
Abstract
Halide perovskite nanocrystals (HPNCs) are currently among the most intensely investigated group of materials. Structurally related to the bulk halide perovskites (HPs), HPNCs are nanostructures with distinct chemical, optical, and electronic properties and significant practical potential. One of the keys to the effective exploitation of the HPNCs in advanced technologies is the development of controllable, reproducible, and scalable methods for preparation of materials with desired compositions, phases, and shapes and low defect content. Another important condition is a quantitative understanding of factors affecting the chemical stability and the optical and electronic properties of HPNCs. Here we review important recent developments in these areas. Following a brief historical prospective, we provide an overview of known chemical methods for preparation of HPNCs and approaches used to control their composition, phase, size, and shape. We then review studies of the relationship between the chemical composition and optical properties of HPNCs, degradation mechanisms, and effects of charge injection. Finally, we provide a short summary and an outlook. The aim of this review is not to provide a comprehensive summary of all relevant literature but rather a selection of highlights, which, in the subjective view of the authors, provide the most significant recent observations and relevant analyses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The first synthetic halide perovskites (HPs) were reported in 1893, when H.L. Wells described preparation of inorganic halide perovskites CsPbX3, CsPb2X5, and Cs4PbX6, (where X is Cl− and Br−) [1]. In the 1950s, C.K. Møller not only identified/confirmed the perovskite structure of these cesium lead halides but also observed photoconductivity in these materials. In the 1990s, D.B. Mitzi and coworkers investigated and described magnetic, electronic, optical, and electroluminescent properties of a variety of inorganic and organic–inorganic halide perovskite materials [2,3,4]. In 2006–2009, in search of more efficient light absorbers, T. Miyasaka and coworkers were the first to use hybrid HP MAPbX3 (MA is CH3NH3+ and X is Br− and I−) as a light-absorbing layer in a dye-sensitized solar cell and demonstrated first perovskite solar cells with ~3.81% (MAPbI3) and ~3.13% (MAPbBr3) Power conversion efficiencies (PCEs) [5]. Shortly after, teams led by T.N. Murakami, T. Miyasaka, H. Snaith, N.G. Park, and M. Grätzel reported first solid-state perovskite solar cells employing MAPbI3−xClx mixed halide perovskite and MAPbI3 with the PCEs between 8% and 10% [6, 7]. These initial advances stimulated vigorous effort in the development of HP solar cells with various architectures and within a span of few years, at the improvement rate unprecedented for photovoltaic technologies, perovskite solar cells have reached PCEs of 26.1% [8,9,10,11,12]. The tandem solar cells combining the HPs with Si have recently reached a record PCE of 32.5% [13]. Despite of the great promise, the commercial exploitation of HP solar cells faces challenges associated with the limited thermal and chemical stability of HPs and toxicity associated with the presence of lead. Intensive research is currently underway to explore solutions for overcoming these challenges and advancing the industrialization of photovoltaics [14, 15].
Shortly after the initial reports describing the preparation and properties of HP solar cells, several studies [16,17,18] showed that unusual excitonic and charge transport properties of HPs could be exploited not only for applications requiring light-induced charge separation and production of current but also for applications where the processes are reversed, i.e., where injected charges recombine radiatively together under an applied external potential. High color purity and low intrinsic rate of nonradiative recombination make HPs very appealing candidates for development of light-emitting diodes (LEDs) and lasers [19]. However, some early studies also revealed that bulk HPs, formed as solid films, have relatively low photoluminescence quantum yields (PLQY), primarily due to the defects forming during the heating and solvent evaporation step of the spin-cast process [20]. The defects at the grain boundaries of the HP crystallites were shown to act as fast nonradiative recombination centers, contributing to observation of a weak photoluminescence (PL) [21]. An important advance in this area was a report by Droseros et al. [22] who showed that PLQY of HPs could be significantly improved simply by reducing the size of the grains from the micrometer- to millimeter-size range crystallites, typical for HP thin films, to the nanometer-size crystals. The improved PLQY in nanocrystals (NCs) was shown to be due to the improved passivation of surface traps and resulting increase in the fraction of excited states decaying radiatively.
The observation of enhanced PL in nanocrystalline HPs and the first reports on solution-based synthesis of HPNCs in 2014 (Fig. 1) [23], have stimulated growing interest in this class of materials. The transition into nanoregime not only helps to increase PLQY but also provides an access to tunability of the electronic structure via the quantum confinement effects. Within a few years, syntheses of high-quality HPNCs with various chemical compositions, sizes, and shapes were reported. The experimental results show that in HPNCs halogen type, structure, size, and phase can be controlled not only during the synthesis but also during the postsynthesis processing (i.e., ion exchange, amine treatment, external photon radiation, etc.). The increased quantum confinement resulting from reduction in size of the crystals at the nanoscale leads to enhanced radiative recombination, which is beneficial for the light-emitting applications. Reduction in the size, however, also leads to an increase in the surface to volume ratio and potentially higher defect concentration. The point defects can also be introduced by doping of HPNCs. Another important challenge is the chemical and phase stability of HPNCs upon exposure to various environmental factors as well as under conditions of applied potential. Understanding of the interplay of these factors is essential for effective exploitation of the benefits of the HPNCs in optoelectronic applications. In this review, we provide a summary of recent literature that, in our view, provides the most relevant analyses and answers to these important challenges and questions.

Reproduced with permission from ref. [23]. Copyright 2014 American Chemical Society
The conventional solution-based synthesis of hybrid perovskite nanoparticles. The green emission of the final product confirms the formation of highly emissive MAPbBr3 NCs.
2 Structure of Halide Perovskites
In bulk HPs, with the general chemical formula ABX3 (A and B are cations and X is a halogen ion), the ions are arranged in a crystal lattice so that the B-site cation is surrounded by six nearest neighbor X anions and the A site cation is located inside the cavity formed by eight corner sharing BX6 octahedra (Fig. 2a). The probability that an ABX3 compound will form a perovskite crystal is related to the sizes of the A, B, and X ions. This is commonly expressed in terms of two parameters: (1) “tolerance factor, (t)” where \(t=({r}_{A}+{r}_{X})/[\sqrt{2\left({r}_{B}+{r}_{X}\right)}]\); (2) “octahedral factor, (μ)” where \(\mu ={r}_{B}/{r}_{X}\). Here rA, rB, and rX are the ionic radii of the A, B, and X ions, respectively. The compound that forms a perovskite phase typically meets the conditions: μ ≥ 0.41 and 0.8 < t < 1.0 [24, 25]. Depending on the type of the A cation, HPs can be categorized into: (a) organic HPs, where A is methylammonium (MA), formamidinium (FA), or other monovalent organic cation or (b) inorganic HPs, where A is Cs+, Rb+, or other monovalent inorganic cation. The B site is typically occupied by divalent ions such as Pb2+, Sn2+, or Ge2+. X is halogen anion, such as Cl−, Br−, or I−. Depending on the temperature, HPs can adopt multiple crystal phases with different symmetry (Fig. 2b). The most studied HP, MAPbI3, forms, in a temperature range of 160–330 K, a tetragonal phase where two of the unit cell lengths are equivalent (a = b ≠ c) [26, 27]. At temperatures below 160 K, it forms an orthorhombic structure where all three unit cell lengths are different (a ≠ b ≠ c). At temperatures above 330 K, the symmetry of the structure increases, with MAPbI3 adopting the cubic crystal structure (a = b = c) [26]. The electronic structure of the bulk HPs is mainly dictated by the extent of mixing of the B cation’s and X anion’s “s” and “p” orbitals (Fig. 2c) [28]. As a result, the optical properties of the HP can be effectively tuned throughout the visible spectral range by partial or complete substitution of the halogen ion. The above principles generally apply to the HPNCs as well. However, when the size of the HP is reduced to the nanoscale level, the electronic structure, optical, and electronic properties are further affected by the size of the NCs due to the quantum confinement effect (Fig. 2d, e). The effects of quantum confinement on the optical properties of HPNCs are discussed in more detail in following sections.

a Three-dimensional (3D) metal halide perovskite structure. In the figure, the green spheres represent halogen atom and black sphere represent organic/inorganic cation sites, inside the eight-corner sharing BX6 octahedra. The spheres inside the octahedra represent the B atoms. b Various crystal phases adopted by MAPbI3 HP. Adopted from ref. [27]. c Molecular orbital diagram for the interaction between the B-site metal atoms and the X-site halogen atoms. Reproduced with permission from ref. [28]. d TEM image of CsPbBr3 HPNCs. e Depiction of the effect of the quantum confinement effect on the photoluminescence of the ABX3 nano cubes
3 Synthetic Strategies for Preparation of HPNCs
3.1 Reprecipitation Method
“Reprecipitation” is an old synthesis method that was first used in the production of crystalline salts in clay pots in south Poland as early as 3500 BC [29]. In this process, ionic compounds are typically dissolved in a suitable solvent until the saturation limit is achieved. The saturated solution is then rapidly transferred into another solvent, in which the ionic compound has a low solubility. This leads to instantaneous reprecipitation and initiation of the crystallization, which continues until the system reaches a thermodynamic equilibrium. This method has been used successfully in the preparation of many crystalline compounds and polymers [30, 31]. In 2012, Papavassiliou et al. [32] reported the first synthesis of hybrid HP via reprecipitation method. (CH3NH3)(C4H9NH3)2Pb2X7, (CH3NH3)(CH3C6H4CH2NH3)2Pb2X7, and CH3NH3PbX3, (X = Cl−, Br−, or I−) with the electronic structure of the bulk material were synthesized by a procedure, where a saturated solution of the precursor salts were first prepared in a highly polar solvent [e.g., dimethylformamide (DMF)], and this solution was subsequently added drop-wise into a nonpolar solvent (e.g., toluene). This led to a rapid precipitation of the HP crystals. Few years later, hybrid (MAPbX3) HPNCs were prepared by the reprecipitation method [33]. The method was further extended to the preparation of other mixed HPNCs, with composition ABX3, where A is MA+, FA+, or Cs+, B is Pb2+ or Sn2+, and X is Cl−, Br−, or I−.[34]
3.2 Ligand-Assisted Reprecipitation (LARP)
When the reprecipitation is done in the presence of ambipolar compounds (surface ligands), which adsorb onto the surfaces of the forming crystals during their growth and help optimize their size, and shape, the method is known as the “ligand-assisted reprecipitation” (LARP). Schmidt et al. [23] were the first to report the solution-based synthesis of MAPbBr3 HPNCs in the presence of ligands. In the synthesis, long-chain ammonium bromide salt with an organic acid was heated (80 °C) in an organic solvent [1-octadecene (1-ODE)]. The subsequent addition of perovskite precursors (PbBr2 and MABr, predissolved in DMF) produced a yellow dispersion, from which the HPNCs immediately precipitated out upon the addition of acetone. The first successful synthesis of various halide hybrid perovskite QDs by the LARP method was reported by Zhang et al. in 2015 [33]. The approach is schematically summarized in Fig. 3a–c. In the process, perovskite precursors (PbX2 and MAX, where X is Cl−, Br−, or I−) are dissolved in a highly polar solvent (DMF), which is followed by an addition of a long-chain amine (e.g., n-octylamine) and an organic acid (e.g., oleic acid). Then, the solution is dropped into vigorously stirred toluene, which leads to the formation of HPNCs. This method was successfully used in the preparation of HPNCs with various halogen compositions and tailored emission properties over the visible spectral range, as shown in Fig. 3d and e. Authors found, that in the absence of ligands, such as n-octylamine, the crystallization is faster, leading to aggregation of the NCs. In addition, it was determined that the addition of oleic acid leads to a significant improvement of the photoluminescence (PL) stability of the prepared HPNCs. In the absence of oleic acid, the reaction yields HPNCs, which rapidly lose emissivity. Thus, oleic acid has multiple functions: it prevents aggregation of HPNCs, improves their chemical stability, and stabilizes their PL. TEM images of the HPNCs prepared by this method are shown in Fig. 3f. In 2016, Levchuk et al. [35] utilized LARP method to synthesize FAPbX3 NCs. The synthesis protocol used was the same as discussed above, except the FAX (FA is CH5N3+ and X is Cl−, Br−, or I−) was substituted with MAX as the perovskite precursor. The significant difference, however, was the use of chloroform, instead of toluene, as the antisolvent for the precipitation of FAPbX3 HPNCs. Synthesis in toluene did not result in the formation of FAPbI3 NCs, while FAPbBr3 or FAPbCl3 immediately formed a turbid suspension of large particles and agglomerates. Similarly, in 2016, Li et al. [36] showed that CsPbX3 HPNCs could also be produced using the LARP approach at room temperature by merely replacing MAX with CsX. It was found that not only hybrid or inorganic perovskite but also other kinds of lead-free perovskites could be synthesized by this method.

Reproduced with permission from Ref. [33]. Copyright 2015 American Chemical Society
a Schematic illustration of the LARP reaction process. Drop-wise addition of a perovskite precursor solution in a polar solvent, such as DMF, into a nonpolar solvent, such as toluene, leads to instantaneous precipitation of the perovskite NPs. b Summary of the starting materials used in the precursor solution. c A photograph of the colloidal MAPbBr3 solution prepared in step 1 of the LARP process. d Digital images of various MAPbX3 (X is Cl−, Br−, I−) NCs under ambient light (top) and a 365 nm UV lamp (bottom). e CIE map indicating the characteristics of various prepared MAPbX3 QDs. f TEM image of MAPbBr3 NCs.
Leng et al. [37] synthesized vacancy-ordered triple halide perovskite, i.e., MA3Bi2X9 (X is Cl−, Br−, or I−) HPNCs by the LARP method. The authors showed that for these nonstandard HPNCs they could tune the NC emission from 360 to 540 nm by varying the halogen composition from Cl− to I via Br and also achieve ~12% PLQY. The same research group also developed a similar method for synthesis of Cs3Bi2X9 perovskites [38, 39]. Since it was found that BiBr3 is only partially soluble in toluene, to fabricate vacancy-ordered triple halide perovskites instead of toluene, octane was used as a “poor solvent” to reprecipitate the HPNCs. In 2017, Zhang et al. [40] showed that more exotic perovskite structures, such as Cs3Sb2Br9, can also be effectively synthesized by the LARP method. They used this method to fabricate blue-emitting Cs3Sb2Br9 perovskite QDs (Fig. 4) with ~46% PLQY at 410 nm. The above examples illustrate that LARP is a convenient and versatile method for preparation of HPNCs. Perovskite NCs with various shapes, sizes, and halogen compositions can be prepared with a simple chemical setup at room temperature. Moreover, LARP is a convenient method for preparation of HPNCs ranging from hybrid to inorganic to alternative perovskite structures (e.g., vacancy-ordered triple halide perovskite, double-perovskite etc.), to structures with mixed halogen compositions. However, the method also has several important drawbacks. In the initial step, highly polar organic solvents, such as DMF/dimethyl sulfoxide (DMSO; “good” solvents) are typically used to effectively dissolve various polar precursors. Even after the recovery of the HPNCs with a nonpolar (“poor”) solvent, the product NCs will usually retain some polar solvent molecules on their surface. The remains of the polar solvents tend to chemically interact with and gradually degrade the freshly formed NPNCs. Zhang et al. [41] have studied the effect of solvent during the synthesis of MAPbI3 NCs. On the basis of Lewis acid–base theory, the authors proposed that the interaction between the DMF/DMSO (coordinating solvents) and the HPNCs precursor PbI2 led to the formation of lead oxide (PbO) as shown schematically in Fig. 5. During the recovery of the HPNCs in nonpolar solvent (toluene), in the open air the authors observed formation of white flocculent precipitate of MAPbI3·H2O. Thus, the interaction between the PbI2 and DMF initially creates iodine vacancies associated with the formation of PbO, which in the presence of moisture from air or solvent subsequently transform into MAPbI3·H2O, which eventually degrades the perovskite structure.

Reproduced with permission from ref. [40]. Copyright 2017 American Chemical Society
a, b Schematic illustration of the reaction process for LARP based chemical synthesis technique for Cs3Sb2Br9 NPs. c Digital images of a Cs3Sb2Br9 NPs colloidal solution with and without 365 nm UV light excitation.

Reproduced with permission from ref. [41]. Copyright 2017 American Chemical Society
Effect of solvent on the crystal structure of MAPbI3 NCs. Schematic illustration of the MAPbI3 formation from MAI and PbI2 in coordinating solvents (top) and noncoordinating solvents (bottom).
3.3 Hot-Injection Method
The hot-injection method was first introduced by Murray et al. [42] in 1993 for the synthesis of cadmium chalcogenide NCs. Rapid injection of organometallic precursor(s) into a hot-solution (> 150 °C) of a high boiling point solvent [(e.g., trioctylphospineoxide (TOPO)], was shown to be a very effective method for preparation of CdE (E is sulfur, selenium, or tellurium) NCs with narrow (< 10%) size dispersions [43]. This is a result of a high control of the nucleation and growth that this approach allows at various stages of the reaction. A rapid injection of a precursor leads to supersaturation of the hot solution by the monomers, followed by a quick nucleation (formation of small NC nuclei) and a subsequent slower NC growth process. The size and shape of the NCs can be effectively controlled via adjustment in the reaction time and temperature, choice of ligands, solvents, etc.
In 2015, Protesescu et al. [44] used for the first time the hot-injection method for the preparation of HPNCs with compositions CsPbX3, where X is Cl−, Br−, or I−. The experimental setup and conditions are schematically shown in Fig. 6a. The HPNCs prepared by the hot-injection method remain stable in terms of their composition and optical properties for a longer period of time (2–3 months) than NCs prepared by the LARP method (few weeks). This has been attributed mostly to the absence of a polar solvents in the hot-injection method. In a typical synthesis of CsPbX3 (X is Cl−, Br−, or I−), first, Cs-oleate was prepared by reaction of Cs2CO3 with oleic acid in 1-octadecene (1-ODE) at 120 °C with vigorous stirring. Separately, PbX2 (X is Cl−, Br−, or I−) and oleic acid (OA) and oleylamine (OAm) were dissolved in 1-ODE and heated up to 140 °C, while being stirred. The reaction yielded Pb-oleates and free Xˉ ions. Rapid injection of Cs-oleate into the hot Pb-solution, followed by rapid (4–10 s) reaction quenching with an ice bath led to the formation of CsPbX3 NCs. The amounts of organic amines, carboxylic acids, and temperature were shown to play a critical role in controlling the shape and size of the NCs. Rapid quenching of the reaction after the injection of Cs-oleate into the Pb and halogen precursor helped to achieve uniformity in the shape, as shown in Fig. 6b. In addition, the halogen composition (from I− to Br− to Cl−) in CsPbX3 was controlled simply by taking the appropriate ratios of PbX2 salts, allowing production of HPNCs with PL tunability across the entire visible spectrum (410–700 nm), as shown in the Fig. 6c–d. The hot-injection approach was also used in the synthesis of less typical HPNCs. For example, Akkerman et al. [45] used this method to prepare Cs4PbBr6 NCs by adding an excess amount of the Cs-oleate precursor. Similarly, an excess of PbBr2 was shown to yield CsPb2Br5 PNCs [46]. Furthermore, a modified hot-injection approach was adopted by Protesescu et al. [47] for the colloidal synthesis of FAPbBr3 PNCs by replacing Cs-oleate with FA-oleate. Lignos et al. [48] extended this approach to the synthesis of mixed-halide FAPb(Cl1–xBrx)3 HPNCs, with a stable blue PL emission. In a separate study, FAPb(I1–xBrx)3 HPNCs with PL tunable from 570 to 780 nm and Cs1–xFAxPbI3 PNCs with a controlled emission from 690 to 780 nm were prepared by Protesescu et al. [49] using a similar methodology. With these and other advances, the hot-injection method has been now well established as a standard approach for preparation of HPNCs ranging from 3D-CsPbX3 to 2D-CsPb2Br5 and 0D-Cs4PbBr6. This method can also be used for synthesis of FA-based hybrid HPNCs, with compositions ranging from Cl− to Br− to I−, with range of sizes and shapes. Until 2015, no report has shown full range halogen composition-controlled synthesis of structurally stable MA+-based hybrid halide perovskite MAPbX3 (X is I−, Br−, or Cl− along with I/Br and Br/Cl mixed halide) via the hot-injection method. This is because, in contrast to Cs+ and FA+ precursors, the MA-oleate is difficult to prepare, as MA-acetate is not readily available. This made MAPbX3 synthesis, via the conventional hot-injection method difficult. In addition, MA+ is volatile in ambient conditions as its boiling temperature is 4 °C. Hence, this molecule is typically stored under THF or alcohols, (e.g., methanol or ethanol) with boiling point in the 60–70 °C range. In 2015, Vybornyi et al. [50] used a modified hot-injection method to synthesize MAPbX3, where X is I− or Br− only. In this approach, the PbX2 was first dissolved, together with organic acid and amine, in 1-ODE heated to 120 °C. Subsequently, after dissolving the temperature was decreased to ~60–50 °C to reduce the risk of MA+ decomposition or evaporation. Injection of MA+ led to rapid formation of MAPbX3 (X is Br− or I−). Although the synthesis of Br or I-based HPNCs was successful, the method was not effective in producing mixed compositions with I/Br and Br/Cl. In 2018, Imran et al. [51] reported a “three-precursor” hot-injection method for the synthesis of HPNCs. This novel approach was based on using benzoyl halides (C7H5OX, X is Cl−, Br−, or I−) as sources of halogen and lead oxide (PbO) as a source of lead ions, instead of the conventional reaction of PbX2 (X is Cl−, Br−, or I−) salts with MA molecule (in THF). Separation of the source of the lead and the halogen into two compounds provided a better control over the relative amounts of the ions in the reaction and, thus, finer tunability of the reaction conditions. In a typical synthesis, the PbO and organic ligands were heated in 1-ODE at 125 °C to solubilize the salts. Subsequently, the temperature was lowered to 65 °C, and MAX was injected, followed by the injection of the benzoyl halide. This approach was successfully used in preparation of APbX3 (A is Cs+, MA+, or FA+ and X is Cl−, Br−, or I−) NCs. In spite of these important advances, the “two-precursor”-based hot-injection synthesis of composition-controlled MA-based hybrid HPNCs has been lacking. In 2019 Roy et al. [52] used a modified hot-injection method by including TOP in the reaction to enhance the solubility of PbX2 in 1-ODE. They used “two-precursor”-based hot-injection method to successfully synthesize all composition-controlled MA-based HPNCs, without the need to use polar solvent or any specialty precursor. On the other side TOPO/TOP classical ligands, which are used to synthesis traditional chalcogenide nanocrystals, are also used in perovskite synthesis to stabilize the perovskite structure. In 2017 Liu et al. [53] reported use of TOP − PbI2 as precursor for synthesis of CsPbI3 NC synthesis via the hot-injection method. In this method presynthesized TOP − PbI2 was injected into the reaction mixture of Cs2CO3, oleic acid, and oleylamine in ODE. They are also able to tune the size of the NC by varying the reaction temperature. They confirm that the use of TOP leads to higher structural stability and enhanced the PLQY by comparing non-TOP caped CsPbI3 NCs. In another example, Wu et al. [54] use TOPO along with oleic acid and oleylamine to synthesis CsPbBr3 NCs in the hot-injection method. Use of TOPO leads to monodisperse CsPbBr3 nanocubes at high temperature, i.e., 260 °C. These TOPO caped NCs exhibited superior stability in ethanol as compared with that of OA/OAm-capped CsPbBr3 nanocubes, regardless of the reaction temperatures at which they were synthesized.

Reproduced with permission from ref. [44] Copyright 2015 American Chemical Society
a Schematic diagram of the hot-injection method used for the synthesis of HPNCs. b TEM image of monodispersed CsPbBr3 nanoparticles obtained using the hot-injection method. c Digital image of CsPbX3 (X is Cl, Br, or I) nanoparticles dispersions (different halide composition in each vial) in toluene under an ultraviolet (UV) lamp (λ = 365 nm). d PL emission spectra of CsPbX3 perovskite NPs with different halogen compositions. PL emission covers the entire visible spectrum (410–700 nm).
3.4 Less Conventional Approaches
The development of the hot-injection and LARP methods allowed for preparation of HPNCs with broad range compositions. Hot injection is very effective method for controlling the HPNC composition, size, and shape, along with the size and shape dispersion in small-scale reactions. Due to the limited stability of HPNCs at ambient conditions, the hot-injection method requires use of strictly anhydrous solvents and inert atmosphere. This means that a relatively complex chemical apparatus must be used, and the syntheses are more time consuming compared with the LARP method. In addition, due to the injection step, the hot-injection method is difficult to scale up to the commercial production scales. Increase in the quantities of the precursor at high temperatures leads to inhomogeneous nucleation and growth, which makes it difficult to control the shape and size of the HPNCs in a large-scale reaction. In addition, using the hot-injection method it is often difficult to produce halogen rich HPNCs, which were shown to have the most desirable PL properties [55]. To address these challenges, several alternative methods were developed to simplify the preparation of composition-controlled HPNCs. These methods are described in the following sections.
3.4.1 Solvothermal Synthesis
One of the methods proposed as an alternative to the hot-injection or LARP approach is solvothermal synthesis. Chen et al. [56] developed a simple solvothermal method for the preparation of HPNCs with uniform halogen composition. In a typical synthesis, cesium acetate and lead halides are mixed in a stainless autoclave containing 1‐ODE, with an organic acid and amines. The autoclave is placed in an oven and heated to 160 °C for 30 min. The reaction leads to the formation of composition-controlled CsPbX3 (X is Cl−, Br−, or I−, along with mixed halides) nanocubes, with PL tunable throughout the visible range. The method provides means not only for tuning of the composition and PL energy but also for morphology of the HPNCs. While the lower initial concentration of the precursors leads to formation of cubic HPNCs, higher precursor concentrations lead to formation of HP Nanorods (HPNRs). Parveen et al. [57] extended this method to the hybrid HPNCs (MAPbX3). In this variation, the amine/acid to precursor ratios were systematically varied to control the size of the NCs. The quantum confinement effects were seen and a systematic shift in the emission peak position is observed from green to blue.
3.4.2 Synthesis Using Microwave Radiation
In 2017, Pan et al. [58] described a new strategy for synthesis of HPNCs using microwave radiation. Cesium acetate (Cs-OAc), lead halide, organic ligands, trioctylphosphine oxide (TOPO), and 1-ODE were mixed together in a quartz tube and transferred into a microwave reactor. Heating the reaction mixture to 160 °C by microwave radiation led to formation of the CsPbX3 HPNCs. This method also allows for control of the halogen composition and the morphology of the HPNCs. The authors of the study found that the shape of the CsPbBr3 NCs varies from nanocubes to a nanoplatelets (NPLs) with change in the reaction temperature from 160 °C to 80 °C. It is also noted that rod-shaped CsPbBr3 HPNRs s can be prepared when the precursors are predissolved prior to the microwave treatment. Shamsi et al. [59] modified the microwave assisted method to prepare blue emitting quantum confined CsPbBr3 NCs. More recently, Liu et al. [60] further extended this approach and formulated a general strategy for synthesis of low-dimensional CsPbBr3 NPs with morphology tunable from QDs to NRs to NPLs. This was achieved by simple adjustment in the ratios of the oleic acid to oleylamine and 1-ODE to diethylene glycol butyl ether (DGBE) and careful variations in the concentration of PbBr2. Together, with the dimensional/morphological changes, the size of the HPNCs could also be optimized by a simple change of the applied microwave power.
3.4.3 Synthesis by Ultrasonication
Jang et al. [61]. reported the preparation of CsPbX3 (X is Cl−, Br−, or I−) and their mixed compositions by ultrasonication. In this approach, ultrasonic irradiation is used to accelerate dissolution of the precursors (AX and PbX2) in nonpolar solvent, such as toluene, and to control the growth rate of the HPNCs. The prolonged sonication was shown to lead to formation of HP nanowires (HPNWs). A similar approach reported by Tong et al. [62] for preparation of CsPbX3 NCs and the characterization of the prepared materials are graphically summarized in Fig. 7. As illustrated in the figure, the method provides a good control over the halogen composition as well as the optical properties of the Cs+, MA+, and FA+-based HPNCs. More recently, Roy et al. [63] developed a method based on ultra-sonication, which allows for control of the halogen composition in already phase-formed HPNCs, exploiting the significant ionic mobility and low activation energy for the ion exchange in these materials. In this method, two compositions of HPNCs (MAPbX3 and MAPbY3) are mixed together in a nonpolar solvent (e.g., toluene) and exposed to ultrasound waves. Upon sonic energy exposure, the Xˉ and Yˉ ions migrate from one HPNC structure to another to ultimately form mixed composition MAPb(X/Y)3. Thus, this method allows for a straightforward preparation of HPNCs with specific mixed halide compositions by appropriate stoichiometric mixing of the phase pure single halogen NCs, followed by a brief period of ultrasonication.

Reproduced with permission from Ref. [62]. Copyright 2016 Willy
a Scheme of the experiment used in the synthesis of the CsPbX3 NCs via the ultrasonication method. b Digital image of perovskite NCs with various halogen (X is Cl−, Br−, or I−) composition in hexane under visible light (top) and UV light (bottom, λ = 367 nm). c Absorption and emission spectra with the PLQY values of the HPNCs. d Products of large-scale synthesis of HPNCs. e PL decay dynamics of the HPNCs (τ ≈ 2.5–34.5 ns).
3.4.4 Mechanochemical Methods
In addition to wet chemical routes, described in previous sections, HPNCs can also be prepared by mechanochemical methods. Mechanochemistry refers to the branch of chemistry where the energy necessary to drive the chemical reaction is provided by a mechanical force (Figs. 8 and 9). The mechanochemical synthesis offers several advantages. The process is entirely solvent-free (or uses small amounts of volatile solvents), which makes it convenient for scale up to industrial production. In addition, the published reports indicate [64, 65] that the mechanochemically synthesized perovskite compounds have better chemical stability than the HPNCs prepared by the conventional polar solvent-based methods. Mechanochemical synthesis of HPNCs is typically carried out either by hand-grinding of the perovskite precursors with mortar-pestle or via ball milling. In a typical synthesis, a stoichiometric amount of the perovskite precursors (AX and PbX2) and long chain ammonium halide salts are transferred into a mortar and ground by hand with a pestle. While hand grinding of perovskite precursors is a simple and convenient approach for preparation of HPNCs, it is also quite tedious and, more importantly, typically leads to formation HPNCs with structural defects. Therefore, a powder ball-milling is often used as an alternative. In this approach, PS precursors are first loaded into a stainless-steel jar, together with several balls made of a high hardness material such as zirconia, corundum, or agate. The balls serve as the grinding medium. The steel jar is transferred into a rotor providing the mechanical energy. The energy provided by rotor is transferred to the reagents by the grinding balls by compression, shear, and/or friction.

Reproduced with permission from Ref. [69]. Copyright 2018 American Chemical Society
Schematic diagram of a Working principle of the planetary ball-mill grinding. b Various stages of the planetary ball-mill grinding process during the mechanochemical synthesis of perovskite NPs.

Reproduced with permission from ref. [69]. Copyright 2018 American Chemical Society
Digital photographs of a a ball mill machine, b zirconia bowl filled with zirconia balls, c bulk CsPbBr3 used as a starting material. d Highly emissive HPNCs produced by the ball-milling process.
Mechanochemical synthesis methods can be subdivided into two approaches. In the first approach, there are two steps, as shown in Eqs. (1) and (2): (1) synthesis of bulk PS by mechanochemical method and (2) reduction of the size of the bulk PS by the mechanochemical method in the presence of a volatile solvent and surface-capping ligand molecules.
The second approach is a direct synthesis of HPNCs from the appropriate precursors in the presence of a solvent and ligands, as shown in Eq. (3):
In 2016, Hintermayr et al. [66] prepared MAPbX3 (X is I−, Br−, Cl−, and their mixtures) NCs by two-step mechanochemical synthesis. In the first step, the bulk PS was prepared by hand grinding an equal amount of MAX and PbX2 with a mortar-pestle method. This was followed by sonication of the reaction mixture in toluene in the presence of ligands. The synthesis yielded HPNCs with controlled chemical composition and PL energies. In this case, the second synthesis step did not involve any mechanical milling. Instead, the ultrasonication was used as an alternative. Zhu et al. [67] followed a two‐step approach for the synthesis of fully inorganic CsPbX3 HPNCs. In the synthesis, the dry ball‐milling step was applied first to produce the desired perovskite compound in a bulk form. In a second step, the product was further milled in a presence of a small amounts of organic ligands, with no added solvent. Authors showed that by this method it is possible to effectively tune the composition of the resulting HPNCs. This method also allows for preparation of HPNCs by grinding of the binary halide precursors directly in wet conditions, i.e., in the presence of solvent and ligands, sometime referred to as liquid‐assisted grinding (LAG) [68]. Protesescu et al. [69] used this method to synthesize hybrid as well as inorganic (FAPbBr3 and CsPbBr3) HPNCs and showed that composition of the perovskites in CsPb(Br/I)3 can be varied through a mechanically induced anion-exchange process. They also demonstrated that prolonged milling causes a blue shift in the PL spectra, which was attributed to the formation of quantum confined HPNCs. Chen et al. [70] extended this method to synthesis of broad range of HPNCs from MA+ to FA+ to Cs+ HPNCs with the range of halogen compositions (Table 1).
4 Composition Modification by Ion Exchange
Controlled exchange of ions at A-, B- or X-sites of the HPNC is an effective method for the preparation of NCs with desired chemical compositions and means for tuning the stability, optical, and properties of HPNCs. The postsynthesis HPNC composition modification by ion exchange was recently covered in several comprehensive reviews [72,73,74]. In short, substitution at the A-site can lead to enhanced phase stability of the PS related to the change in the tolerance factor. Similarly, B-site doping can also increase the PS stability by modification of the B–X bond length, whereas the X-site substitution is primarily used to modify the bandgap of the HPNCs. We note that this oversimplified generalization, while conceptually useful, is not entirely accurate in describing all the effects of the dopant ions in the HPNCs. Various ion substitutions often lead to more subtle variations in the band alignment, PL energy, or carrier mobilities etc. Partial substitution of the B-site cation was shown to be thermodynamically less favorable than the substitution of ions at the A and X sites, because of the larger formation energy of the former [75]. Based on theory, [52] the conduction band of ABX3 perovskites is mainly composed of Pb2+ 6p orbitals, with a minor contribution from X− ns orbitals (n = 5, 4, and 3 for I−, Br−, and Cl−, respectively), whereas the valence band is primarily comprised of Pb2+ 6p–X ns antibonding states. As a result, the electronic band gap of HPs is not significantly affected by the A+ cations, but a direct substitution of the B-site cation leads to dramatic changes in the electronic structure of HPNCs and their optical and electronic properties. On the other hand, removal of any atom from the lattice leads to formation of point defects. Various experimental observations indicate that defects due to halogen vacancy and B-site defects lead to reduced PLQY. In contrast, the vacancies or substitutions at the organic/inorganic A-site do not significantly affect the HP electronic structure or optical properties. However, they do affect the chemical and thermal stability of the HP thin films and NCs [76]. Due to its prominent effect on the electronic structure of the HPNCs and its potential to address the Pb-associated toxicity of the HPNCs, in the following sections we review most common approaches used in B-site substitution and the effect they have on the electronic properties of HPNCs. For a detailed overview of the methods used for A- and X-site ion exchanges and the effects of these substitutions on the properties of HPNCs, we refer the reader to the recent comprehensive reviews dedicated to this topic [72, 73].
4.1 Homovalent B-Site Ion Exchange
The main motivation in search for effective strategies for the B-site substitution is the reduction of the toxic lead content. The tin (Sn2+) ion has so far been the most explored because of its low toxicity and chemical similarity with Pb2+ [77, 78]. Both partial and a complete substitution of Pb2+ by Sn2+ were demonstrated [79, 80]. In one example, Zhang et al. [81] synthesized Sn2+-doped CsPbBr3 NCs by the hot-injection method. In this synthesis, stoichiometric amount of PbBr2 and SnBr2 were taken in 1-ODE, along with equal ratio of OA and OAm, and heated to 100 ℃, followed by addition of cesium stearate at ~140 ℃, which led to the formation of Sn-doped CsPbBr3 NCs. A blue shift in the absorption edge from 520 to 496 nm, along with a blue shift in the PL peak, was observed, correlating with the increase in the Sn2+ content in the structure (Fig. 10a, b). It was also shown that with increasing Sn2+ content the PLQY decreases (71–37%). Deng et al. [82] synthesized Sn2+-doped CsPbBr3 NCs via the LARP method [82]. They found that CsPb1–xSnxBr3 NCs show enhanced PLQY (for x = 0.1), but the PLQY decreased sharply for x > 0.1. This suggests that higher content of Sn2+ leads to formation of defect states and increases the efficiency of the nonradiative excited state relaxation. It was also observed that with increasing concentration of Sn2+, Cs(Pb2+/Sn2+)Br3 perovskite gradually converts into Cs2Sn4+Br4. This is because Sn in its +2-oxidation state is unstable under ambient conditions, spontaneously oxidizing into Sn4+. The associated change in the ionic radius destabilizes the perovskite lattice. A complete replacement of Pb2+ with Sn2+ has also been studied. In 2016, Jellicoe et al. [83] synthesized CsSnX3 (where X is Cl−, Br−, I−, or I−/Br− and Br−/Cl− mixed halides) HPNCs via the hot-injection method. The conventional hot-injection method used for synthesis of CsPbX3 was unsuccessful in preparation of CsSnX3 and ultimately led to precipitation of CsX. To prevent the formation of CsX and to aid formation of CsSnX3, initially, 1 M stock solution of SnX2 in the presence of tri-n-octylphosphine (TOP) was prepared at 170℃, and then stock solution is injected into the solution of Cs-oleate, which leads to the formation of CsSnX3 NCs. The electronic structure of CsSnX3 NCs was shown to be tunable with absorption and PL ranging from 500 to 900 nm (Fig. 10c–e), depending on the halogen composition. The authors observed that Sn2+-based ASnX3 NCs have smaller bandgap than the Pb2+-based APbX3 NCs. This is due to the higher electronegativity of the Sn2+ ions occupying the “B” site in the ABX3 structure. Although the Sn-based HPs have smaller bandgaps than their Pb-based analogs, they are chemically unstable under the ambient conditions. XPS and PL lifetime investigations revealed that under ambient conditions the Sn-based PSs undergo spontaneous gradual oxidation of Sn from oxidation state +2 to +4 . Consequently, the Sn-based PSs have limited practical potential for photovoltaic as well as other optoelectronic applications. In addition, a recent study has also shown that the starting material and the decomposition product SnI2 are more toxic than PbI2 [84].

Steady state a absorption and b emission spectra of CsPb1−xSnxBr3 NCs, where x = 0–0.7 and as x increases, the absorption band is blue shifted from 520 nm (x = 0) to 496 nm (x = 0.7). Reproduced with permission from ref. [81]. Copyright 2016 Elsevier. c XRD pattern of CsSnI3–xBrx perovskite NPs shows that these NPs belong to the orthorhombic phase. d TEM image of CsSnI3 perovskite NCs. e Steady-state absorption and emission spectra of CsSnI3−xBrx and CsSnBr3−xClx NCs. Reproduced with permission from ref. [83]. Copyright 2016 American Chemical Society
Transition metal doping in semiconductors is widely explored as means for optimization of optical, magnetic, and electronic properties. In the case of lead-based HPNCs, the effect of substitution in the B-site was explored with a broad range of transition metal ions (Co2+, Cu2+, Fe2+, Mg2+, Mn2+, Ni2+ Sr2+, Zn2+ etc.) [75]. Of the explored metals, the doping with Mn2+ was perhaps the most widely studied. Chlorine-based CsPbCl3 is the most suitable material for studies of doping effects, thanks to its relatively wide bandgap, which provides an opportunity for introduction of new intraband states via doping. Parobek et al. [85, 86] and Liu et al. [85, 86] were the first to investigate the doping of Mn2+ into CsPbCl3 via the hot-injection approach. In this approach, MnCl2 and PbCl2 were dissolved together in 1-ODE as a solvent at a moderate temperature (160 °C) in the presence of equal amount of organic amine and an acid, such as oleylamine and oleic acid. Then, an independently prepared Cs-oleate was injected into the precursor mixture solution to initiate the crystallization of the Mn2+-doped CsPbCl3. While the undoped CsPbCl3 has a characteristic narrow PL with peak at ~402 nm, Mn2+-doping leads to observation of a new PL peak at ~586 nm (Fig. 11a–c). The new PL changes the color of the emitted light from blue to bright orange. The low energy excited state of the Mn-doped CsPbCl3 has a long emission life time of ~1.6 ms due to the spin forbidden nature of the 4T1–6A1 transition [87]. The substitution of the halogen ion from Cl− to Br− ions leads to an increase in the PL intensity of the band-to-band transition, while the PL intensity of the peak associated with the Mn2+ dopant gradually declines with increasing Br− content. This observation indicates that the excitonic energy relaxation through the Mn2+ dopant states is reduced in the Br-based PSs, which means that the Mn-doping plays a less significant role in the relaxation of excited state in CsPbBr3 than in CsPbCl3 PSs.

a UV–visible light absorption spectra of Mn:CsPbCl3, CsPbCl3, Mn:CsPb(Cl/Br)3, and CsPb(Cl/Br)3 NCs. b PL emission spectra of Mn-doped and undoped CsPbCl3 and CsPb(Cl/Br)3 NCs (inset: digital image of Mn2+ doped and undoped perovskite NCs). c Emission lifetime of Mn2+-doped and undoped CsPbCl3 and CsPb(Cl/Br)3 NCs. Reproduced with permission from ref. [85]. Copyright 2016 American Chemical Society. d Digital image of Sn, Cd, and Zn doped perovskite NCs. e Absorption and emission spectra of Sn-doped CsPbBr3 NPs show a change from 512 to 479 nm. f Absorption and emission spectra of Cd2+ and Zn2+ doped CsPbBr3 show a change from 512 to 483 nm and 512 nm to 462 nm, respectively. Reproduced with permission from ref. [88]. Copyright 2016 American Chemical Society
In a related study, Stam et al. [88] incorporated Cd2+ and Zn2+ ions into the CsPbBr3 NCs to tune their optical properties. They initially synthesized CsPbBr3 NCs by method described by Protesescu et al. [44]. Next, 1 mmol of metal bromide salt (MBr2, with M being Sn2+, Zn2+, or Cd2+) in 10 mL of toluene (0.1 M MBr2), in the presence of 100 μL of OAm, was prepared. Doping of metal cation was achieved by ion exchange in the presence of 0.5 mL of cation precursor (with concentrations ranging from 0.125 to 1.67 mM), injected into 1.5 mL of diluted NCs in toluene (concentration ∼0.01 μM) and mixed and stirred at room temperature for ∼16 h. Cd2+ or Zn2+ doping in CsPbBr3 leads to a blue shift in the absorption and PL spectra, as shown in Fig. 11d–f. PL peak position was tuned from ~452 to ~512 nm by Cd2+-doping and from ~462 to ~512 nm via Zn2+-doping. In both cases the doped NCs showed narrow PL (FWHM ≈ 80 meV) with high PLQYs (> 60%). Shen et al. [89] showed that Zn2+ doping in CsPbI3 NCs leads to a small change in the PL energy (690–678 nm), but PLQY increases from 61.3% to 98.5%. Likewise, successful Cu2+ doping in CsPbBr3 NCs was reported by Bi et al. [90]. In this report, they used the hot-injection method to synthesize Cu-doped CsPbBr3 NCs. PbBr2 and mixture of copper acetate Cu(OAc)2 and CuBr2 in stoichiometric ratio in 1-ODE, in the presence of oleic acid oleylamine, were heated up to 185 ℃. Previously prepared Cs-oleate was injected into the reaction mixture, resulting in the formation of Cu-doped CsPbBr3. The Cu2+ doping was shown to cause a small blue shift in the PL from 517 to 499 nm. The authors also reported enhanced PLQY and improved thermal stability of Cu2+-doped NCs. De et al. [91] also doped Cu2+ in the CsPbCl3 NCs. The hot-injection method was used to prepare Cu-doped CsPbCl3 as discussed by Protesescu et al. [44]. PbCl2 and CuCl2 in stoichiometric ratio were dissolved in 1-ODE in the presence TOP and heated up to 185 °C. Previously prepared Cs-oleate was injected into the reaction mixture producing Cu-doped CsPbCl3. The authors found no significant shift in the PL energy but observed an improved PLQY. It was shown that an optimal amount of Cu2+ dopant in CsPbCl3 can boost the PLQY of the NCs from 0.5% to 60% for the PL at 403 nm. Variations in the PLQY for PL at 430 nm (8–92%) and 460 nm (22–98%) were observed in Cu:CsPb(Br/Cl)3. Thawarkar et al. [92] synthesized Ni-doped CsPbBr3. PbBr2, NiBr2, and oleic acid were added to round-bottom flask and heated up to 200 °C, and previously prepared Cs-oleate was injected into the reaction mixture yielding Ni-doped CsPbBr3 NCs. They observed that Ni2+ doping of CsPbBr3 leads to an increase in the PLQY without an observable shift in the PL peak position [92]. Similarly, Yong et al. [93] found that Ni2+ doping in CsPbCl3 does not affect the PL energy but boosts the PLQY from 2.4% to 96.5%. However, the isovalent doping with Mn2+ and other metals led to enhanced PLQY due to improved passivation of the defect states associated with the halogen vacancies (VX, X is Cl−, Br−, or I−). These studies showed that the homovalent substitution at the B-site can alter the PL properties of the HPNCs in various ways, depending on composition, while retaining the original electronic structure relatively intact. This has important implications for exploitation of these materials in LED applications.
4.2 Heterovalent B-Site Ion Exchange
Bismuth (Bi3+) is another suitable ion for the Pb2+ replacement in the ABX3 PS structure. The Bi is located next to the Pb in the periodic table (group 5A) and the Bi3+ and Pb2+ have isoelectronic structures. Heterovalent doping presents an opportunity to modify the electronic structure of the perovskite via the B-site substitution. Since the Pb2+ and Bi3+ have similar ionic radii (Bi3+ 1.03 Å and Pb2+ 1.19 Å), the Bi2+ doping in ABX3 perovskite does not significantly alter its tolerance factors. Zhou et al. [94] were the first to report doping of MAPbI3 perovskite thin films with Bi3+. The perovskite precursor solution was prepared by mixing MAI and PbI2 in a 1:1 molar ratio in DMF and additional BiI3 in DMF was added to the mixture to achieve desired Bi-doped MAPbI3. Then the solution was spin coated on a glass substrate to form the Bi-doped MAPbI3 thin film. They showed that with Bi3+ doping, the PL shifts from visible to NIR spectral range from 850 to 1600 nm. Abdelhady et al. [95] demonstrated that doping of Bi3+ in the single crystal (SC) MAPbBr3 PS leads to a dramatic change in its electric and optical properties. The heterovalent Bi3+ ions act as an electron donor and increase the electric conductivity (from ∼10–8 Ω–1 cm–1 for the undoped crystal to ∼10–4 Ω–1 cm–1 for the 10% Bi-doped MAPbBr3 crystals). The Bi3+ doping increases the carrier densities and also converts the PS into p-type. They also observed that with increasing Bi3+ content the band edge of the doped crystal shifts towards lower energies. For example, for MAPbBr3 with no doping the band edge is located at 570 nm and doping with ~10% Bi3+ shifts the band edge to 680, which leads to change in the bandgap from 2.17 eV (for undoped MAPbBr3) to 1.89 eV (for 10% Bi-doped MAPbBr3). Later, spectroscopic ellipsometry measurements by Nayak et al. [96] in Bi3+-doped MAPbBr3 showed that bandgap energy decreases from 2.18 to 1.95 eV and that the red shift seen in the absorption spectra is due to the increase in the Urbach energy (from 19 meV to 50–55 meV), not due to the shift in the conduction band or valance band edge positions. This increase in the Urbach energy is related to the increase in the structural disorder, which directly affects the absorption onset of the Bi3+-doped perovskites. Lozhkina et al. [97] observed similar effect in Bi3+-doped CsPbBr3 single crystals. Pristine and Bi3+-doped CsPbBr3 single crystals were grown using antisolvent vapor-assisted crystallization method. In a typical procedure, CsBr, PbBr2, and BiBr3 were dissolved in DMSO in various rations then transferred to another container, covered with cellulose membrane, and placed into a vessel with 20% ethanol solution as antisolvent. Solution was kept for 48 h at 40 °C to form the Bi-doped CsPbBr3 single crystals. Though Bi3+ doping in CsPbBr3 single crystal leads to a significant redshift in the absorption spectra, low temperature PL studies indicate that the excitonic peak position does not change, suggesting that the bandgap of the PS remains unaltered. Begum et al. [98] observed that Bi3+ doping in CsPbBr3 HPNCs shows a small blue shift in the bandgap value (from 2.43 to 2.398 eV) along with a change in excited state carrier relaxation dynamics in CsPbBr3 by reducing radiative transitions. Zhang et al. [99] reported substitution of Pb2+ ions in CsPbBr3 HPNCs with Sb3+. In this process, CsBr and PbBr2 were dissolved in DMF with oleic acid and oleylamine. In another vessel, various stoichiometric amounts of SbBr3 were dissolved in toluene, then the DMF solution containing perovskite precursor was added to the toluene, resulting in the formation of Sb-doped CsPbBr3. While Sb3+ doping of CsPbBr3 QDs in the 2.2–2.9 nm size had no impact on the bandgap, the PLQY was found to increase upon substitution. Liu et al. [100] synthesized Al-doped CsPbBr3 via the hot-injection method. AlBr3, PbBr2, oleic acid, and oleylamine are taken on 1-ODE. Then, the temperature was raised to 150 °C and already prepared Cs-oleate solution was injected into the reaction mixture, resulting in formation of Al-doped CsPbBr3. As a result of doping, a blue shift in the PL energy was observed. The above results indicate that while the effect of heterovalent B-site doping on the bulk APbX3 PSs is relatively well understood, the understanding is less clear for the single crystal and HPNC systems. The effects of doping on the properties of HPNCs are summarized in Fig. 12.

Overview of the effects of doping on the properties of HPNCs
5 Postsynthesis Phase Modification
In addition to composition, the phase and optical properties of the HPs can also be controlled by various postsynthesis modifications. These include surface modification/functionalization of the perovskite NCs via ligands exchange and specific interactions, irradiation by light, application of external pressure, water-assisted phase control strategies, etc.
5.1 Transformations via Chemical Treatment
Postsynthesis surface treatment by ligands is very useful for tuning the optical properties of the HPNCs. The ligand exchange or addition of new ligands can lead not only to improvement in the optical properties but also to changes in the phase and/or the shape of the HPNCs. For example, 3D ABX3 PS can be transformed into 2D AB2X5 PS via appropriate postsynthesis chemical treatment. In 2017, Balakrishna et al. [101] reported preparation of 2D CsPb2Br5 nanosheets via an addition/treatment of 3D CsPbBr3 NCs with dodecyl dimethylammonium bromide (DDAB) spontaneously at room temperature. The transformation was experimentally monitored by electronic absorption spectroscopy. Within 10 min after addition of the DDAB, the authors observed a drop in the absorbance in the 400–520 nm region and an increase in the absorbance at 320 nm. The increase in the absorbance at 320 nm was attributed to the formation of a [PbBr3ˉ] complex resulting from the exfoliation of the Cs+ ions from the CsPbBr3 by DDAB. Subsequently, emergence of another absorbance peak at 345 nm was observed. This was attributed to the formation of the [Pb2Br5ˉ] complex, which gradually transforms into CsPb2Br5 nanosheets. During the transformation, the PL spectra showed a blue shift and the PLQY decreased from 70% to 4%. Another example of the post synthesis phase transformation is the conversion of bulk (3D) CsPbBr3 HPNCs into zero-dimensional (0D) Cs4PbBr6 [102,103,104]. In this transformation, the externally added amines extracted the PbBr2 from the CsPbBr3 NCs, leading to the formation of the Cs4PbBr6. Palazon et al. [102] showed that addition of tetramethyl-ethylenediamine (TMEDA) to orthorhombic CsPbBr3 8-nm nanocubes in toluene at room temperature transforms them into 50-nm rhombohedral shaped Cs4PbBr6 in 2 min. During the transformation, the absorption peak shifted from 490 to 317 nm, with a complete loss of PL. Liu et al.[104] demonstrated that when alkyl thiol ligands are added to the CsPbBr3 NCs hexanes or toluene solution, the 3D-CsPbBr3 PS are transformed into 0D-Cs4PbBr6 NCs (Fig. 13). Interestingly, the transformation was found to be reversible. Chemical reactions of Cs4PbBr6 with organic acids [103], PbBr2 [45] and Prussian blue [105] in hexane room temperature, were found to leads to the formation of 3D CsPbBr3. It was also found that not only amines but also heat [105, 106], water [107] and pressure [108] can be used to accomplish this type of phase transformation. This also applies to hybrid perovskites. Recently, Huisman et al. [109] showed that thermal annealing of 0D-MA4Pb(Br1–xIx)6·2H2O in air at ~75 °C triggers a dramatic transformation of the 0D phase to the 3D MAPbI1–xBrx PS. They also demonstrated that this transformation can be reversed by simply cooling down the compound in air. The absorbance spectra revealed two sharp absorption peaks at 288 nm and 364 nm, which were attributed to the formation of MA4Pb(Br1–xIx)6·2H2O. Upon heating the compound to ~70–100 °C, an onset in the absorption spectra was observed at 800 nm, attributed to the formation of MAPbI3 NCs.

Reproduced with permission from ref. [104]. Copyright 2017 American Chemical Society
Schematic crystal structure of a 3D-CsPbBr3 and b 0D Cs4PbBr6. c TEM image of CsPbBr3 NCs along with the digital image of the CsPbBr3 after phase transformation. d TEM image of Cs4PbBr6 NCs along with the digital image of the Cs4PbBr6 after phase transformation. The chemical equation shows the reaction pathway for the 3D to 0D perovskite phase transformation.
5.2 Transformations Induced by Irradiation
The postsynthesis phase transformation of PSs by chemical routes has been the most studied process for control of the PS phases. However, the number of studies of HPSs revealed that irradiation by visible/UV light can also lead to their postsynthesis phase and composition transformation. Photo-activated phase changes were previously studied in the CdSe quantum dots [110]. More recently, it was found that in the case of APbBr1–xIx, PS thin films, the irradiation by 400 nm pulsed laser light leads to a halogen phase separation with formation of “I”-rich and “Br”-rich domains [111]. The formation of the domains is associated with narrowing of the band gap in the domains with increased iodine content and widening of the bandgap in the domains with higher bromine content. In the case of single halide perovskite thin films in ambient atmosphere, the 532 nm laser light irradiation of the surface was found to lead to a phase degradation [112] or alteration [113]. Due to the large surface to volume ratio, NCs are much more sensitive to light exposure than their bulk counterparts. Several reports showed that a thin films of few‐monolayer thick CsPbBr3 nanoplatelets can be transformed from 2D to 3D materials when irradiated with 325 nm continuous-wave (CW) laser radiation of 20 mW cm−2 [114]. This process is generally referred to as the photo-induced phase transformation process (PDPT). From the mechanistic standpoint, the PDPT can be divided into two steps. In the first step, the surface ligands desorb from the NC surface, thanks to their relatively small binding energy [36]. In the second step, the ions diffuse between the nanoplatelets, which coalesce into larger structures (Fig. 14). The phenomenon was first observed by Wang et al. [114], followed by a report by Shamsi et al. [59]. The authors of these reports concluded that this kind of PDPT is not possible for hybrid PSs systems. Ha et al. [115] later contradicted this conclusion by observing transition from n = 2 to n = 3 layers in the MAPbBr3 upon irradiation with 365 nm LED. Later, Roy et al. [116] showed that full conversion of quasi 2D MAPbBr3 halide perovskite nanoplatelets (HPNPLs) to 3D MAPbBr3 HPNPLs is possible by excitation of a 340 nm UV light source (12 mW) in hexanes. Similar transformations were also observed in HPNRs. In 2022 Sen et al. [117] showed UV-assisted (365 nm UV excitation) conversion of 2D Ruddlesden–Popper MAPbI3 HPNPLs into stable 3D MAPbI3 nanorods. Since understanding of PDPTs is essential for development of HP optoelectronic applications, more studies are needed to fully understand this phenomenon.

Reproduced with permission from ref. [114]. Copyright 2017 Wiley
a Time evolution of PL spectra of few layers thick CsPbBr3 nanoplatelet. b PL variation mapping of few layers thick CsPbBr3 nanoplatelet throughout the whole transformation process. The inset shows the PL photographs of the sample with varied exposure time. c Schematic illustration of PDPT; CsPbBr3 nanoplatelets gradually coalesce and convert to 3D or bulk phase CsPbBr3 under continuous photon irradiation.
6 Size and Shape Control
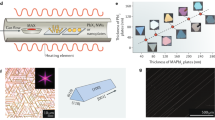
The ability to effectively control the size and shape of the HPNCs is an important pre-requirement for their effective practical exploitation. Therefore, significant efforts have been devoted to the development of various strategies for synthesizing size- and shape-controlled hybrid or inorganic HPNCs. In the most commonly used synthetic approaches, the HPNCs are prepared as nanocubes. The cubic CsPbBr3 HPNCs were first synthesized via the hot-injection method [44], where lead halide precursors were dissolved in the 1-ODE, organic amine, and an acid. Cs-oleate was injected at 140–180 °C, and the reaction was quenched using an ice bath. Depending on the injection temperature, the size of the nanocubes could be controlled in the range of 4–15 nm. Interestingly, the injection of Cs-oleate at a lower temperature (~130 °C) was shown to lead to the formation of halide perovskite nanoplatelets (HPNPLs) [118]. It was also observed that extended heating leads to formation of HP nanowires as a side product [119]. Almeida et al. [120] have shown that the size of the nanocubes can be controlled by systematic variations in the ratio of the oleic acid (OA) and oleylamine (OAm). The nanocubes in the size range from 4 to 16.4 nm were prepared by changing the OA/OAm ratio in the range 1:1 to 1:10. Protesescu et al. [47] synthesized FAPbBr3 nanocubes by the hot-injection method, where presynthesized oleylammonium bromide (OAmBr) (halide source) was injected at 130℃ into the dissolved FA+ and Pb2+ acetate precursor. This is slightly different from the conventional hot-injection method where PbBr2 acts as a both Pb2+ and Br− source. By adjusting the amount of OAmBr, the size of the nanocubes was tuned from 5 to 50 nm.
Pan et al. [121] have varied the composition of carboxylic acids and amines to systematically investigate the effect of the acid and amine on the size and shape of the HPNCs. The results are schematically summarized in Fig. 15. The results show that an equal amount of amine and acid leads to a formation of monodisperse HPNCs. They also showed that by changing the Cs-oleate injection temperature (140–200 °C) size of the CsPbBr3 PNCs can be accurately controlled in the range 5–12 nm [44]. The initial efforts to prepare the layered HPNCs have focused on replacing small size organic cation with a long chain hydrocarbon cations. In the resulting structure the lead halide inorganic octahedral layers are spatially separated by the organic molecules. The chemical formula for this type of compound can be written as L2[ABX3]n–1BX4, where n = 1,2,3,4,5,… ∞ is the number of the octahedral layer. For example, for n = 1, the structure becomes L2BX4, which is a fully 2D PS structure. On the other hand, for n = ∞, the structure becomes ABX3, which is a 3D structure. For n = 2 to few-layers-thick PSs are formed, known as the quasi-2D perovskites [116]. The optical properties of the layered PSs are primarily determined by the composition and thickness of the inorganic layer. The PSs with different thicknesses of the inorganic layer exhibit different optical properties, due to the confinement effects similar to those observed for 3D perovskites. Compared with the 3D PSs, the layered HPNCs show higher structural stability due to the more negative enthalpy of formation. Around 2015, various research groups synthesized the first colloidal PS NPLs [71, 118, 122]. Initially, perovskite NPLs were identified as a by-product of the synthesis of the 3D perovskite NCs via the hot-injection method, particularly for the cases where injection was done at relatively lower temperatures (< 120 °C) [118]. Multiple efforts were also made to prepare NPLs by the LARP method. In the LARP method, perovskite precursors are first dissolved in a polar solvent such as DMF or DMSO and subsequently transferred to a nonpolar solvent for crystallization. Sichert et al. [71] were the first to report the preparation of MAPbBr3 NPLs via the LARP method. In the reported procedure, they dissolved MABr, PbBr2 and oleylammonium bromide (OAmBr) in DMF and transferred the solution into toluene for crystallization. By varying the MAX to OAmBr ratio, they were able to produce MAPbBr3 NPLs with various numbers of inorganic layers. Subsequently, Akerman et al. [123] reported the synthesis of CsPbBr3 NPLs (n = 3–5) via the LARP method. Berkenstine et al. [118] showed that injection of Cs-precursor into lead halide solution at relatively low temperatures (~120 °C) leads to a formation of HPNPLs. Depending on the injection temperature, they were able to synthesize CsPbBr3 NPLs with n = 1–5. Vybornyi et al. [50] reported preparation of MAPbBr3 NPLs via the hot-injection method, whereby the MA+ precursor was injected at very low temperature (60 °C). Interestingly, it was observed that PS NPLs synthesized via the hot-injection method had a smaller lateral dimension (10–100 nm) [50, 118, 124] compared with the NPLs synthesized via LARP method (100–1000 nm) [71, 125, 126]. Shamsi et al. [127] showed that by adjusting the ratio of short and long ligands, lateral size of the HPNPLs can be systematically adjusted. The optical properties of HPNPLs possess some unique features. They are characterized by strong optical quantum confinement as well as dielectric confinement. HPNCs have an exciton Bohr radius of ~3 nm or larger, depending on the halogen [44, 71, 123, 128]. Since a single layer of PbX6 octahedra is ~6 Å thick [116], even few layers thick HPNPLs are spatially confined along the axis perpendicular to the NPL layers. Moreover, due to the long chain amines, these structures are also dielectrically confined, which leads to large exciton binding energies with values up to several hundred meV [129,130,131] (Table 2).

Reproduced with permission from ref. [121]. Copyright 2017 American Chemical Society
Size and shape control approach during the hot-injection method. High reaction temperature with shorter carboxylic acid results in larger size nano cubes and shorter chain length amines result in thinner CsPbBr3 nanoplatelets. Lower reaction temperature with shorter carboxylic acid results in nanoplatelets and shorter, long chain length amines result in thinner, and thicker CsPbBr3 nanoplatelets, respectively.
In addition to NPLs, significant effort has been dedicated to synthesizing perovskite nanowires (NWs). In 2015, Zhang et al. [119] reported the first synthesis CsPbBr3 and CsPbI3 NWs via the hot-injection method. In this process, PbX2 along with organic ligands were dissolved in 1-ODE and heated up to 150 °C. After the injection of previously prepared Cs-oleate into PbBr2 solution, within t < 10 min, formation of nanocubes (NCs) with size ranging from 3 to 7 nm was observed by TEM. As reaction time increased to 10 min, a few thin NWs (9 nm in diameter) were found and with increasing time (40 min), reaction products were fully converted to HPNWs. Specifically, it was shown that by extending the reaction time after the injection of Cs-oleate, the lead and halogen source transform the HPNCs to HPNWs. Later, Tong et al. [132] synthesized various halide perovskite composition NWs by ultrasonication method. They found that during the sonication, initially cubic HPNCs are formed. They took Cs2CO3 and PbX2 in 1-ODE with 1:1 volume ratio of oleic acid and oleylamine, and then, the reaction mixture was subjected to tip sonication at a power of 30 W for 10 min, which led to the initial formation of HPNCs. However, prolonged ultrasonication (up to 60 min) under similar reaction conditions lead to the formation of HPNWs. One of the challenges in the preparation of HPNWs is that they are typically prepared in a mixture with cubic 3D NCs. More recently, Zhang et al. [133] developed a new purification method, which increases the yield of HPNWs by using ethyl acetate (EA) as the antisolvent. This approach allows more effective separation of the ultrathin HPNWs from 3D NCs and other impurities.
7 Defects in HPNCs
In the cubic PS crystal structure, the “A” cation is in a 12-fold coordination, surrounded by four BX6 octahedra. While this represents an ideal, equilibrium arrangement, real crystals often contain defect sites (e.g., at the surface and grain boundaries) where the structure deviates from this ideal arrangement. The types of defect found in the PS crystals depend on the details of the synthesis, such as precursor stoichiometry, methodology (LARP or hot-injection) and temperature (in bulk as well in the case of NCs) [20]. In addition, the irradiation with light, exposure to heat, moisture, and oxygen can also lead to formation of defect states and degradation or decomposition of the PS structure [134, 135]. Although PSs have unusually high defect tolerance in terms of optical and charge transport properties, detailed understanding of the defect formation and effects on the PS properties is critical for development of PS-based devices and applications. In general, the probabilities of the formation of various defects are related to their formation energies. Some information about these energies can be obtained from theoretical calculations, which then can provide guidance for the optimization of experimental preparation conditions [20, 136].
One of the characteristic features of HPS are low excitonic binding energies (Eb) (20 meV to 80 meV for the MAPbX3 (X = I and Br) at room temperature. While a small Eb is an advantage for some applications, such as photovoltaics, they are less desirable in other optoelectronic applications, such as light emitting devices, where effective radiative recombination of carriers is desirable [137]. One way this problem can be addressed is by exploring the reduced dimensionality effects, as the Eb typically increases in quantum confined NCs. However, a significant reduction in the structure size also means significant increase in the surface-to-volume ratio, which typically translates into high probability of defects formation. Finding the optimal balance requires a thorough understanding of the parameters affecting the formation of defects in HPSs.
The defects typically observed in a semiconductor can be divided into two categories: (1) crystallographic defects, i.e., interruption in the perfect lattice of the semiconductor and (2) impurities, i.e., presence of a foreign element/ion within the semiconductor lattice. The defects can form as point defects (missing of an atom at a specific crystal position) or site substitution (atoms occupying the wrong position in the lattice) [138]. The probability of the internal defect formation varies with growth conditions and depends on the choice of solvent, precursor, temperature, and dopant [139]. In the case of ABX3 PSs, the most commonly studied defect is the point defect. There have been 12 types of point defects reported in MAPbI3 PSs. They are typically categorized into three vacancy-induced defects: VMA, VPb, and VI; three interstitial addition defects: MAi, Pbi, and Vi; and six antisite occupation defects: MAPb, MAI, PbMA, PbI, IMA, and IPb [20, 136, 140,141,142,143,144]. Various theoretical studies have established that in PSs point defects have higher formation energies and contribute to formation of deep-level defect states, whereas point defects have lower formation energies and contribute to formation of shallow-defect states. It was also found that point defects, which contribute to formation of deep level defect states, do not produce nonradiative recombination centers [20, 142, 144]. Theoretical calculations also showed that IPb, IMA, Pbi, PbI, Vi, and PbMA contribute mainly to the formation of deep level defect states; however, the understanding here is still evolving [145]. It was also observed that depending upon the formulation of the perovskite structure, the formation energy of PbI, VI, and IMA could be sufficiently low so as to lead to creation of nonradiative recombination centers. The shallow point defects can be classified into two categories: (1) acceptor defects (VMA, VPb, Ii, MAPb, IMA, and IPb) and (2) donor defects (VI, MAi, Pbi, PbMA, MAI, and PbI), which can be viewed as an unintentional doping in perovskite structure. Hence, by controlling the formation of defects in the PS structure, it is possible to prepare n-type or p-type materials. In the typical PS nanomaterial synthesis, depending on the synthesis method, various types of defects can be generated [146].
7.1 X-Site Defects
In 2015, Protesescu et al. [44] synthesized high quality HPNCs and observed that CsPb(I/Br)3 NCs had significantly higher PLQY (~90–95%) than CsPb(Br/Cl)3 NCs (~10–25%). Similar observation was made for the hybrid MAPbBr3−xClx HPNCs listed in Table 3 [147]. In the Br–Cl perovskites, deep level trap states formed due to the formation of Cl‾ vacancies (VCl). These deep-level traps act as nonradiative relaxation centers, contributing to reduction in the PLQY [148]. The formation of the vacancies can also be observed in the absorption spectra of the HPNCs. Urbach energy of the I–Br series is significantly higher than the Br–Cl series as a result of the higher level of disorder and density of defect states, leading to an increase in the probability of sub-bandgap transitions. Number of researchers explored various techniques to overcome this problem. Zhang et al. [33] synthesized the hybrid HPNCs via LARP method where the use of polar solvent is essential for formation of NCs. A strong interaction between the PbI2 and the coordinating solvent (DMF/DMSO) led to formation of PbI2·DMF/DMSO intermediates, which promote formation of the I vacancies (VI). Under the ambient conditions, these vacancies are filled by oxygen molecules, which react to form PbO as an initial step in a gradual chemical degradation of the HPNCs [41]. In contrast, the HPNCs prepared by the hot-injection two-in-one precursor method (i.e., PbX2, where X is Cl−, Br−, or I− is the source of lead and halogen simultaneously) typically suffer from halogen deficiency, which leads to the formation of halogen vacancies. These halogen vacancy defect states increase the efficiency of the nonradiative relaxation and lower the PLQY of the HPNCs [44, 55]. Dutta et al. [149] demonstrated that annealing of HPNCs with long-chain ammonium halides at higher reaction temperatures (250 °C) reduces the formation of defect states and increases the PLQY to ~50%. To minimize the halogen defects, halogen-rich conditions are often used in the synthesis of HPNCs. Liu et al. [55] developed a three-precursor method to achieve halogen-rich conditions, for example by replacing PbX2 with PbO and NH4X. As shown in Fig. 16, this approach leads to high PLQY in the HPNCs [55]. NH4X provides excess halogen during the reaction which reduces the halogen vacancy and hence increases the PLQY of the HPNCs. This kind of understanding of halogen defects and their effect in the optical properties is essential for development of effective device applications.

Reproduced with permission from ref. [55]. Copyright 2017 American Chemical Society
a Steady-state UV and PL spectra of perovskite NCs synthesized via the three-precursor method. b Time-resolved PL decay and respective curve-fitting for CsPbBr3 NCs. c Average PLQY values of various perovskite NCs synthesized in different halide circumstances. d Schematic diagram of the effect of rich/poor halide conditions on the optical properties of perovskite NCs.
7.2 B-Site Defects
Various reports demonstrated that partial substitution/doping of the B-site of the perovskite can reduce point defects, structural disorder or distortion in the perovskite octahedra. It was also observed that the PLQY of pristine CsPbCl3 can be significantly enhanced by the postsynthesis treatment with CuCl2, suggesting that this treatment reduces the Cl‾ vacancies [150]. In one example, Yong et al. [93] showed that doping of CsPbBr3 perovskite NCs with Ni2+ significantly reduces the halogen defects (VX) and increases the lattice order of the PS structure, which leads to enhanced exciton radiative recombination. Specifically, 2–3% substitution of Pb2+ with Ni2+ led to increase of PLQY from ~1.2 to ~93% (see Fig. 17a, b) [93]. Similarly, doping with Zn2+ ions was also found to lead to defect passivation in CsPbBr3 perovskite NCs and a significant increase in the PLQY from 54% to 74% (see Fig. 17c, d) [151]. The defect passivation by Zn2+ was attributed to the ability of the ZnBr2 to produce Br-rich conditions in the PS by filling the halogen vacancies. Mondal et al. [152] showed that postsynthesis doping of Cd2+ into CsPbCl3 NCs leads to formation of NCs with PLQY near unity (96 ± 2%). The defect passivation by the CdCl2 post-treatment was investigated by time-resolved photoluminescence (TRPL) and ultrafast transient absorption spectroscopy (TAS) at various temperatures [152]. The results showed that the suppression of the nonradiative transition in the doped CsPbCl3 NCs can be attributed to the passivation of the defect states associated with the VX vacancies. The conclusions of the study are schematically summarized in Fig. 17e. In other studies, it was shown that doping of the B-site in the PS structures can also lead to formation of new defect states. For example, doping of MAPbI3 single crystals by Bi3+ was found to enhance the electrical conductivity of the material by factor of three [153]. Increase in the measured Urbach energy together with a significant blue shift in the PL spectra were attributed to the formation of Bi3+-related donor states. These donor states release more electrons to the conduction band, which results in the observed increase in conductivity. First-principles modeling by Mosconi et al. [154] showed that the presence of the Bi3+ dopants in the MAPbI3, induces formation of deep trap states. Similar observation was also made in the case of Bi3+-doped MAPbBr3. Abdelhady et al. [95] found that by doping MAPbBr3 single crystals with Bi3+, the conductivity of the crystals increased by four orders of magnitude. While pristine MAPbBr3 is a p-type semiconductor, Hall effect measurements showed that after Bi3+ majority doping carriers switch from holes to electrons. The authors of the study also reported that Bi3+ doping reduces the bandgap by 300 meV. However, later Nayak et al. [96] showed that the appearance of a band gap shift can also be explained in terms of the increase in the Urbach energy associated with the Bi3+ doping. The doping of CsPbBr3 PSNCs by Bi3+ was found to also lead to an overall increase in the amount of defects and a decrease in the PLQY from 78% to 8% [98]. In spite of the extraordinary success and progress, in the understanding of the effects of B-site doping on defects, there are many questions that remain unanswered as far as the effective passivation strategies or controlled generation of new defects in the HPSs.

a UV–visible light and PL spectra of undoped and Ni-doped CsPb(Cl/Br)3 NCs showing the change in the PLQY of the HPNCs. b PL decay spectra of undoped and Ni-doped CsPb(Cl/Br)3 NCs. Reproduced with permission from ref. [93]. Copyright 2018 American Chemical Society. c Emission spectra and PLQYs of pristine and Zn-doped CsPbBr3 NCs. d PLQY of CsPbBr3 NPs after exposure to ambient atmosphere as a function of time. Reproduced with permission from Ref. [151]. Copyright 2017 American Chemical Society. e Schematic depiction of the effect of Cd2+ doping on the exciton relaxation in CsPbBr3 NCs. (NR is a nonradiative recombination process). Reproduced with permission from ref. [152]. Copyright 2018 American Chemical Society
7.3 A-Site Defects
In the PS family of materials, conduction band minimum (CBM) is mainly formed by the 6p orbitals of Pb2+, with a minor contribution from 5s orbitals of I−, 4s orbitals of Br−, or the 3s orbitals of Cl−, whereas the valence band maxima (VBM) are comprising mainly the 6p antibonding orbitals of Pb2+ and the 5s orbitals of I−, 4s orbitals of Br−, or the 3s orbitals of Cl−. Theoretical modeling showed that there is no contribution from the orbitals of organic cations to the CBM and VBM [52]. This is schematically shown in Fig. 2. The analysis also shows that A-site cationic bonding states are located deep inside the VB and do not hybridize with PbI6 octahedra near the VBM or the CBM. While the organic cations do not contribute to the formation of states near the VBM or CBM, they play a major role in providing structural stability of the PS materials [155]. Since the electronic contributions from the A-site cations lie deep inside the band structure, changing of the A-site cation does not directly influence the formation of defects. Rather, the substitution of the A-site affects the electronic structure and optical properties indirectly. For example, doping of MAPbX3 (MA ionic radius 0.18 nm) with a larger cation such as FA+ (ionic radius 0.22 nm) or smaller cation such as Cs+ (ionic radius 0.167 nm) or Rb+ (ionic radius 0.152 nm), increases (FA+)/decreases (Cs+, Rb+) the B−X bond length and causes the lattice expansion (FA+)/contraction (Cs+, Rb+), leading a decrease(FA+)/increase (Cs+, Rb+) in the bandgap energy [156].
8 Degradation of HPNCs
8.1 Oxygen and Moisture-Induced Degradation
Although, brief exposure of the PS films to water can enhance their thin film quality during the perovskite crystallization [157, 158], extended contact of PSs to an atmosphere with high relative humidity (RH) leads to their irreversible degradation. Leguy et al. [159] showed that when HPSs are exposed to water vapor, due to the hygroscopic nature of the MA+ ion, a hydrated MAPbI3 PS phase (MAPbI3‧H2O) is formed. This phase form can be fully reverted to MAPbI3 perovskite (nonhydrated) upon drying [159]. Ahn et al. [160] showed that it is the charges trapped in the defect states located at the grain boundaries or surfaces that are primarily responsible for this reversible degradation. Though the excess amount of water eventually leads to complete degradation of the perovskite structure. In addition to water, molecular oxygen is another important source of degradation of HPSs. In 2015, Aristidou et al. [161] showed that when MAPbI3 PS is exposed to O2 in the presence of light, it decomposes rapidly into MA, PbI2, and I2. It was proposed that degradation process is initiated by a light induced conversion of O2 into O2‾ super oxides, which react with MA+ cations. The theoretical analysis showed that O2‾ likely forms by the reaction of the ambient O2 with the negatively charged iodine in the (VI) defect sites (Fig. 18). In the proposed mechanism, the VI defects serve as traps for the atmospheric O2, which upon photoexcitation and interaction with I− turns into highly reactive superoxide, which gradually degrades the PS. This can be experimentally observed as gradual reduction in absorbance spectra in HPS exposed to dry air (including oxygen). In contrast, it was found that a controlled, short-term exposure of a HPS to moisture passivates the surface traps, which increases the observed PL. Consistently, with this assessment, Brenes et al. [162] showed that PL intensity of the HPSs depends not only on the PS morphology but also on the environment to which the HPS is exposed (see Fig. 19a–c) [162]. The authors also found that when exposed to O2 and/or H2O molecules the PL of the PS grains, which initially show weak PL, is significantly enhanced, while the grains, which are initially bright, remain relatively unaffected by the exposure to O2 and/or H2O. To understand these results, density functional theory calculations were performed. Consistent with the studies mentioned above, the analysis showed that O2 binds strongly to the surface iodide vacancies. These oxygen molecules passivate the carrier trap states, which are generated from the iodine vacancies. This passivation of defect states by oxygen molecules not only enhances the emission properties of the perovskite but also increases the electric conductivity of the MAPbI3. Huang et al. [163] observed similar effect in the CsPbBr3 NCs. They reported that when CsPbBr3 NCs thin films are exposed to relative humidity of 60–80% for 8 h, in the absence of O2, no PL intensity decrease was observed. When the same films were exposed to the oxygen atmosphere, PL intensity increased because of the formation of superoxide with surface halogen vacancies. This phenomenon is known as the oxygen boost effect. However, oxygen boost effect can be seen only with short-time illuminations. Upon continuous illumination, the highly emissive green color HPNCs thin film turn into nonemissive, yellow-colored film. Mukherjee et al. [164] reported similar observation in studies of single crystals of MAPbBr3 NPLs. They observed an initial increase in the PL intensity when MAPbBr3 NPLs were exposed to O2 atmosphere. Upon continuous exposure to O2, the NPLs eventually lost the PL completely. They also observed two-state blinking pattern (with less emissivity) under inert conditions and photo-brightening blinking pattern (with more emissivity) in O2 and moist air atmosphere. These observations indicate that under inert conditions HPNCs are stabilized but they lose the PL. On the other hand, in O2 and moist air atmosphere the HPNCs are more emissive but have reduced stability. Stoeckel et al. [165] studied the electrical response of organohalide PSs in the presence of oxygen. They found a 3000‐fold increase in the electric conductivity when HPS films were exposed to the oxygen atmosphere (Fig. 19d, e). The studies discussed in this section show that oxygen and moisture can, on a short-term, passivate the defects at surfaces or the grain boundaries, but in the long term, they cause the degradation of the PSs. Better understanding of these processes is essential for development of high-performance HPS devices.

Reproduced with permission from ref. [161]. Copyright 2017 Nature
a Schematic representation of the interaction of the O2 with MAPbI3 during the superoxide formation. b Binding and reduction of O2 at various sites of MAPbI3 along with the calculated superoxide formation energy of different sites.

a PL mapping of a MAPbI3 perovskite film in dry nitrogen. b PL emission from a bright grain [blue circle in (a)] shows photo brightening under O2 and H2O along with a steady emission in inert atmosphere. c A dark grain [pink circle in (a)] under dry nitrogen shows steady emission and shows photo brightening under humid environment (≈45% relative humidity). Reproduced with permission from ref. [162]. Copyright 2018 Wiley. Current–voltage measurement of (d) two‐step deposited perovskite thin film (e) and single‐step deposited perovskite thin film at different O2 concentrations. Reproduced with permission from ref. [165]. Copyright 2017 Wiley
8.2 Light-Induced Degradation
Experimental studies have shown that hybrid/inorganic ABX3 HPSs are very sensitive to light, particularly in the ultraviolet light spectral range, regardless of whether the material is in the bulk form or in the form of NC. With respect to phase and composition stability of HPSs, light can be viewed as a reagent. Numerous examples consistent with this assessment can be found in literature. In one example, Merdasa et al. [166] showed that exposure of MAPbI3 to 514-nm CW laser light leads to a large blue-shift (60 nm) of the PL spectrum with a sharp decrease in the PL intensity, indicating structural degradation of the PS to PbI2 form. This was attributed to the light-induced migration of the MA ions, distorting the lattice structure of the PS and altering the Pb–I–Pb bond angle, which changed the bandgap and led to the formation of the PbI2 phase. Similar effect was observed for CsPbI3 HPNCs [167], by single crystal spectroscopy, which upon a visible light irradiation of 466 nm pulsed laser diode underwent an irreversible, photo-induced blue shift of the PL and ultimately to a complete PL quenching. They found that both light and water can independently degrade CsPbI3 NCs. Light acts as a catalyst speeding up the degradation. Light induced effects resemble the vacancy-mediated A-site ion migration [168], as the photoexcitation reduces the barrier to ion migration. As a result of the ion migration, in a short-term, a capacitance builds up in the PS, which leads to an observation of hysteresis in the J–V characteristics of the HP solar cells [169, 170]. In the case of mixed halide PSs, a continuous irradiation of the mixed HPSs (thin film or NCs) leads to phase segregation due to ion migration [113]. Hoke et al. [171,172,173] were first to report on the ion migration behavior in the mixed HPSs. Halide ion migration produces in the mixed halide MAPb(Br/I)3 PSs ‘I’ rich and “Br”-rich domains. The resulting phase segregation leads to formation of regions with a narrower bandgap in the case of iodine and wider bandgap in the case of bromine. A reversible red shift in the PL spectra is observed in the mixed halide perovskite thin films due to the halogen ion migration caused by photoinduction. The photoexcitation was also found to lead to generation of a lattice strain [174,175,176]. To minimize the irradiation generated lattice strain, ions migrate to reach an equilibrium position and a halogen rich domain are formed, which lead to the observed in the PL spectra. On the other hand, Zhang et al. [177] showed that CsPbBr1.2I1.8 NCs (on a single-particle level) undergo a nonreversible blue shift from 630 to 520 nm and then eventually lose the emission leads as they photodegrade. Although, the formation of halogen rich domains is a reversible process and the interruption of irradiation can restore the system to its original state, the photoinduced ion migration is an undesirable effect from the standpoint of development of optoelectronic applications. In addition, it was found that iodine-rich regions can act as nonradiative recombination centers, which can trap the photo-generated charge carriers and significantly reduce important photovoltaic parameters such as open-circuit voltage and quantum efficiency [174, 178].
9 Electrochemistry and Spectroelectrochemistry of HPNCs
Charged HPNCs play an important role in technologies such as photovoltaics, light-emitting diodes, photodetectors, and others. These devices rely on efficient NC-to-NC hopping of carriers to or from electrodes. Significant populations of charged NCs can form during the device operation. Spectroelectrochemistry is a useful tool for studies of how perovskite NCs can be charged, how the charge migrates in HPNC structures and how charging affects their optical properties. In spite of its practical importance, this understanding is so far limited. This is mainly due to the instability of the HPNCs upon charging. In 2016, Vikash et al. [179] used electrochemistry to investigate the absolute energy offsets of the conduction band minima (CBM) and valance band maxima (VBM) in the CsPbX3 (X is Cl−, Br−, or I−) NCs, while systematically varying the halogen composition. Cyclic voltammetry (CV) results showed that as the halogen was changed from X = Cl− to Br− to I−, the VBM systematically shifted from ~1.8 to ~1.4 V to ~1.0 V (versus NHE), whereas the CBM shifted from −1.3 to −1.2 V to −1.1 V, respectively. Samu et al. [180] investigated the charging of CsPbBr3 and MAPbI3 thin films by electrochemistry and spectroelectrochemistry, coupled with X-ray diffraction (XRD) and X-ray photoelectron (XPS) spectroscopies. In the case of CsPbBr3, two oxidation peaks, one at ~0.8 V and one at ~1.5 V, and one reduction peak at ~ −1.0 V, with respect to NHE electrode, were observed. During the oxidation, the absorbance at 518 nm was observed to first increase at the first oxidation potential, primarily due to the changes in PS film scattering, and then gradually decrease again following the second oxidation (Fig. 20a–c). During the reduction cycle, the absorption decreased dramatically at the potentials more negative than −1.0 V (−1.2 V versus Ag/AgCl) (Fig. 20b). The observed changes were assigned to the chemical processes summarized in Eqs. 1−3 below:

Spectroelectrochemical data was recorded for FTO/TiO2/CsPbBr3 films during the a oxidation and b reduction half cycle together with the absorbance change at the excitonic peak. c Photographs of perovskite deposited electrodes after CV cycle. Reproduced with permission from ref. [180]. Copyright 2018 American Chemical Society. d CV of a CsPbBr3 NC electrode. The estimated VB edge position is marked by the black line. e PL of a NC electrode as a function of the applied potential during three CV scans ranging from −0.10 to +1.20 V. Hole injection into the VB edge of the NCs, starting at +0.9 V, quenches the PL, which largely recovers when the applied potential is lowered to below +0.9 V. f Differential absorbance of a NC electrode as a function of the applied potential, ranging from −0.10 to +1.05 V. The initial absorption spectrum is plotted in black. Reproduced with permission from ref. [182]. Copyright 2021 American Chemical Society
Oxidation:
Reduction:
Similar observations and conclusions were reached by Lee et al. [181] in studies of CsPbBr3 nanoparticulate thin films. The reasons for the observations can be understood by referring to the energy diagram shown in Fig. 21a. Because the oxidation reactions (1) and (2) are thermodynamically more favorable than the injection of a hole into the VB of the CsPbBr3, the application of the potential required for injection of a hole leads to irreversible chemical changes in the composition of the material according to reactions (1) and (2). Similarly, on the reductive side, the thermodynamically favorable reaction (3) effectively competes with the electron injection into CsPbBr3 CB. The oxidation of iodine and reduction of Pb2+ are thermodynamically even more favorable in the case of CsPbI3. Therefore, application of potentials required for hole/electron into the VB/CB leads to even more rapid chemical degradation of the HPNCs. In contrast, CsPbCl3 was shown to have significantly better stability with respect to chemical degradation under the conditions of electrochemical charging. More recently, Mulder et al. [182] showed that the reversible p-doping of CsPbBr3 NCs can be achieved by modification of their surface properties by synthetic means. In the preparation of CsPbBr3 NCs, instead of PbBr2, benzoyl bromide was used as the halide source. This led to the formation of halogen-rich CsPbBr3 NCs, thermodynamically stabilized with respect to the electrochemical oxidation. In the stabilized HPNCs, the spectroelectrochemical studies (Fig. 20d–f) showed that observed changes in the emission and absorption are due to the reversible injection of hole into the core of the NCs. These results can be understood in terms of the energy diagram shown in Fig. 21b. The use of the halogen-rich precursor led to stabilization of the VBM (~0.9 V versus NHE). As a result, the competing oxidation processes, oxidation of Br− and Pb2+ are thermodynamically less favorable. The injection of electron into the CB was not successful due to the rapid reduction of the HPNCs, according to reaction (3). These results show that surface modification can be used as an effective tool in modifying the electronic structure of HPNCs, which leads to their stabilization with respect to carrier doping.

a Comparison of the band edge positions and the potentials of various redox reactions observed in lead halide perovskites. Reproduced with permission from ref. [180]. Copyright 2021 American Chemical Society. b Energy diagram, indicating the energy level of the VB edge, CB edge, and VOC of the measured CsPbBr3 NCs. Reproduced with permission from ref. [182]. Reproduced with permission. Copyright 2018 American Chemical Society
In a separate study, Wong et al. [183] investigated electrochemiluminescence (ECL) of CsPbCl3 and Mn-doped CsPbCl3 in the presence of benzoyl peroxide as a coreactant. In the electrochemical studies of the undoped CsPbCl3 two oxidation peaks were observed, one at 0.8 V and 1.2 V and one reduction peak at −1.7 V. The authors attributed these peaks to injection of first and second hole into the VB and injection of electron into the CB, respectively. When attempting to generate ECL using a rapid potential step past the potentials for hole or electron injection, no ECL was observed in the undoped CsPbCl3. This suggests that the observed electrochemical response is likely dominated by the electrochemical degradation of the CsPbCl3 NCs, similar to the processes shown in reactions (1)–(3), rather than hole or electron injection. A weak ECL was observed in Mn-doped CsPbCl3 following the potential step past 0.7 V, attributed to partial stabilization of the charged HPNC by the Mn dopant [184].
10 Summary and Outlook
The advances summarized in previous sections clearly show that over the past decade HPNCs were established as a distinct new class of materials with unique properties and great practical potential. Diverse set of approaches for the preparation of HPNCs were elaborated, with the hot-injection method representing arguably the most important advance. Good progress was made in identifying the key parameters influencing the size and shape of the produced HPNCs. Development of approaches for effective doping of HPNCs opened path to additional tuning of their electronic structure and optical properties, further extending their utility. Important advances were made in understanding of the relationship between the composition, defects and optical properties of HPNCs, chemical processes responsible for their degradation, as well as the effects of charge injection. These advances represent the necessary early steps towards the development of stable materials suitable for exploitation in applications.
Despite, and in many instances thanks to this significant progress, important questions and challenges remain, or have emerged, that need to be answered before HPNCs will enjoy broad acceptance as materials of choice for optoelectronic, optical, electrical, chemical, medical, or other applications. One example is the mechanism and the dynamics of the NC growth. While for chalcogenide NCs the dynamics of NC growth is reasonably well described by La Mer type diagrams with various distinct steps, this is not the case for HPNCs. Due to the ionic nature of the perovskite structure, in the hot-injection or LARP methods HPNCs are formed nearly instantaneously, which makes it difficult to resolve various stages of the crystal growth process (e.g., nucleation, growth, and ripening). Hence, for the HPNCs the general crystal growth kinetics and mechanism were not yet clearly established. For majority of reactions, the mechanism is specific for a single type of reaction or for a specific size or shape, with dimension tuning primarily achieved through thermodynamic control. Another area, where more advances are needed is the understanding of the HPNC surface chemistry. Due to the highly dynamic nature of HPNC surface ligands, the HPNCs are very sensitive to surface treatment and/or change of environment. During purification, PLQY of HPNCs can be reduced by 20–40% due to removal/substitution of surface ligands, yet our understanding of the origins of the high lability of the surface ligands and potential solutions are quite limited. Previously proposed use of bidentate ligands may be a step in a right direction, but more systematic studies are needed in this area. Another challenge to be addressed is the long-term chemical stability of the HPNCs. Though it is observed that CsPbBr3 NCs are relatively stable over a long period of time, preparation of CsPbI3 NCs with stable phase and PLQY is quite challenging. Moreover, while significant progress was made in the synthesis of inorganic halide perovskite nanocubes, the examples of organic halide perovskite nanocubes are much more limited. Significant efforts were dedicated to development of methods for synthesis of perovskite NPLs, but our understanding of how to reproducibly grow HP NPLs with control over thickness and shape are limited. Additionally, the extent to which perovskite NPLs exist as isolated sheets in a colloidal solution rather than as small crystallites of the Ruddlesden–Popper phase remains unclear. Therefore, systematic studies of the behavior of HP NPLs in colloidal solutions are needed. Moreover, since the PLQY of these relatively low as compared with the cubic analogues, there is a need for studies aimed at improving the PLQY the perovskite NPLs.
To date, the hot-injection and LARP methods, the most utilized approaches for synthesis HPNCs, utilized long chain alkylamines to stabilize the NCs. These types of ligands represent an electronic energy barrier, hindering interparticle electronic coupling and thereby constraining the potential applications of the HPNCs in optoelectronics and electronics. To address this challenge, new synthetic or ligand-exchange approaches need to be developed that will allow preparation of HPNCs with more conductive or smaller ligands, without compromising the quality and stability of the HPNCs. An additional problem from the standpoint of electronic application of HPNCs is the ion migration, which leads to hysteresis in the device performance. New approaches need to be identified that will limit the negative impact of the ion migration on the performance of devices.
In the case of the doped perovskite structures, knowledge of the dopant location within the HPNCs is important from a fundamental as well as practical standpoint. However, currently we have limited information about whether the available doping approaches yield NCs with dopants located on the surface of the HPNC or inside its lattice. This is in part due to the low stability of HPNCs under the standard electron microscopy conditions. Creative solutions to overcoming this problem are needed to better understand the structure of doped HPNCs. More studies are also needed to better understand the origins of the defects in HPNCs during the synthesis and as well as during the interaction with the ambient environment and their effect on optical and electrical properties of HPNCs. Presence of toxic lead in the structure of HPNCs remains a challenge from the standpoint of their potential commercial utility. While some advances were made in this area, the optical properties of prepared lead-free HPNCs are much less appealing than the lead-based counterparts. Thus, more effort is needed in development of lead-free HPNCs. The intrigue of these and similar questions and challenges guarantees that HPNCs will remain an intense area of research in the foreseeable future.
References
Wells HL (1893) Z Anorg Chem 3:195. https://doi.org/10.1002/zaac.18930030124
Mitzi DB (1999) In: Karlin KD (ed) Progress in Inorganic Chemistry, Vol 48 p 1
Mitzi DB, Feild CA, Harrison WTA et al (1994) Nature 369:467. https://doi.org/10.1038/369467a0
Mitzi DB, Wang S, Feild CA et al (1995) Science 267:1473. https://doi.org/10.1126/science.267.5203.1473
Kojima A, Teshima K, Shirai Y et al (2009) J Am Chem Soc 131:6050. https://doi.org/10.1021/ja809598r
Kim H-S, Lee C-R, Im J-H et al (2012) Sci Rep 2:591. https://doi.org/10.1038/srep00591
Lee MM, Teuscher J, Miyasaka T et al (2012) Science 338:643. https://doi.org/10.1126/science.1228604
Correa-Baena J-P, Saliba M, Buonassisi T et al (2017) Science 358:739. https://doi.org/10.1126/science.aam6323
Ball JM, Lee MM, Hey A et al (2013) Energy Environ Sci 6:1739. https://doi.org/10.1039/C3EE40810H
Yang WS, Noh JH, Jeon NJ et al (2015) Science 348:1234. https://doi.org/10.1126/science.aaa9272
Yang WS, Park B-W, Jung EH et al (2017) Science 356:1376. https://doi.org/10.1126/science.aan2301
Liang Z, Zhang Y, Xu H et al (2023). Nature. https://doi.org/10.1038/s41586-023-06784-0
Mariotti S, Köhnen E, Scheler F et al (2023) Science 381:63. https://doi.org/10.1126/science.adf5872
Khenkin MV, Katz EA, Abate A et al (2020) Nat Energy 5:35. https://doi.org/10.1038/s41560-019-0529-5
Katz EA (2020) Nat Energy 5:1. https://doi.org/10.1038/s41560-020-0552-6
D’Innocenzo V, Srimath Kandada AR, De Bastiani M et al (2014) J Am Chem Soc 136:17730. https://doi.org/10.1021/ja511198f
Deschler F, Price M, Pathak S et al (2014) J Phys Chem Lett 5:1421. https://doi.org/10.1021/jz5005285
Tan Z-K, Moghaddam RS, Lai ML et al (2014) Nat Nanotechnol 9:687. https://doi.org/10.1038/nnano.2014.149
Stranks SD, Snaith HJ (2015) Nat Nanotechnol 10:391. https://doi.org/10.1038/nnano.2015.90
Yin W-J, Shi T, Yan Y (2014) Appl Phys Lett 104:063903. https://doi.org/10.1063/1.4864778
de Quilettes DW, Vorpahl SM, Stranks SD et al (2015) Science 348:683. https://doi.org/10.1126/science.aaa5333
Droseros N, Longo G, Brauer JC et al (2018) ACS Energy Lett 3:1458. https://doi.org/10.1021/acsenergylett.8b00475
Schmidt LC, Pertegás A, González-Carrero S et al (2014) J Am Chem Soc 136:850. https://doi.org/10.1021/ja4109209
Manser JS, Christians JA, Kamat PV (2016) Chem Rev 116:12956. https://doi.org/10.1021/acs.chemrev.6b00136
Travis W, Glover ENK, Bronstein H et al (2016) Chem Sci 7:4548. https://doi.org/10.1039/C5SC04845A
Whitfield PS, Herron N, Guise WE et al (2016) Sci Rep 6:35685. https://doi.org/10.1038/srep35685
Thomson S (2018) Application Note: Edinburgh Instruments.
Lee J-H, Bristowe NC, Lee JH et al (2016) Chem Mater 28:4259. https://doi.org/10.1021/acs.chemmater.6b00968
Kurlansky M, Schindler S (2006) The story of salt. G.P. Putnam's Sons
Kasai H, Nalwa HS, Oikawa H et al (1992) Jpn J Appl Phys 31:L1132. https://doi.org/10.1143/jjap.31.l1132
Zhao YS, Fu H, Peng A et al (2008) Adv Mater 20:2859. https://doi.org/10.1002/adma.200800604
Papavassiliou GC, Pagona G, Karousis N et al (2012) J Mater Chem 22:8271. https://doi.org/10.1039/C2JM15783G
Zhang F, Zhong H, Chen C et al (2015) ACS Nano 9:4533. https://doi.org/10.1021/acsnano.5b01154
Xing J, Yan F, Zhao Y et al (2016) ACS Nano 10:6623. https://doi.org/10.1021/acsnano.6b01540
Levchuk I, Osvet A, Tang X et al (2017) Nano Lett 17:2765. https://doi.org/10.1021/acs.nanolett.6b04781
Li X, Wu Y, Zhang S et al (2016) Adv Funct Mater 26:2435. https://doi.org/10.1002/adfm.201600109
Leng M, Chen Z, Yang Y et al (2016) Angew Chem Int Ed 55:15012. https://doi.org/10.1002/anie.201608160
Yang B, Chen J, Hong F et al (2017) Angew Chem Int Ed 56:12471. https://doi.org/10.1002/anie.201704739
Leng M, Yang Y, Zeng K et al (2018) Adv Funct Mater 28:1704446. https://doi.org/10.1002/adfm.201704446
Zhang J, Yang Y, Deng H et al (2017) ACS Nano 11:9294. https://doi.org/10.1021/acsnano.7b04683
Zhang F, Huang S, Wang P et al (2017) Chem Mater 29:3793. https://doi.org/10.1021/acs.chemmater.7b01100
Murray CB, Norris DJ, Bawendi MG (1993) J Am Chem Soc 115:8706. https://doi.org/10.1021/ja00072a025
Chang J, Waclawik ER (2014) RSC Adv 4:23505. https://doi.org/10.1039/C4RA02684E
Protesescu L, Yakunin S, Bodnarchuk MI et al (2015) Nano Lett 15:3692. https://doi.org/10.1021/nl5048779
Akkerman QA, Park S, Radicchi E et al (2017) Nano Lett 17:1924. https://doi.org/10.1021/acs.nanolett.6b05262
Han C, Li C, Zang Z et al (2017) Photon Res 5:473. https://doi.org/10.1364/PRJ.5.000473
Protesescu L, Yakunin S, Bodnarchuk MI et al (2016) J Am Chem Soc 138:14202. https://doi.org/10.1021/jacs.6b08900
Lignos I, Protesescu L, Emiroglu DB et al (2018) Nano Lett 18:1246. https://doi.org/10.1021/acs.nanolett.7b04838
Protesescu L, Yakunin S, Kumar S et al (2017) ACS Nano 11:3119. https://doi.org/10.1021/acsnano.7b00116
Vybornyi O, Yakunin S, Kovalenko MV (2016) Nanoscale 8:6278. https://doi.org/10.1039/C5NR06890H
Imran M, Caligiuri V, Wang M et al (2018) J Am Chem Soc 140:2656. https://doi.org/10.1021/jacs.7b13477
Roy M, Vikram, Banerjee S et al (2019) Chem Eur J 25:9892. https://doi.org/10.1002/chem.201805859
Liu F, Zhang Y, Ding C et al (2017) ACS Nano 11:10373. https://doi.org/10.1021/acsnano.7b05442
Wu L, Zhong Q, Yang D et al (2017) Langmuir 33:12689. https://doi.org/10.1021/acs.langmuir.7b02963
Liu P, Chen W, Wang W et al (2017) Chem Mater 29:5168. https://doi.org/10.1021/acs.chemmater.7b00692
Chen M, Zou Y, Wu L et al (2017) Adv Func Mater 27:1701121. https://doi.org/10.1002/adfm.201701121
Parveen S, Paul KK, Giri PK (2020) ACS Appl Mater Interfaces 12:6283. https://doi.org/10.1021/acsami.9b20896
Pan Q, Hu H, Zou Y et al (2017) J Mater Chem C 5:10947. https://doi.org/10.1039/C7TC03774K
Shamsi J, Rastogi P, Caligiuri V et al (2017) ACS Nano 11:10206. https://doi.org/10.1021/acsnano.7b04761
Liu H, Wu Z, Gao H et al (2017) ACS Appl Mater Interfaces 9:42919. https://doi.org/10.1021/acsami.7b14677
Jang DM, Kim DH, Park K et al (2016) J Mater Chem C 4:10625. https://doi.org/10.1039/C6TC04213A
Tong Y, Bladt E, Aygüler MF et al (2016) Angew Chem Int Ed 55:13887. https://doi.org/10.1002/anie.201605909
Roy M, Vikram, Bhawna et al (2021) J Phys Chem Lett 12:1189. https://doi.org/10.1021/acs.jpclett.0c03426
Leupold N, Schötz K, Cacovich S et al (2019) ACS Appl Mater Interfaces 11:30259. https://doi.org/10.1021/acsami.9b09160
Dou B, Wheeler LM, Christians JA et al (2018) ACS Energy Lett 3:979. https://doi.org/10.1021/acsenergylett.8b00305
Hintermayr VA, Richter AF, Ehrat F et al (2016) Adv Mater 28:9478. https://doi.org/10.1002/adma.201602897
Zhu Z-Y, Yang Q-Q, Gao L-F et al (2017) J Phys Chem Lett 8:1610. https://doi.org/10.1021/acs.jpclett.7b00431
Baláž P, Achimovičová M, Baláž M et al (2013) Chem Soc Rev 42:7571. https://doi.org/10.1039/C3CS35468G
Protesescu L, Yakunin S, Nazarenko O et al (2018) ACS Appl Nano Mater 1:1300. https://doi.org/10.1021/acsanm.8b00038
Chen D, Li J, Chen X et al (2019) ACS Appl Mater Interfaces 11:10059. https://doi.org/10.1021/acsami.8b19002
Sichert JA, Tong Y, Mutz N et al (2015) Nano Lett 15:6521. https://doi.org/10.1021/acs.nanolett.5b02985
Sandeep K, Padmakumar K, Ambili KU et al (2022). Physica Status Solidi B-Basic Solid State Phys. https://doi.org/10.1002/pssb.202100600
Byranvand MM, Otero-Martinez C, Ye JZ et al (2022). Adv Opt Mater. https://doi.org/10.1002/adom.202200423
Jiang H, Cui S, Chen Y et al (2021) Nano Select 2:2040. https://doi.org/10.1002/nano.202100084
Swarnkar A, Mir WJ, Nag A (2018) ACS Energy Lett 3:286. https://doi.org/10.1021/acsenergylett.7b01197
Qiu Z, Li N, Huang Z et al (2020) Small Methods 4:1900877. https://doi.org/10.1002/smtd.201900877
Ogomi Y, Morita A, Tsukamoto S et al (2014) J Phys Chem Lett 5:1004. https://doi.org/10.1021/jz5002117
Hao F, Stoumpos CC, Chang RPH et al (2014) J Am Chem Soc 136:8094. https://doi.org/10.1021/ja5033259
Hao F, Stoumpos CC, Cao DH et al (2014) Nat Photonics 8:489. https://doi.org/10.1038/nphoton.2014.82
Liu C, Fan J, Li H et al (2016) Sci Rep 6:35705. https://doi.org/10.1038/srep35705
Zhang X, Cao W, Wang W et al (2016) Nano Energy 30:511. https://doi.org/10.1016/j.nanoen.2016.10.039
Deng J, Wang H, Xun J et al (2020) Mater Des 185:108246. https://doi.org/10.1016/j.matdes.2019.108246
Jellicoe TC, Richter JM, Glass HFJ et al (2016) J Am Chem Soc 138:2941. https://doi.org/10.1021/jacs.5b13470
Babayigit A, Duy Thanh D, Ethirajan A et al (2016) Sci Rep 6:18721. https://doi.org/10.1038/srep18721
Parobek D, Roman BJ, Dong Y et al (2016) Nano Lett 16:7376. https://doi.org/10.1021/acs.nanolett.6b02772
Liu W, Lin Q, Li H et al (2016) J Am Chem Soc 138:14954. https://doi.org/10.1021/jacs.6b08085
Mir WJ, Jagadeeswararao M, Das S et al (2017) ACS Energy Lett 2:537. https://doi.org/10.1021/acsenergylett.6b00741
van der Stam W, Geuchies JJ, Altantzis T et al (2017) J Am Chem Soc 139:4087. https://doi.org/10.1021/jacs.6b13079
Shen X, Zhang Y, Kershaw SV et al (2019) Nano Lett 19:1552. https://doi.org/10.1021/acs.nanolett.8b04339
Bi C, Wang S, Li Q et al (2019) J Phys Chem Lett 10:943. https://doi.org/10.1021/acs.jpclett.9b00290
De A, Das S, Mondal N et al (2019) ACS Mater Lett 1:116. https://doi.org/10.1021/acsmaterialslett.9b00101
Thawarkar S, Rana PJS, Narayan R et al (2019) Langmuir 35:17150. https://doi.org/10.1021/acs.langmuir.9b02450
Yong Z-J, Guo S-Q, Ma J-P et al (2018) J Am Chem Soc 140:9942. https://doi.org/10.1021/jacs.8b04763
Zhou Y, Yong Z-J, Zhang K-C et al (2016) J Phys Chem Lett 7:2735. https://doi.org/10.1021/acs.jpclett.6b01147
Abdelhady AL, Saidaminov MI, Murali B et al (2016) J Phys Chem Lett 7:295. https://doi.org/10.1021/acs.jpclett.5b02681
Nayak PK, Sendner M, Wenger B et al (2018) J Am Chem Soc 140:574. https://doi.org/10.1021/jacs.7b11125
Lozhkina OA, Murashkina AA, Shilovskikh VV et al (2018) J Phys Chem Lett 9:5408. https://doi.org/10.1021/acs.jpclett.8b02178
Begum R, Parida MR, Abdelhady AL et al (2017) J Am Chem Soc 139:731. https://doi.org/10.1021/jacs.6b09575
Zhang X, Wang H, Hu Y et al (2019) J Phys Chem Lett 10:1750. https://doi.org/10.1021/acs.jpclett.9b00790
Liu M, Zhong G, Yin Y et al (2017) Adv Sci 4:1700335. https://doi.org/10.1002/advs.201700335
Balakrishnan SK, Kamat PV (2018) Chem Mater 30:74. https://doi.org/10.1021/acs.chemmater.7b04142
Palazon F, Almeida G, Akkerman QA et al (2017) Chem Mater 29:4167. https://doi.org/10.1021/acs.chemmater.7b00895
Udayabhaskararao T, Houben L, Cohen H et al (2018) Chem Mater 30:84. https://doi.org/10.1021/acs.chemmater.7b02425
Liu Z, Bekenstein Y, Ye X et al (2017) J Am Chem Soc 139:5309. https://doi.org/10.1021/jacs.7b01409
Palazon F, Urso C, De Trizio L et al (2017) ACS Energy Lett 2:2445. https://doi.org/10.1021/acsenergylett.7b00842
Palazon F, Dogan S, Marras S et al (2017) J Phys Chem C 121:11956. https://doi.org/10.1021/acs.jpcc.7b03389
Turedi B, Lee KJ, Dursun I et al (2018) J Phys Chem C 122:14128. https://doi.org/10.1021/acs.jpcc.8b01343
Xiao G, Cao Y, Qi G et al (2017) J Am Chem Soc 139:10087. https://doi.org/10.1021/jacs.7b05260
Huisman BAH, Palazon F, Bolink HJ (2021). Inorg Chem. https://doi.org/10.1021/acs.inorgchem.1c00212
Cordero SR, Carson PJ, Estabrook RA et al (2000) J Phys Chem B 104:12137. https://doi.org/10.1021/jp001771s
Motti SG, Patel JB, Oliver RDJ et al (2021) Nat Commun 12:6955. https://doi.org/10.1038/s41467-021-26930-4
Yuan H, Debroye E, Janssen K et al (2016) J Phys Chem Lett 7:561. https://doi.org/10.1021/acs.jpclett.5b02828
Hoke ET, Slotcavage DJ, Dohner ER et al (2015) Chem Sci 6:613. https://doi.org/10.1039/C4SC03141E
Wang Y, Li X, Sreejith S et al (2016) Adv Mater 28:10637. https://doi.org/10.1002/adma.201604110
Ha SK, Mauck CM, Tisdale WA (2019) Chem Mater 31:2486. https://doi.org/10.1021/acs.chemmater.8b05310
Roy M, Vikram, Bhawna et al (2021) Phys Chem Chem Phys 23:27355. https://doi.org/10.1039/D1CP03529K
Sen A, Chatterjee S, Sen P (2022) J Phys Chem C 126:18057. https://doi.org/10.1021/acs.jpcc.2c05987
Bekenstein Y, Koscher BA, Eaton SW et al (2015) J Am Chem Soc 137:16008. https://doi.org/10.1021/jacs.5b11199
Zhang D, Eaton SW, Yu Y et al (2015) J Am Chem Soc 137:9230. https://doi.org/10.1021/jacs.5b05404
Almeida G, Goldoni L, Akkerman Q et al (2018) ACS Nano 12:1704. https://doi.org/10.1021/acsnano.7b08357
Pan A, He B, Fan X et al (2016) ACS Nano 10:7943. https://doi.org/10.1021/acsnano.6b03863
Tyagi P, Arveson SM, Tisdale WA (2015) J Phys Chem Lett 6:1911. https://doi.org/10.1021/acs.jpclett.5b00664
Akkerman QA, Motti SG, Srimath Kandada AR et al (2016) J Am Chem Soc 138:1010. https://doi.org/10.1021/jacs.5b12124
Bertolotti F, Nedelcu G, Vivani A et al (2019) ACS Nano 13:14294. https://doi.org/10.1021/acsnano.9b07626
Shamsi J, Kubicki D, Anaya M et al (2020) ACS Energy Lett 5:1900. https://doi.org/10.1021/acsenergylett.0c00935
Weidman MC, Seitz M, Stranks SD et al (2016) ACS Nano 10:7830. https://doi.org/10.1021/acsnano.6b03496
Shamsi J, Dang Z, Bianchini P et al (2016) J Am Chem Soc 138:7240. https://doi.org/10.1021/jacs.6b03166
Weidman MC, Goodman AJ, Tisdale WA (2017) Chem Mater 29:5019. https://doi.org/10.1021/acs.chemmater.7b01384
Saidaminov MI, Mohammed OF, Bakr OM (2017) ACS Energy Lett 2:889. https://doi.org/10.1021/acsenergylett.6b00705
Koutselas IB, Ducasse L, Papavassiliou GC (1996) J Phys: Condens Matter 8:1217. https://doi.org/10.1088/0953-8984/8/9/012
Ishihara T, Takahashi J, Goto T (1989) Solid State Commun 69:933. https://doi.org/10.1016/0038-1098(89)90935-6
Tong Y, Bohn BJ, Bladt E et al (2017) Angew Chem Int Ed 56:13887. https://doi.org/10.1002/anie.201707224
Zhang D, Yu Y, Bekenstein Y et al (2016) J Am Chem Soc 138:13155. https://doi.org/10.1021/jacs.6b08373
Fang H-H, Adjokatse S, Wei H et al (2016) Sci Adv 2:e1600534. https://doi.org/10.1126/sciadv.1600534
Gordillo G, Otálora CA, Reinoso MA (2017) J Appl Phys 122:075304. https://doi.org/10.1063/1.4999297
Agiorgousis ML, Sun Y-Y, Zeng H et al (2014) J Am Chem Soc 136:14570. https://doi.org/10.1021/ja5079305
Kim Y-H, Wolf C, Kim Y-T et al (2017) ACS Nano 11:6586. https://doi.org/10.1021/acsnano.6b07617
Queisser HJ, Haller EE (1998) Science 281:945. https://doi.org/10.1126/science.281.5379.945
Freysoldt C, Grabowski B, Hickel T et al (2014) Rev Mod Phys 86:253. https://doi.org/10.1103/RevModPhys.86.253
Kim J, Lee S-H, Lee JH et al (2014) J Phys Chem Lett 5:1312. https://doi.org/10.1021/jz500370k
Eames C, Frost JM, Barnes PRF et al (2015) Nat Commun 6:7497. https://doi.org/10.1038/ncomms8497
Buin A, Pietsch P, Xu J et al (2014) Nano Lett 14:6281. https://doi.org/10.1021/nl502612m
Xu J, Buin A, Ip AH et al (2015) Nat Commun 6:7081. https://doi.org/10.1038/ncomms8081
Buin A, Comin R, Xu J et al (2015) Chem Mater 27:4405. https://doi.org/10.1021/acs.chemmater.5b01909
Ball JM, Petrozza A (2016) Nat Energy 1:16149. https://doi.org/10.1038/nenergy.2016.149
Walsh A, Scanlon DO, Chen S et al (2015) Angew Chem Int Ed 54:1791. https://doi.org/10.1002/anie.201409740
Wang Y-N, Ma P, Peng L-M et al (2017) Acta Phys Chim Sin 33:2099. https://doi.org/10.3866/PKU.WHXB201705115
Leng M, Yang Y, Chen Z et al (2018) Nano Lett 18:6076. https://doi.org/10.1021/acs.nanolett.8b03090
Dutta A, Behera RK, Dutta SK et al (2018) J Phys Chem Lett 9:6599. https://doi.org/10.1021/acs.jpclett.8b02825
Das Adhikari S, Behera RK, Bera S et al (2019) J Phys Chem Lett 10:1530. https://doi.org/10.1021/acs.jpclett.9b00599
Woo JY, Kim Y, Bae J et al (2017) Chem Mater 29:7088. https://doi.org/10.1021/acs.chemmater.7b02669
Mondal N, De A, Samanta A (2019) ACS Energy Lett 4:32. https://doi.org/10.1021/acsenergylett.8b01909
Li C, Chen X, Li N et al (2020) J Mater Chem C 8:3694. https://doi.org/10.1039/C9TC06854F
Mosconi E, Merabet B, Meggiolaro D et al (2018) J Phys Chem C 122:14107. https://doi.org/10.1021/acs.jpcc.8b01307
Lu C-H, Biesold-McGee GV, Liu Y et al (2020) Chem Soc Rev 49:4953. https://doi.org/10.1039/C9CS00790C
Ono LK, Juarez-Perez EJ, Qi Y (2017) ACS Appl Mater Interfaces 9:30197. https://doi.org/10.1021/acsami.7b06001
Zhou H, Chen Q, Li G et al (2014) Science 345:542. https://doi.org/10.1126/science.1254050
Ling L, Yuan S, Wang P et al (2016) Adv Funct Mater 26:5028. https://doi.org/10.1002/adfm.201601557
Leguy AMA, Hu Y, Campoy-Quiles M et al (2015) Chem Mater 27:3397. https://doi.org/10.1021/acs.chemmater.5b00660
Ahn N, Kwak K, Jang MS et al (2016) Nat Commun 7:13422. https://doi.org/10.1038/ncomms13422
Aristidou N, Eames C, Sanchez-Molina I et al (2017) Nat Commun 8:15218. https://doi.org/10.1038/ncomms15218
Brenes R, Eames C, Bulović V et al (2018) Adv Mater 30:1706208. https://doi.org/10.1002/adma.201706208
Huang H, Chen B, Wang Z et al (2016) Chem Sci 7:5699. https://doi.org/10.1039/C6SC01758D
Mukherjee A, Roy M, Pathoor N et al (2021) J Phys Chem C 125:17133. https://doi.org/10.1021/acs.jpcc.1c02207
Stoeckel M-A, Gobbi M, Bonacchi S et al (2017) Adv Mater 29:1702469. https://doi.org/10.1002/adma.201702469
Merdasa A, Bag M, Tian Y et al (2016) J Phys Chem C 120:10711. https://doi.org/10.1021/acs.jpcc.6b03512
Yuan G, Ritchie C, Ritter M et al (2018) J Phys Chem C 122:13407. https://doi.org/10.1021/acs.jpcc.7b11168
Yang J-H, Yin W-J, Park J-S et al (2016) J Mater Chem A 4:13105. https://doi.org/10.1039/C6TA03599J
Kim H-S, Jang I-H, Ahn N et al (2015) J Phys Chem Lett 6:4633. https://doi.org/10.1021/acs.jpclett.5b02273
Li C, Tscheuschner S, Paulus F et al (2016) Adv Mater 28:2446. https://doi.org/10.1002/adma.201503832
Yoon SJ, Kuno M, Kamat PV (2017) ACS Energy Lett 2:1507. https://doi.org/10.1021/acsenergylett.7b00357
Hentz O, Zhao Z, Gradečak S (2016) Nano Lett 16:1485. https://doi.org/10.1021/acs.nanolett.5b05181
Yoon SJ, Draguta S, Manser JS et al (2016) ACS Energy Lett 1:290. https://doi.org/10.1021/acsenergylett.6b00158
Bischak CG, Hetherington CL, Wu H et al (2017) Nano Lett 17:1028. https://doi.org/10.1021/acs.nanolett.6b04453
Barker AJ, Sadhanala A, Deschler F et al (2017) ACS Energy Lett 2:1416. https://doi.org/10.1021/acsenergylett.7b00282
Mosconi E, De Angelis F (2016) ACS Energy Lett 1:182. https://doi.org/10.1021/acsenergylett.6b00108
Zhang H, Fu X, Tang Y et al (2019) Nat Commun 10:1088. https://doi.org/10.1038/s41467-019-09047-7
Unger EL, Kegelmann L, Suchan K et al (2017) J Mater Chem A 5:11401. https://doi.org/10.1039/C7TA00404D
Ravi VK, Markad GB, Nag A (2016) ACS Energy Lett 1:665. https://doi.org/10.1021/acsenergylett.6b00337
Samu GF, Scheidt RA, Kamat PV et al (2018) Chem Mater 30:561. https://doi.org/10.1021/acs.chemmater.7b04321
Lee C, Kim K, Shin Y et al (2021). Front Energy Res. https://doi.org/10.3389/fenrg.2020.605976
Mulder JT, du Fossé I, Alimoradi Jazi M et al (2021) ACS Energy Lett 6:2519. https://doi.org/10.1021/acsenergylett.1c00970
Wong JM, Xu J, Zhang R et al (2021) J Phys Chem C 125:13696. https://doi.org/10.1021/acs.jpcc.1c04335
Tang L, Wei J, Cai Z et al (2021) Electrochem Commun 131:107123. https://doi.org/10.1016/j.elecom.2021.107123
Acknowledgements
M.R. and M.S. acknowledge financial support from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 810701, Slovak Research and Development Agency under grant agreement no. APVV-19-410, and Slovak Ministry of education under grant agreement no. 1/0892/21. M.R. acknowledges partial support by Comenius University postdoctoral fellowship. M.A. would like to acknowledge NCPRE-02, and NCPRE-03 from MNRE for the financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Roy, M., Sykora, M. & Aslam, M. Chemical Aspects of Halide Perovskite Nanocrystals. Top Curr Chem (Z) 382, 9 (2024). https://doi.org/10.1007/s41061-024-00453-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-024-00453-0