were evaluated, and the selectivity was confirmed since no interferences were observed in the blank extract when compared to the fortified water samples. Moreover, the chromatograms of the extracts presented satisfactory chromatographic resolution (Fig. 4). (2) Linearity Linearity of the proposed method was investigated by analyzing six dilutions for each EDC in the range of 0.01–10 µg/mL. Analytical curves were constructed for the selected compounds, and good linearity was observed with R2 higher than 0.99 for the seven EDCs (Table 7). (3) Precision Precision was considered at two levels: repeatability and intermediate precision. The repeatability (intra-day precision) study of each EDC was carried out by estimating the correspondence responses six times on the same day with 10 µg/mL concentration. Inter-day precision study of each EDCs was carried out by estimating the responses to correspondence three times on three different days. The precision articulated as inter- and intra-day relative standard deviation (% RSD) < 2, which indicates that there was no significant difference for the assay which was tested within 1 day and between days. The extraction recovery percentages (% R) and RSD obtained are presented in (Table 7). (4) Accuracy The accuracy of the method was evaluated with five repetitions by recovery tests at three concentration levels (10, 30 and 50 µg/mL). Therefore, the recovery tests were performed by extraction of the compound under study, present in water matrices, according to the proposed technique. The results obtained for extraction of the seven EDCs from water, as well as the respective coefficients of variation, are presented in (Table 7).

Chromatograms obtained from the analysis of water samples by a combine of SPE and GC–MS. a Blank extract, b spiked water sample
(5) Detection and quantification limits The detection limit (LOD) of the proposed method was determined as being equal to at least three times the baseline signal (noise) obtained for water samples free of EDCs (blank), fortified with EDCs, submitted to the SPE technique and analyzed by GC–MS. The quantification limit (LOQ) was determined as being the signal at least ten times greater than the noise signal. Detection limits were between 0.3 and 2.0 ng/L and quantification limits were between 1 and 10 ng/L (Table 7). Several authors developed methods for the extraction and detection of some EDCs in liquid samples (Table 6) with LODs and recovery similar to those determined in this work. However, these methods examine only one family of EDCs at a time. For the simultaneous detection of different EDCs, the LOD and recovery founds in the present work showed better results compared to those reported previously (Sghaier et al. 2017).
Application
The developed method was applied to the samples collected from two WWTPs both the influent and the effluent in Tunis, capital of Tunisia. Table 8 presents the concentration of every individual EDC detected in each sampling station. Among the selected hormones, E1 was detected in all sampling points. The concentration of E1 was found to be 10.3–23.6 ng/L, which was similar to other research reported in Australia, Canada and Japan 17.3–19.6 ng/L (Sun et al. 2014) and was slightly lower than that founded in China 69.3–280 in ng/L (Xu et al. 2014). It should be mentioned that in some cases the concentration of EE2 and E1 detected in the effluent was higher than that detected in the influents, which could be explained either by deconjugation or retransformation of conjugated compounds during treatment into the original compounds. αE2 and TST were not detected in the two samples, this may be because there instability and degradation in the WWTP. The concentration of BPA and NP detected in S1 was higher than those founded in S2. It may be due to the type of wastewater. The efficiencies of treatment were for ranged between 8 and 100%. Above all, the result showed that global eliminations yield were ≤ 45% for both Tunis WWTPs with 14 and 45%, respectively, for Chotrana WWPT (S) and Médina Jadida-Ben Arous (S2). Médina Jadida-Ben Arous WWTP showed better treatment than Chotrana WWPT. However, surprisingly for the case of EE2 detected in Médina Jadida-Ben Arous WWTP, the contamination of EE2 in the output (11.3 ± 1.8 ng/L) was higher than that present in the input (Table 8). This is likely a result of drug residues entering the WWTP in their conjugated form and becoming microbially deconjugated, thus releasing the parent compounds into the treated wastewater (Evgenidou et al. 2015).
Conclusion
An analytical method for the simultaneous extraction, preconcentration, and determination of estrogens (estrone, 17α-estradiol and 17α-ethinylestradiol), androgens (testosterone), progestogens (progesterone) and phenolic compound (bisphenol A and 4-nonylphenol) in wastewater has been optimized. The SPE has been used for the extraction/preconcentration step and combined with GC–MS for quantification in SIS mode. Doehlert matrix was used to build a mathematical model. The optimized method proved to be effective for the seven endocrine disrupter compounds. The LOQs were satisfied with the value between 1 and 10 ng/L of sample. In addition, the method presented high recoveries, up to 91.9%, with RSD below 2%, demonstrating good accuracy and precision. Linearity values were adequate with the values of r2 higher than 0.996. The application of the method to samples from two different WWTPs showed that the concentrations of EDCs found ranged from 4.5 to 36.8 ng/L and some of them (17α-estradiol, 17α-ethinylestradiol and testosterone) were not detected in the two wastewater samples, and other, such as progesterone only in one sample.
References
Aydin E, Talinli I (2013) Analysis, occurrence and fate of commonly used pharmaceuticals and hormones in the Buyukcekmece Watershed, Turkey. Chemosphere 90:2004–2012. doi:10.1016/j.chemosphere.2012.10.074
Ballesteros-Gómez A, Rubio S, Pérez-Bendito D (2009) Analytical methods for the determination of bisphenol A in food. J Chromatogr A 1216:449–469. doi:10.1016/j.chroma.2008.06.037
Belhaj D, Baccar R, Jaabiri I et al (2015) Fate of selected estrogenic hormones in an urban sewage treatment plant in Tunisia (North Africa). Sci Total Environ 505:154–160. doi:10.1016/j.scitotenv.2014.10.018
Evgenidou EN, Konstantinou IK, Lambropoulou DA (2015) Occurrence and removal of transformation products of PPCPs and illicit drugs in wastewaters: a review. Sci Total Environ 505:905–926. doi:10.1016/j.scitotenv.2014.10.021
Facco JF, Martins ML, Bernardi G et al (2015) Optimization and validation of a multiresidue method for pesticide determination in maize using gas chromatography coupled to tandem mass spectrometry. Anal Methods 7:359–365. doi:10.1039/C4AY01970A
Friedrich MT, Martins ML, Prestes OD, Zanella R (2016) Use of factorial design in the development of multiresidue method for determination of pesticide residues in wheat by liquid chromatography-tandem mass spectrometry. Food Anal Methods 9:2541–2551. doi:10.1007/s12161-016-0447-0
Gao L, Sun L, Wan S et al (2013) Degradation kinetics and mechanism of emerging contaminants in water by dielectric barrier discharge non-thermal plasma: the case of 17β-Estradiol. Chem Eng J 228:790–798. doi:10.1016/j.cej.2013.05.079
Gorga M, Petrovic M, Barceló D (2013) Multi-residue analytical method for the determination of endocrine disruptors and related compounds in river and waste water using dual column liquid chromatography switching system coupled to mass spectrometry. J Chromatogr A 1295:57–66. doi:10.1016/j.chroma.2013.04.028
Grover DP, Zhang ZL, Readman JW, Zhou JL (2009) A comparison of three analytical techniques for the measurement of steroidal estrogens in environmental water samples. Talanta 78:1204–1210. doi:10.1016/j.talanta.2008.12.049
Guedes-Alonso R, Sosa-Ferrera Z, Santana-Rodríguez JJ (2013) Simultaneous determination of hormonal residues in treated waters using ultrahigh performance liquid chromatography-tandem mass spectrometry. J Anal Methods Chem 2013:1–8. doi:10.1155/2013/210653
Hibbert DB (2012) Experimental design in chromatography: a tutorial review. J Chromatogr B 910:2–13. doi:10.1016/j.jchromb.2012.01.020
Huang B, Pan X-J, Wan X et al (2011) Simultaneous determination of steroid endocrine disrupting chemicals in water by solid phase extraction-derivatization- gas chromatographic-mass spectrometry. Chin J Anal Chem 39:449–454. doi:10.1016/S1872-2040(10)60431-8
Jiang J-Q, Zhou Z, Sharma VK (2013) Occurrence, transportation, monitoring and treatment of emerging micro-pollutants in waste water—a review from global views. Microchem J 110:292–300. doi:10.1016/j.microc.2013.04.014
Jin W, Huang B, Wang B et al (2013) Simultaneous determination of androgens and progestogen in surface water and sediment by gas chromatography-mass spectrometry. Chin J Anal Chem 41:205–209. doi:10.1016/S1872-2040(13)60628-3
Karnjanapiboonwong A, Suski JG, Shah AA, Cai Q, Morse AN, Anderson TA (2011) Occurrence of PPCPs at a wastewater treatment plant and in soil and groundwater at a land application site. Water Air Soil Pollut 216:257–273. doi:10.1007/s11270-010-0532-8
Kemmerich M, Rizzetti TM, Martins ML et al (2015) Optimization by central composite design of a modified QuEChERS method for extraction of pesticide multiresidue in sweet pepper and analysis by ultra-high-performance liquid chromatography–tandem mass spectrometry. Food Anal Methods 8:728–739. doi:10.1007/s12161-014-9951-2
Labadie P, Budzinski H (2005) Determination of steroidal hormone profiles along the Jalle d’Eysines River (near Bordeaux, France). Environ Sci Technol 39:5113–5120. doi:10.1021/es048443g
Latrous El Atrache L, Ben Sghaier R, Bejaoui Kefi B et al (2013) Factorial design optimization of experimental variables in preconcentration of carbamates pesticides in water samples using solid phase extraction and liquid chromatography–electrospray-mass spectrometry determination. Talanta 117:392–398. doi:10.1016/j.talanta.2013.09.032
Laurenson JP, Bloom RA, Page S, Sadrieh N (2014) Ethinyl estradiol and other human pharmaceutical estrogens in the aquatic environment: a review of recent risk Assessment Data. AAPS J 16:299–310. doi:10.1208/s12248-014-9561-3
Lee I-S, Lee S-H, Oh J-E (2010) Occurrence and fate of synthetic musk compounds in water environment. Water Res 44:214–222. doi:10.1016/j.watres.2009.08.049
Munaretto JS, Ferronato G, Ribeiro LC et al (2013) Development of a multiresidue method for the determination of endocrine disrupters in fish fillet using gas chromatography–triple quadrupole tandem mass spectrometry. Talanta 116:827–834. doi:10.1016/j.talanta.2013.07.047
Net S, Rabodonirina S, Sghaier RB et al (2015) Distribution of phthalates, pesticides and drug residues in the dissolved, particulate and sedimentary phases from transboundary rivers (France–Belgium). Sci Total Environ 521–522:152–159. doi:10.1016/j.scitotenv.2015.03.087
Nie Y, Qiang Z, Zhang H, Ben W (2012) Fate and seasonal variation of endocrine-disrupting chemicals in a sewage treatment plant with A/A/O process. Sep Purif Technol 84:9–15. doi:10.1016/j.seppur.2011.01.030
Nielsen S (2014) Food analysis. Springer Science & Business Media, New York
Rabodonirina S, Net S, Ouddane B et al (2015) Distribution of persistent organic pollutants (PAHs, Me-PAHs, PCBs) in dissolved, particulate and sedimentary phases in freshwater systems. Environ Pollut 206:38–48. doi:10.1016/j.envpol.2015.06.023
Reinen J, Suter MJ-F, Vögeli AC et al (2010) Endocrine disrupting chemicals—linking internal exposure to vitellogenin levels and ovotestis in Abramis brama from Dutch surface waters. Environ Toxicol Pharmacol 30:209–223. doi:10.1016/j.etap.2010.07.004
Rice SL, Hale RC (2009) Parameters for ultra-performance liquid chromatographic/tandem mass spectrometric analysis of selected androgens versus estrogens in aqueous matrices. Anal Chem 81:6716–6724. doi:10.1021/ac900134m
Sghaier RB, Net S, Ghorbel-Abid I et al (2017) Simultaneous detection of 13 endocrine disrupting chemicals in water by a combination of SPE-BSTFA derivatization and GC–MS in transboundary rivers (France–Belgium). Water Air Soil Pollut. doi:10.1007/s11270-016-3195-2
Sim W-J, Lee J-W, Oh J-E (2010) Occurrence and fate of pharmaceuticals in wastewater treatment plants and rivers in Korea. Environ Pollut 158:1938–1947. doi:10.1016/j.envpol.2009.10.036
Stafiej A, Pyrzynska K, Regan F (2007) Determination of anti-inflammatory drugs and estrogens in water by HPLC with UV detection. J Sep Sci 30:985–991. doi:10.1002/jssc.200600433
Sun Y, Huang H, Sun Y et al (2014) Occurrence of estrogenic endocrine disrupting chemicals concern in sewage plant effluent. Front Environ Sci Eng 8:18–26. doi:10.1007/s11783-013-0573-5
Thurman EM, Writer JH, Ferrer I (2013) Injection port derivatization for GC/MS–MS. In: Comprehensive analytical chemistry. Elsevier, pp 115–141
Vandenberg LN, Colborn T, Hayes TB et al (2012) Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev 33:378–455. doi:10.1210/er.2011-1050
Xu Y, Xu N, Llewellyn NR, Tao H (2014) Occurrence and removal of free and conjugated estrogens in wastewater and sludge in five sewage treatment plants. Env Sci Process Impacts 16:262–270. doi:10.1039/C3EM00199G
Yang S, Hai FI, Nghiem LD et al (2013) Understanding the factors controlling the removal of trace organic contaminants by white-rot fungi and their lignin modifying enzymes: a critical review. Bioresour Technol 141:97–108
Yang Y, Cao X, Zhang M, Wang J (2015) Occurrence and distribution of endocrine-disrupting compounds in the Honghu Lake and East Dongting Lake along the Central Yangtze River, China. Environ Sci Pollut Res 22:17644–17652. doi:10.1007/s11356-015-4980-y
Yu Y, Wu L (2015) Determination and occurrence of endocrine disrupting compounds, pharmaceuticals and personal care products in fish (Morone saxatilis). Front Environ Sci Eng 9:475–481. doi:10.1007/s11783-014-0640-6
Zhang R, Li N, Wang C et al (2011) Ionic liquid foam floatation coupled with solid phase extraction for separation and determination of hormones by high-performance liquid chromatography. Anal Chim Acta 704:98–109. doi:10.1016/j.aca.2011.08.016
Zoeller RT, Brown TR, Doan LL et al (2012) Endocrine-disrupting chemicals and public health protection: a Statement of Principles from The Endocrine Society. Endocrinology 153:4097–4110. doi:10.1210/en.2012-1422
Acknowledgements
The authors are grateful for the support provided by CPER CLIMBIO project. We acknowledge the financial support from the Tunisian Minister of Higher Education and Scientific Research, which provided a PhD scholarship for Rafika Ben Sghaier.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ben Sghaier, R., Tlili, I., Latrous El Atrache, L. et al. A Combination of Factorial Design, Off-line SPE and GC–MS Method for Quantifying Seven Endocrine Disrupting Compounds in Water. Int J Environ Res 11, 613–624 (2017). https://doi.org/10.1007/s41742-017-0054-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41742-017-0054-y