
Abstract
Direct reductive N-methylation of inexpensive and readily available nitro compounds as raw material feedstocks is more attractive and straightforward compared with conventional N-methylation of amines to prepare biologically and pharmaceutically important N-methylated amine derivatives. This strategy for synthesis of N-methylamines avoids prepreparation of NH-free amines and therefore significantly shortens the separation and purification steps. In recent years, numerous methylating agents and catalytic systems have been reported for this appealing transformation. Thus, it is an appropriate time to summarize such advances. This review elaborates on the most important discoveries and advances in this research arena, with special emphasis on the mechanistic aspect of reactions that may provide new insights into catalyst improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
N-Methylated amines represent important fine and bulk chemicals as they occur in many medicines [14], dyes [5], and pesticides [6, 7] (Scheme 1). These structural motifs also serve as valuable intermediates in organic synthesis [8,9,10,11]. Moreover, they are common motifs in surfactants [12]. The development of convenient and cost-effective methods for their preparation is thus of scientific interest. This special class of amines has traditionally been synthesized by N-methylation of NH-free amines using methylating reagents such as methyl halides methanol, dimethyl carbonate, dimethyl sulfate, dimethyl sulfoxide (DMSO), formaldehyde, etc. [13, 14]. As is well known, one of the main methods to generate primary amines is reduction of nitro compounds [15,16,17,18]. Therefore, without question, the development of new and efficient approaches to construct N-methylated amines directly from corresponding nitro compounds is highly desirable in economic terms.

Examples of N-methylamines: a medicines, b dyes, and c pesticides
Fortunately, in recent years, a number of unique methodologies have been developed for one-pot synthesis of N-methylated amines from nontoxic, low-cost, and readily available nitro compounds through a sequential hydrogenation and methylation process. Despite the remarkable progress that has been achieved in this appealing research arena, a comprehensive overview of this domain has not appeared in literature to date. Our aim in this review is to try to provide an up-to-date overview of the most important advances and developments regarding direct reductive N-methylation of nitro compounds, with particular emphasis on the mechanistic aspects of such reactions. For clarity, the review is structured based on the type of methylating agent applied (i.e., MeOH, DMSO, HCHO, HCOOH, and CO2/H2).
2 Methylations with Methanol
Direct N-methylation of nitro compounds using methanol as the methyl source is probably the area of this field that has experienced most growth over the past few years. The story of synthesis of N-methylamines through reductive methylation of nitro compounds with methanol started with Li and coworkers, who reported that heating (170 °C) of nitrobenzene in methanol in the presence of a pretreated Raney-Ni catalyst under an inert atmosphere afforded N,N-dimethylaniline in up to 98% yield [19]. In this reaction, methanol plays a dual role, as substrate and solvent. Moreover, it acts not only as a methyl source but also as a hydrogen source. Although only one example was provided, and under harsh conditions, that paper represents the first example of direct N-methylation of nitro compounds with methanol. Six years later, Shi and colleagues developed a practical Pd-catalyzed N,N-dimethylation of a library of electron-rich nitrobenzenes 1 with methanol under mild conditions [20]. The reactions proceeded in the presence of a catalytic amount of TiO2-supported palladium nanoparticles (Pd0.8/TiO2) under irradiation by ultraviolet (UV) light at room temperature to give the corresponding N,N-dimethylaniline products 2 in moderate to excellent yields (Scheme 2). The results indicated that increasing or decreasing the Pd loading reduced the product yields. Noteworthily, other TiO2-supported metal catalysts (e.g., Au/TiO2, Pt/TiO2, and Pt/TiO2) failed to promote this reaction. Based on the quantitation of intermediates by the gas chromatography–mass spectrometry (GC–MS) method for the reaction of aniline with methanol, the authors proposed that this reductive methylation reaction proceeds though the following key steps (Scheme 3): (1) photoexcitation of TiO2 to form electron (e−) and positive hole (h+) pairs; (2) oxidation of methanol with h+ to furnish formaldehyde and H+; (3) reduction of H+ on the surface of Pd particles by the e− transferred from the TiO2 conduction band to produces a hydride on the particles (H–Pd species); (4) condensation of formaldehyde and aniline A (generated in situ by reduction of nitrobenzene 1) with the help of Lewis-acid site on the TiO2 surface to afford imine B; (5) hydrogenation of imine B by H–Pd species on the surface of TiO2 to yield N-methylaniline C; (6) coupling of formaldehyde and N-methylaniline C to provide aminium F through intermediates D and E; and (7) hydrogenation of aminium F by H–Pd species to generate the final N,N-dimethylaniline products 2.

Shi’s synthesis of N,N-dimethylanilines 2

Proposed mechanism for formation of N,N-dimethylanilines 2
In this context, Kundu’s group developed an efficient conversion of nitrobenzene derivatives 4 to the corresponding N-methylanilines 5, using a homogeneous ruthenium [NNN]-type pincer complex 3 as catalyst and NaOMe as base under open air (Scheme 4a) [21]. This selective N-monomethylation reaction was experimentally simple, performed by heating the substrates at 110 °C in MeOH, and was applicable to both electron-rich and electron-poor nitroarenes. Moreover, the optimized condition was also successfully applied for N-monomethylation of a nitroheteroaromatic compound and a series of α,β-unsaturated nitro compounds. Interestingly, when aliphatic nitro compounds 6 were used as substrates under the identical conditions, the corresponding N,N-dimethylamines 7 were obtained as the sole products (Scheme 4b). The authors ascribed the diselectivity of aliphatic nitro compounds to more nucleophilic intermediates.

Ru-catalyzed N-methylation of a nitrobenzenes 4; b aliphatic nitro compounds 6 developed by Kundu
In 2019, Xu’s research team reported another example of selective N-monomethylation of nitroarenes with methanol using an encapsulated iridium nanocatalyst in combination with tBuOK base at 170 °C [22]. The reaction showed good tolerance for nitroarenes with electron-donating functional groups. However, this catalytic system was unfruitful with nitro compounds bearing a strongly electron-withdrawing groups (e.g., CN), and lower yields were obtained with nitro compounds bearing halogen groups due to side-reactions. Concurrently, Beller and coworkers implemented selective N-monomethylation of a number of electron-rich and electron-poor nitroarenes 8 with methanol catalyzed by easily available Pd(OAc)2 in the presence of a specific phosphine ligand containing nitrogen atoms L1 (Scheme 5) [23]. A relatively wide range of sensitive functional groups such as hydroxyl, amino, and ether functionalities in different positions on phenyl rings of nitroarenes were well tolerated by this synthetic methodology, but some drawbacks still existed, such as high reaction temperature (80–110 °C) and narrow application (only aromatic nitro compounds). A similar N-monomethylation of nitroarenes with methanol was also reported by Zhang, Ibrahimu, and Yang, employing [RuCl2(p-cymene)2]2 as catalyst and an NNN pincer (amine–pyridine–imine, API) as ligand [24]. Notably, a diverse set of important functional groups (e.g., OMe, Cl, Br, CO2Me, OH, CN, and NHSO2Me) were compatible with the reaction conditions employed, and other aliphatic and benzylic alcohols such as ethanol, propan-2-ol, butan-1-ol, cyclohexanol, and (4-methoxyphenyl)methanol were also successfully subjected to this reaction.

Selective N-monomethylation of nitroarenes 8 with methanol catalyzed by Pd(OAc)2
In the same year, Siddiki and Shimizu and their coworkers rertpoed a general protocol for selective N-monomethylation of nitroarenes with methanol in presence of molecular hydrogen under basic conditions using carbon-supported Pt nanoparticles (Pt/C) as heterogeneous catalyst [25]. Choice of an appropriate base had a substantial effect on the facility of the reaction. Screening of a number of common bases such as Et3N, DBU, K2CO3, Na2CO3, Cs2CO3, NaOEt, NaOMe, NaOtBu, KOtBu, NaOH, and KOH revealed KOtBu as the most suitable for this conversion. Both aromatic and heteroaromatic nitro compounds 10 took part in the reaction easily and provided the expected N-methylamines 11 in high to excellent yields ranging from 76% to 92% (Scheme 6). However, the requirement for high temperature and hydrogen atmosphere may limit the range of application of this protocol in organic synthesis. Concurrently, in a closely related study, Natte’s research group synthesized 12 N-methylaniline derivatives in good to excellent yields (up to 95%) via reductive methylation of the corresponding nitroarenes with methanol as both carbon and hydrogen source using commercially available Pd/C in combination with KOtBu as a catalytic system under open air [26]. Interestingly, the use of the reaction conditions developed for N-monomethylation for an extended time (36 h) at slightly higher temperature (150 °C) led to N,N-dimethylated products in fair to excellent yields.

Pt/C-catalyzed N-monomethylation of nitroarenes 10 with methanol in presence of molecular hydrogen
Several metal-based catalytic systems have also been developed and have showed high catalytic performance for N-methylation of nitroarenes with methanol (Tables 1). These include Cu/Al2O3-DH [27], [IrBr(CO)2(ĸC-tBuImCH2PyCH2OMe)] (12) [28], N-heterocyclic carbene-iridium complex 13 [29], and bench-stable Mn PN3P pincer precatalyst 14 [30]. Notably, Cu/Al2O3-DH also displayed good catalytic activity in selective N,N-dimethylation of aliphatic nitro compounds. However, only one nitroalkane was subjected to the reaction using this catalyst. N-Methylation of aliphatic nitro compounds with methanol was also investigated by using Mn-catalyst 14. Unfortunately, in this case, starting materials were completely decomposed.
Despite the remarkable accomplishments over the last few years in this appealing research arena, there is still further need for new catalytic systems that are responsive to milder reaction conditions and shorter reaction times.
3 Methylations with Carbon Dioxide
The use of carbon dioxide (CO2) as an abundant, renewable, nonflammable, and environmentally friendly one-carbon (C1) feedstock has attracted increasing attention [31,32,33,34,35,36]. Methylation of N–H bond with CO2 in the presence of a reductant (e.g., H2, hydrosilanes, hydroboranes) is one of the well-established CO2-fixation reactions [37]. Although amines have been diversely utilized as N-nucleophiles in the title reaction, direct methylation of nitro compounds with CO2 in a single click remains largely underdeveloped. The first report of direct methylation of nitrobenzenes with CO2 can be found in a 2014 paper by Shi et al. [38], who showed that treatment of a small series of electron-rich nitrobenzenes 15 with catalytic amounts of CuAlOx (nano-CuOx particles deposited on the plane of AlOx) under CO2–H2 atmosphere at 170 °C afforded the corresponding N,N-dimethylaniline derivatives 16 in good yields via a domino reduction/methylation process (Scheme 7a). Although high conversion was obtained for all four nitrobenzenes tested, the requirement for elevated temperature may prevent the application of this protocol to some degree. A mechanism that explains this transformation starts with the hydrogenation of nitrobenzene 15 with H2 in the presence of catalyst to form aniline A, which after carbonylation with CO2/H2 generates N-phenylformamide B. Next, the quick hydrogenation of this intermediate with H2 leads to the N-methylaniline C. Finally, N-methylaniline C again reacts with CO2/H2 to afford N-methyl-N-phenylformamide D, which after hydrogenation results in the formation of the observed N,N-dimethylaniline products 16 (Scheme 7b). On the basis of these results, the same group reported the first example of one-pot methylation of aromatic nitriles with CO2 into N,N-dimethylbenzylamines utilizing their heterogeneous catalyst.

a Cu-catalyzed direct methylation of nitrobenzenes 15 with CO2 and H2; b proposed mechanistic pathways for formation of N,N-dimethylaniline derivatives 16
Shortly thereafter, the same authors reported selective N-monomethylation of the same set of nitrobenzene 15 employing Pd/ZrCuOx as catalyst, in which Pd was coloaded on the ZrCuOx support and the ratio of Pd to Cu and Zr was 1:1.7:2.1 [39]. The reactions were run under 1.0 and 2.5 MPa pressures of CO2 and H2, respectively, in octane and generally provided the respective N-methylanilines 17 in good yields within 30 h (Scheme 8). However, the reaction conditions were still very harsh (150 °C), and there is further need to development novel catalytic systems that allow N-methylation reactions of nitroarenes using CO2/H2 as methyl source under milder conditions.

Selective N-monomethylation of nitrobenzenes 15 with CO2 and H2 catalyzed by Pd/ZrCuOx
Drawing inspiration from those works, Han et al. developed an elegant electrochemical approach for N,N-dimethylation of nitroarenes 18 using CO2/H2O as methyl source under ambient conditions [40]. They identified Pd2.2/Co–N/carbon and 1-amino-methylphosphonic acid (AMPA) as the optimal catalyst and cocatalyst, respectively, and 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Bmim]Tf2N) as the ideal supporting electrolyte. The established electrochemical reaction was conducted in a typical H-type cell consisting of a cathode (working electrode), an anode (platinum gauze auxiliary electrode), and an Ag/Ag+ reference electrode, tolerating several important functional groups (e.g., F, Cl, Br, OMe, and SMe), and providing the expected N,N-dimethylanilines 19 in good to excellent yields, ranging from 71% to 90% (Scheme 9). It should be mentioned that the presence of the cocatalyst was crucial for the success of this conversation, since in the absence of AMPA only NH-free anilines were formed and could not be further converted to the methylated products even when extending the reaction time. Experiments indicated that AMPA acts as a Brønsted base and could activate the proton on the anilines to promote the formation of N,N-dimethylanilines. Detailed mechanistic studies showed that nitrosobenzenes, N-arylhydroxylamine, formamides, and formyl radical are the key intermediates of this CO2-fixation reaction (Scheme 10).

Electrochemical synthesis of N,N-dimethylanilines 19 from nitroarenes 18 using CO2/H2O as methyl source

Plausible mechanistic pathway for the reaction in Scheme 9
4 Methylations with (Para)Formaldehyde
After pioneering work by the groups of Rhee [41] and Isobe [42] on the synthesis of N-methylaniline and ethyl 2-(4-methoxy-3-(methylamino)phenyl)acetate, respectively, through selective N-monomethylation of the corresponding nitroaryls with formaldehyde (HCOH), the first general report of direct N-methylation of nitro compounds utilizing this cheap and readily available methylating reagent was published in 2013 by Rong et al. [43]. In that study, 11 N,N-dimethylaniline derivatives 21 were efficiently prepared by treatment of the respective nitrobenzenes 20 with 3 equiv. HCOH in the presence of a catalytic amount of skeletal copper in refluxing methanol under molecular hydrogen atmosphere (Scheme 11). The optimized protocol tolerated both electron-rich and electron-poor substrates bearing various sensitive functional groups such as chloro, methoxy, hydroxyl, ketone, and amide functionalities, and generally provided the target N,N-dimethylated products 21 in high to quantitative yields. Needless to say that the amino group (NH2) was also converted to NMe2 during the reaction. Scheme 12 outlines a plausible mechanism for this conversion. Firstly, nitrobenzene 20 undergoes reduction by H2 over S-Cu to produce the aniline A, which after condensation reaction with formaldehyde leads to the formation of imine intermediate B. Subsequently, hydrogenation of this intermediate with H2 forms the N-methylaniline C, which after reaction with another molecule of formaldehyde produces (N-methylanilino)methanol intermediate D. Finally, dehydration of alcohol moiety of this intermediate and subsequent hydrogenation of the generated carbon–carbon double bond by H2 afford the observed N,N-dimethylaniline 21 (Scheme 12, path a). In another possibility, addition of methanol to imine intermediate B gives N-(methoxymethyl)aniline E, which undergoes nucleophilic addition with formaldehyde to yield N-ethyl-N-(methoxymethyl)aniline F. Next, some addition and elimination reactions occur to form the desired N,N-dimethylated product 21 (Scheme 12, path b).

Rong’s synthesis of N,N-dimethylanilines 21

Mechanistic proposal for the generation of N,N-dimethylanilines 21
Four years later, an iron-catalyzed version of the title reductive amination was investigated by Beller et al. [44]. They designed and prepared the nonnoble iron oxide-based nanocatalyst (Fe2O3/NGr@C = pyrolyzed Fe-phenanthroline complex on carbon), which enables efficient N,N-dimethylation of a diverse range of nitroarenes 22 using paraformaldehyde as both methylating and reducing agent. The reactions were performed in the absence of external hydrogen in a binary solvent DMSO/H2O with ratio 1:1, tolerated both aromatic and heteroaromatic nitro compounds, and provided the respective N,N-dimethylated (hetero)aromatic amines 23 in good to excellent yields within 1–2 days (Scheme 13). However, an elevated temperature (130 °C) was necessary for successful transformation. It is noteworthy that this process can be easily scaled up to provide multigram quantities of the target N,N-dimethylated amines without sacrificing the yield or outcome of the methodology. Remarkably, the authors successfully applied their protocol to post-modification of several pharmaceuticals (e.g., nimodipine, cilnidipine, nicardipine, and nimesulide) and fluorescent molecules (e.g., fluorenone, rhoamine derivatives). They also nicely expanded their methodology to selective monomethylation of nitro compounds by controlling the concentration of paraformaldehyde and the reaction time. Of note, recycling experiments indicated that their catalyst could be recovered and reused up to five times without significant loss in activity. Concurrently, Jagadeesh’s research group identified a related cobalt material (Co3O4/NGr@C) as catalyst for selective N,N-dimethylation of a diverse range of functionalized nitroarenes with aqueous formaldehyde in presence of formic acid in t-butanol [45]. Importantly, this catalytic system was also active for aliphatic nitro compounds.

Selected examples of Fe-catalyzed N,N-dimethylation of nitroarenes 22 with paraformaldehyde
Subsequently, Shi and colleagues reported a TiO2-supported nano-Pd catalyst (Pd/TiO2) could enable kinetically controlled synthesis of N-monomethylamines 25 from nitrobenzenes 24 with paraformaldehyde in presence of molecular hydrogen under mild conditions (Scheme 14a) [46]. Replacing TiO2 with some other supports (e.g., Al2O3, SiO2, Fe2O3, CuO, and C) led to much lower yield and monoselectivity, or even no desired product at all. The superior catalytic activity of Pd/TiO2 over the other tested catalysts could be associated with the good H2 activation ability and high amine adsorbing capacity of this catalyst, as elucidated by NH3-temperature-programmed desorption (TPD) and H2-temperature-programmed reduction (TPR) analysis, while the high monoselectivity of this catalyst should be attributed to preferential adsorption of the primary amine over N-monomethylamine on the Pd/TiO2 surface, as elucidated by NH3/Me2NH-TPD. Very recently, Yang’s research team reported efficient reductive N,N-dimethylation of a library of functionalized and structurally diverse nitrobenzenes 26 to afford the desired N,N-dimethylanilines 27 in fair to almost quantitative yields (35–98%), employing paraformaldehyde as CH3 source and Cu nanoparticles (20 nm) supported on amorphous Al2O3 (Cu/Al2O3) as heterogeneous catalyst in the absence of external molecular hydrogen (Scheme 14b) [47]. Cal-Cu/Al2O3 and ip-Cu/Al2O3 were also found to be relatively effective catalysts, whereas Ni/Al2O3, Co/Al2O3, CuAl-LDH, NiAl-LDH, and CoAl-LDH proved to be completely ineffective. Regarding the influence of substituents, nitroarenes bearing electron-donating substituents on the phenyl ring delivered the products in better yields compared with those incorporating electron-withdrawing groups.

a Pd/TiO2-catalyzed synthesis of N-monomethylamines 25 from nitrobenzenes 24 and paraformaldehyde in presence of molecular hydrogen; b N,N-Dimethylation of nitrobenzenes 26 with paraformaldehyde catalyzed by Cu/Al2O3
5 Methylations with Formic Acid
Despite the remarkable advances in hydrogenation and formylation of nitroaromatics using formic acid over the last few years [48, 49], reported examples of direct N-methylation of nitrocompounds using formic acid as methylating agent are scarce. To the best of the authors’ knowledge, only one example of this kind of reaction has been reported in literature to date. In this preliminary work, eight N,N-dimethylanilines 29 were synthesized in excellent isolated yields by reaction of nitroarenes 28 with formic acid with 4 mol.% gold-based solid catalyst (Au/rutile) in refluxing toluene under hydrogen atmosphere (Scheme 15) [50]. Interestingly, the efficiency of this synthetic strategy was not dependent on the electronic and steric effects of the group directly bonded onto the phenyl ring. It is noteworthy that, when the reaction was carried out with o-dinitrobenzenes under inert atmosphere, the corresponding benzimidazoles were obtained in almost quantitative yields. Although those authors did not propose a reaction mechanism for the formation of N,N-dimethylanilines, based on literature [51], a N-formylamine intermediate A might be involved in this transformation. Of note, at the beginning of the 1990s, in a related investigation, Jenner and Taleb reported the synthesis of a small library of N,N-dimethylanilines by direct methylation of corresponding nitroarenes utilizing methyl formate (HCO2Me) as methyl group source in the presence of a combination of Ru3(CO)12 and Bu4PBr as a catalytic system under solvent-free conditions at 220 °C [52].

Synthesis of N,N-dimethylanilines 29 by reaction of nitroarenes 28 with formic acid
6 Methylations with Dimethyl Sulfoxide
Dimethyl sulfoxide (DMSO) is an inexpensive and low-toxicity organosulfur compound that is widely used as a polar aprotic solvent, radical scavenger, oxidant, and source of –Me, –CHO, –CN, –SMe, –SO2Me, and –O [53]. Despite the variety of protocols for methylation of amines using DMSO as methyl group source, reported examples of direct methylation of nitro compounds using this reagent are very scarce. In 2014, Wang et al. communicated the first and only example of direct N-methylation of nitro compounds with DMSO as methyl group source, in presence of formic acid as hydrogen source [54]. They showed that heating of nitroarenes 30 in DMSO in the presence of a catalytic amount of FeCl2·7H2O and over-stoichiometric amounts (20-fold excess) of formic acid and trimethylamine afforded the N,N-dimethylanilines 31 in good to high yields within 12 h (Scheme 16). The reaction proved to be efficient on various nitrobenzenes bearing electron-withdrawing groups (e.g., Cl, Br, and I) and electron-donating groups (e.g., OMe and SMe), affording exclusively dimethylated products. Of note, primary and secondary amines as well as nitrogen-containing heterocycles could also be easily N-methylated under the identical conditions. This methodology was also successfully applied in the high-yielding synthesis of galipinine, an alkaloid used against fever, from its corresponding tetrahydroquinoline precursor. The results of deuterium labeling experiments demonstrated that, in each CH3 group of dimethylated amines, two protons come from DMSO and one from HCO2H. Regarding the plausible mechanistic course for this transformation (Scheme 17), the authors speculated that the reaction starts with the generation of acylated intermediate A through the reaction of DMSO with formic acid anhydrides that, after Pummerer rearrangement, affords key intermediate C. In another possibility, protonation of DMSO by HCO2H leads to the formation of intermediate B, which after dehydration under high temperature provides intermediate C. Subsequently, intermediate C intercepts amine D (generated in situ by reduction of nitro compound 30 under the reaction condition) to form intermediate E, which after elimination of methanethiol furnishes imine F. Finally, reduction of imine F by formic acid yields the methylated amine product G.

Fe-catalyzed methylation of nitroarenes 30 with DMSO

Plausible mechanism for the reaction in Scheme 16
7 Miscellaneous Reactions
In 2020, the innovative research group of Radosevich established a robust P(III)/P(V)-catalyzed C–NMe bond formation reaction through reductive coupling of inexpensive and easy-to-handle nitromethane (MeNO2) as methylamine surrogate with arylboronic acid derivatives 32, affording N-methylanilines 33 by using a small ring organophosphorus-based catalyst (1,2,2,3,4,4-hexamethylphosphetane P-oxide, 34) and a mild terminal reductant hydrosilane (PhSiH3) in nitrogen atmosphere [55, 56]. The broad synthetic scope of this process was established by using a library of various (hetero)aromatic boronic acids and esters bearing both electron-donating and electron-withdrawing functional groups (Scheme 18). This synthetic strategy was also successfully applied for fabrication of isotopically labeled N-methylanilines from various stable isotopologues of nitromethane (i.e., CD3NO2, CH315NO2 and 13CH3NO2), indicating that this compound is a versatile precursor for direct installation of the methylamino group. The authors speculated that this C–N bond-forming reaction proceeds via formation of reactive phosphorane B through (3 + 1) cheletropic addition of phosphetane A and nitromethane, followed by its simultaneous fragmentation to give nitrosomethane (MeNO) and phosphetane P-oxide 34, which after reduction with hydrosilane regenerate the catalyst. Next, (2 + 1) addition of phosphine catalyst and MeNO forms oxazaphosphirane intermediate C, which undergoes reaction with boronic acid 32 to furnish betaine intermediate D. Finally, 1,2-metallate rearrangement of D delivers the desired N-methylaniline 33 and regenerates the catalyst (Scheme 19).

P(III)/P(V)-catalyzed reductive coupling of NO2 with (hetero)aromatic boronic acid derivatives 32

Proposed mechanistic pathway for formation of N-methylanilines 2
8 Conclusion
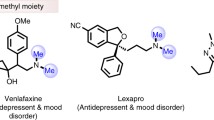
N-Methylamines belong to a highly important class of amines that are present in many natural and synthetic drugs (e.g., calcimycin, oxycontin, venlafaxine, and Lexapro), dyes (e.g., toluylene red, methyl red, methyl yellow, and methyl violet), and pesticides (e.g., bromethalin). There is therefore special scientific interest in the development of straightforward and efficient protocols for their preparation. Recently, direct reductive N-methylation of nitro compounds with various methylating agents (e.g., MeOH, DMSO, HCHO, HCOOH, and CO2/H2) has emerged as a concise and convenient strategy to access N-methyl- and N,N-dimethylamines that, beside high step economy, offers main advantages such as inexpensive and easily available starting materials, simplicity, and the avoidance of prepreparation of NH-free amines. Interestingly, most of the procedures covered in this review exhibit high selectivity for challenging N-monomethylation reactions. However, this new page of N-methylated amine synthesis generally required high temperature. Thus, the development of new catalytic systems that allow this transformation under milder conditions would be desirable. Moreover, the substrate scope is mainly limited to aromatic nitro compounds. Therefore, without question, there is still further need to study the scope and limitations of this synthetic procedure (Fig. 1).

Synthesis of N-methylamines through direct reductive N-methylation of nitro compounds
References
Martinez GR, Grieco PA, Williams E, Kanai K, Srinivasan C (1982) J Am Chem Soc 104:1436–1438
Pergolizzi JV Jr, Taylor R Jr, LeQuang JA, Raffa RB (2018) J Pain Res 11:301–311
Holliday SM, Benfield P (1995) Drugs 49:280–294
Masilamani S, Ruppelt SC (2003) Am Fam Physician 68:2235–2236
Zollinger H (2003) Color chemistry: syntheses, properties, and applications of organic dyes and pigments. Wiley
Peterson ME (2013) Top Companion Anim Med 28:21–23
Nakao T, Banba S (2016) Bioorg Med Chem 24:372–377
Li H, He Z, Guo X, Li W, Zhao X, Li Z (2009) Org Lett 11:4176–4179
Wang X, Wang Y, Yuan Y, Xing C-H (2014) Tetrahedron 70:2195–2202
Du Y-D, Tse C-W, Xu Z-J, Liu Y, Che C-M (2014) ChemComm 50:12669–12672
Das P, Begam HM, Bhunia SK, Jana R (2019) Adv Synth Catal 361:4048–4054
Cross J, Singer EJ (2019) Cationic surfactants: analytical and biological evaluation. CRC Press
Chen Y (2019) Chem Eur J 25:3405–3439
Yan G, Borah AJ, Wang L, Yang M (2015) Adv Synth Catal 357:1333–1350
Wienhöfer G, Sorribes I, Boddien A, Westerhaus F, Junge K, Junge H, Llusar R, Beller M (2011) J Am Chem Soc 133:12875–12879
Sharma S, Kumar M, Kumar V, Kumar N (2014) J Org Chem 79:9433–9439
Orlandi M, Tosi F, Bonsignore M, Benaglia M (2015) Org Lett 17:3941–3943
Hosoya H, Misal Castro LC, Sultan I, Nakajima Y, Ohmura T, Sato K, Tsurugi H, Suginome M, Mashima K (2019) Org Lett 21:9812–9817
Xu L, Li X, Zhu Y, Xiang Y (2009) New J Chem 33:2051–2054
Zhang L, Zhang Y, Deng Y, Shi F (2015) RSC Adv 5:14514–14521
Paul B, Shee S, Chakrabarti K, Kundu S (2017) Chemsuschem 10:2370–2374
Fu A, Liu Q, Jiang M, Xu G (2019) Asian J Org Chem 8:487–491
Wang L, Neumann H, Beller M (2019) Angew Chem Int Ed 131:5471–5475
Zhang S, Ibrahim JJ, Yang Y (2019) Org Chem Front 6:2726–2731
Jamil MA, Touchy AS, Rashed MN, Ting KW, Siddiki SH, Toyao T, Maeno Z, Shimizu K-i (2019) J Catal 371:47–56
Goyal V, Gahtori J, Narani A, Gupta P, Bordoloi A, Natte K (2019) J Org Chem 84:15389–15398
Zhang H, Wang J, Liu M, Liu D (2020) Appl Surf Sci 20:146708
González-Lainez M, Jiménez MV, Passarelli V, Pérez-Torrente JJ (2020) Catal. Sci Technol 10:3458–3467
Wang J, Wu J, Chen Z-N, Wen D, Chen J, Zheng Q, Xu X, Tu T (2020) J Catal 389:337–344
Reed-Berendt BG, Mast N, Morrill LC (2020) Eur J Org Chem 20:1136–1140
Sarvestani MRJ, Mert N, Vessally E (2020) J Chem Lett 1:93–102. https://doi.org/10.22034/jchemlett.2020.120304
Li M, Abdolmohammadi S, Hoseininezhad-Namin MS, Behmagham F, Vessally E (2020) J CO Util 38:220–231
Feng L, Li X, Liu B, Vessally E (2020) J CO2 Util 40:101220
Xu W, Ebadi AG, Toughani M, Vessally E (2020) J CO2 Util 20:101358
Wang L, Que S, Ding Z, Vessally E (2020) RSC Adv 10:9103–9115
Daghagheleh M, Vali M, Rahmani Z, Sarhandi S, Vessally E (2018) Chem Rev Lett 1:23–30. https://doi.org/10.22034/crl.2018.85117
Li Y, Cui X, Dong K, Junge K, Beller M (2017) ACS Catal 7:1077–1086
Cui X, Dai X, Zhang Y, Deng Y, Shi F (2014) Chem Sci 5:649–655
Cui X, Zhang Y, Deng Y, Shi F (2014) ChemComm 50:13521–13524
Sun X, Zhu Q, Hu J, Kang X, Ma J, Liu H, Han B (2017) Chem Sci 8:5669–5674
Byun E, Hong B, De Castro KA, Lim M, Rhee H (2007) J Org Chem 72:9815–9817
Sydnes MO, Isobe M (2008) Tetrahedron Lett 49:1199–1202
Rong Z, Zhang W, Zhang P, Sun Z, Du W, Wang Y (2013) Catal Commun 41:115–118
Natte K, Neumann H, Jagadeesh RV, Beller M (2017) Nat Commun 8:1–9
Senthamarai T, Murugesan K, Natte K, Kalevaru NV, Neumann H, Kamer PC, Jagadeesh RV (2018) ChemCatChem 10:1235–1240
Wang H, Yuan H, Yang B, Dai X, Xu S, Shi F (2018) ACS Catal 8:3943–3949
Dong X, Wang Z, Yuan Y, Yang Y (2019) J Catal 375:304–313
Orlandi M, Brenna D, Harms R, Jost S, Benaglia M (2016) Org Process Res Dev 22:430–445
Nasrollahzadeh M, Motahharifar N, Sajjadi M, Aghbolagh AM, Shokouhimehr M, Varma RS (2019) Green Chem 21:5144–5167
Yu L, Zhang Q, Li SS, Huang J, Liu YM, He HY, Cao Y (2015) Chemsuschem 8:3029–3035
Baig RBN, Verma S, Nadagouda MN, Verma RS (2016) Green Chem 18:1019–1022
Jenner G, Taleb AB (1992) J Mol Catal 77:247–255
Wu XF, Natte K (2016) Adv Synth Catal 358:336–352
Jiang X, Wang C, Wei Y, Xue D, Liu Z, Xiao J (2014) Chem Eur J 20:58–63
Li G, Qin Z, Radosevich AT (2020) J Am Chem Soc 142:16205–16210
Lipshultz JM, Li G, Radosevich AT (2021) J Am Chem Soc 143:1699–1721
Acknowledgements
This research was funded by the Shaanxi Science and Technology project (Grant No. 2021JQ-861).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Dr. Morteza Abdoli for his distinguished scientific efforts for our research group.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiang, Z., Mahmood, E.A., Harofteh, N.Z. et al. Methods for Direct Reductive N-Methylation of Nitro Compounds. Top Curr Chem (Z) 380, 27 (2022). https://doi.org/10.1007/s41061-022-00382-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-022-00382-w