
Abstract
Since the pioneering independent reports of Akiyama and Terada, the use of chiral phosphoric acids (CPAs) and derivatives as a versatile tool for asymmetric synthesis with good reactivity, regioselectivity, diastereoselectivity and enantioselectivity has emerged, forming an important part of the implementation of asymmetric counteranion-directed catalysis reported to date. In these achievements, the combination of metals with CPAs has enabled various catalytic modes beyond the scope of typical acid catalysis, such as relay catalysis, ion-pairing catalysis, and binary acid catalysis. The first-row transition metals (Sc–Zn) are considered to be sustainable transition metals and have received a great deal of attention. These naturally abundant metals display excellent Lewis acidity and function as powerful redox catalysts in synthesis involving both one and two-electron transfers. Hence, in this chapter, we summarize recent advances in the development of asymmetric reactions using a combination of first-row transition metals and CPAs. Furthermore, we provide a detailed discussion of the mechanisms involved in order to understand the interaction of the metal/phosphate and the origins of the asymmetric control of the transformations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chiral Brønsted acids, in particular 1,1′-bi-2-naphthol (BINOL)-derived phosphoric acids, have emerged as an increasingly prominent tool for asymmetric synthesis [1, 2]. The chiral phosphoric acids (CPAs, Fig. 1) containing acid/base dual function simultaneously, have been widely recognized as effective organocatalysts [3,4,5], and significant progress has been made in their utilization since the seminal reports by the groups of Akiyama [6] and Terada [7] individually in 2004 [8,9,10,11]. Besides BINOLs, chiral diols bearing C2-symmetry, e.g., H8-BINOL, SPINOL, VAPOL, VANOL, and TADDOL, have been used as variants of CPAs [5]. The majority of catalyst modifications for CPAs aim towards tuning the substituents at the 3,3′-positions of binaphthyl skeletons to achieve high selectivity. A new family of planar CPAs has also been reported recently, including ferrocene-bridged paracyclophane [12,13,14,15] and 1,8-biphenylene-tethered paracyclophane [16] frameworks.

Chiral phosphoric acid (CPA) analogs
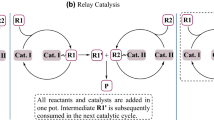
In view of their application as chirality-inducing agents, CPAs conventionally provide hydrogen-bonding interactions to form a contact ion pair with electrophilic components, through their relatively strong, yet appropriate, acidity [17, 18]. Further, the combination of metals and CPAs has exhibited multiple and peculiar reactivity beyond the single acid effect for asymmetric reactions, and many review articles covering this area have been published [19,20,21,22,23,24,25,26,27,28,29,30,31,32]. Up to now, various transition-metals such as Pd [33,34,35], Ag [4, 36, 37], Rh [38,39,40], Ir [41,42,43], Au [44,45,46,47], Ru [48,49,50,51], Fe [52], Cu [53, 54], and main metals e.g., Mg [55,56,57], Ca [58,59,60], and In [61, 62], have been employed in such dual catalytic systems. According to literature reports and catalytic principles, these systems are generally divided into four catalytic modes: relay catalysis (or cascade catalysis, sequential catalysis, Fig. 2a), counteranion-directed catalysis (CDC) (Fig. 2b) [27, 29, 47], chiral phosphate catalysis (Fig. 2c), and binary-acid catalysis (Fig. 2d) [19,20,21,22,23,24, 26, 32]. The resulting ion-pairing between the chiral anion (i.e., phosphate anion) and cationic metal complex (Fig. 2b), or metal cation (Fig. 2c) allows high efficiency and stereocontrol of the reactions [29]. In the binary-acid catalysis [32], the free phosphoric acid serves as a dual neutral ligand and Brønsted acid catalyst, resulting in a single binary complex bearing a bi-/multi-activation site (e.g., proton and metal center). The counter anion or ligand in the metal cation also shows a dramatic effect on catalytic performance. Besides protons, a second metal species, such as lithium and calcium, etc., can also have a synergistic effect in catalysis.

Catalytic modes combining phosphoric acids and metals. a Relay catalysis, b anion-directed catalysis, c metal phosphate catalysis, d binary-acid catalysis
Among the wide range of transition metals, first-row transition metals (Sc–Zn) display unique advantages (Fig. 3), such as affordability, less toxicity, environment-friendliness, and abundance, ranging from 16 ppm (Sc) to 43,200 ppm (Fe) in the Earth’s continental crust [63,64,65,66,67,68,69,70]. Further, these metal complexes have proven to be powerful Lewis acid catalysts, as well as redox catalysts for reactions via either one- or two-electron transfers [66, 71]. For example, copper [72, 73] and nickel [74,75,76,77] have been well established as single-electron transfer (SET) catalysts to initiate radical reactions. Based on a statistical analysis of the literature, the combination of first-row transition metals (Mn, Fe, Cu, Zn, etc.) with CPAs has received continuous attention and great progress has been made in this field in recent years. In this current review, we summarize recent advances in catalytic asymmetric reactions promoted by the combination of first-row transition metals with CPAs (Fig. 3). According to the metal catalysis involved, the content is divided into six sections, consisting of Mn, Fe, Cu, Zn, Sc and miscellaneous metals with CPAs.

Combining first-row transition-metals and CPAs for asymmetric catalysis
2 Combination of Mn with CPAs
In 2010, List and co-workers developed a novel ion-pairing catalyst for the epoxidation of olefins 1 with PhIO 3 as a terminal oxidant [78]. As shown in Scheme 1, the ion-pairing catalyst contains an achiral Mn(III)–salen cation complex 4 and a chiral phosphate counteranion. Under optimized oxidative conditions, both acyclic and cyclic olefins react rapidly, furnishing the expected optically active oxiranes 2 in excellent yields and enantioselectivities (up to 96% ee). Remarkably, even styrenes bearing ether, nitro, ester, and cyano group were well applicable. This variant of Jacobsen–Katsuki epoxidation of alkenes [79] provides an efficient implementation of the concept of asymmetric counteranion-directed catalysis (ACDC) [29]. Mechanistically, the phosphate anion acts as a stereocontroller via communication with cationic intermediate, significantly stabilizing enantiomorphic conformation of the cationic catalyst [e.g., MnIII(salen) and the oxidation state O = MnV(salen)] [80].

Enantioselective epoxidation of olefins with Mn–salen phosphate complexes
Recently, Schneider and co-workers reported an asymmetric protocol for 4H-chromenes 7 synthesis via a relay manganese(III)/Brønsted acid catalysis (Scheme 2) [81]. The precatalyst Mn(dbm)3 (Hdbm = dibenzoylmethane) provided a superior catalytic system for the conversion of 2-alkyl-substituted phenols 5 to ortho-quinone methide (o-QM, 8) intermediates under an atmosphere of pure oxygen, followed by chiral BINOL phosphoric acid-promoted Michael addition with β-dicarbonyl compounds. The resulting chiral manganese monophosphate complex was identified as an effective catalyst in the addition process. Finally, products 7 were obtained via para-toluenesulfonic acid (TsOH)-promoted cyclodehydration sequence. The method was limited to the electron-rich phenols and the acyclic β-dicarbonyl compounds (including β-ketoesters and acetylacetone), and rigid β-dicarbonyls [82] were inapplicable because they could not act as a bidentate ligands.

Asymmetric addition of β-dicarbonyls to ortho-quinone methides (o-QMs) via relay Mn(III) phosphate catalysis
Metal–organic frameworks (MOFs) have attracted increasing interest in recent years as a new family of porous crystalline hybrid materials as heterogeneous catalysts. In 2017, Liu and Cui [83] demonstrated that the chemical stability, catalytic activity, and enantioselectivity of chiral MOFs can be tuned simultaneously by changing the steric and electronic effect of the ligand. Three porous chiral MOFs with the framework formula were prepared from different chiral CPA. As shown in Scheme 3, under both batch and flow reaction conditions, the CF3-containing MOF 12 from (R)-L3 displayed excellent reactivity in the enantioselective alkylations of indoles and pyrroles 9 with electron-poor alkenes 10. In contrast, the corresponding homogeneous catalysts gave targets 11 with low enantioselectivities.

Enantioselectivity of metal–organic frameworks (MOFs) for alkylation of indoles and pyrroles
3 Combination of Fe with CPAs
3.1 Enantioselective Oxidations
Inspired by the study of ACDC chemistry [29] and the Mn(III)–salen system, in 2012, List and co-workers [84] reported an asymmetric oxidation of sulfides 13 with the combination of achiral Fe-salen cation 15 (Metallosalen) and chiral phosphate counteranion as an efficient oxygen-transfer catalyst (Scheme 4). The strategy utilized PhIO 3 as an oxidant and chiral iron-salen from (S)-L1 as a catalyst. Thioethers 13, especially the electron-poor and sterically bulky ones, underwent the oxidation reaction smoothly, resulting in chiral sulfoxides 14 with wide substrate scope, good yields and excellent enantioselectivities (up to 96% ee). This protocol disclosed the first application of asymmetric counteranion-directed catalysis to iron catalysis [52].

Enantioselective iron-salen-catalyzed sulfoxidation
In 2016, Pappo, Toste and co-workers [85] designed a novel chiral iron(III)-BINOL phosphate complex Fe[(R)-L1]3 as the catalyst for the enantioselective oxidative homo- and cross-couplings of 2-naphthols 16 and 16′, allowing expedited access to 1,1′-bi-2-naphthols (R)-17 with moderate-to-excellent yields and enantioselectivities (54–92% ee) (Scheme 5). The approach provided the first method for the synthesis of C1- and C2-symmetric BINOLs 17, in which the 3- and 3′-positions are ready for further chemical transformations. Assisted by di-t-butyl peroxide (DTBP) 18 as an oxidant, a redox mechanism was proposed involving SET iron species, and a key radical-anion coupling step between the electrophilic naphthoxyl radical (19) and a second nucleophilic 2-naphthol(ate) partner (16′) to form species 20 (Scheme 5). Further oxidation and deprotonation provided product 17 efficiently with the aid of tBuO radicals.

Enantioselective iron-catalyzed oxidative coupling of 1,1′-bi-2-naphthols
In 2017, the Pappo group further used the resulting chiral iron phosphate generated from (R)-L4 for asymmetric cross-dehydrogenative coupling reactions (Scheme 6) [86]. Current oxidation between 2-naphthols 16 and (–)-menthol-derived β-ketoesters 21 provided polycyclic hemiacetals 22 with good yields and diastereoselectivities (64–80% de). It is worth noting that the reaction temperature of 50 °C was crucial for inducing the combination of β-ketoesters 21. Otherwise, a competitive oxidative radical-anion coupling with a second nucleophilic 2-naphthol(ate) partner 16 takes place to afford the homo-coupling product 17. The chiral iron monophosphate Fe[(R)-L4]3 acts as the active redox catalyst to control stereoselectivity. As shown in Scheme 6, coupling takes place between two associated ligands via a radical-anion coupling mechanism. The formation of a persistent bounded naphthoxyl radical 23 followed by an intramolecular coupling afforded the expected polycyclic hemiacetal product 22.

Chiral iron phosphate-catalyzed counteranion-directed catalysis (CDC) reactions between 2-naphthols and β-ketoesters
3.2 Enantioselective Reductions
Recently, non-noble metal catalysts for the homogeneous hydrogenation of simple ketones and imines have been reported [87, 88]. The dual catalysis of achiral transition metal with chiral Brønsted acid offers an alternative route to promote asymmetric hydrogenation reactions [87, 88]. In 2011, Beller and co-workers [89] combined Knölker’s iron complex 26 containing a cyclopentadienone ligand with a CPA, such as (S)-TRIP L5, to start an asymmetric hydrogenation of imine substrates 24 with molecular hydrogen as hydrogen donor (Scheme 7a) [90]. Based on the chiral anion-directed catalysis strategy, the expected amines 25 were obtained with 60–94% yields and excellent enantioselectivities (67–96% ee).

Cooperative iron and chiral phosphoric acid (CPA)-catalyzed asymmetric hydrogenation of imines
A general asymmetric hydrogen mechanism was proposed in Scheme 7b [90]. Knölker’s iron complex 26 may coordinate with CPA (S)-L5 to form a complex that possesses an iron hydride and an acidic hydrogen available for transfer to the polar imine moiety. The resulting reactive intermediate 27 is trapped by H2 to regenerate the iron hydride 26/(S)-L5 complex and release 25, thus accomplishing the asymmetric hydrogenation cycle.
Taking into account the limitations in purification of the unstable ketimines 24, the authors further developed the direct asymmetric reductive amination of ketones 29 with anilines 28 under a similar chiral iron phosphate catalysis (Scheme 8) [91]. Various aromatic, heteroaromatic and aliphatic ketones were converted to the corresponding chiral amines 25 in good yields and good-to-excellent enantioselectivities (up to > 99% ee). Electron-rich/neutral anilines 28 have proved to be feasible, while the ortho-substituted aromatic ketones and ortho-substituted anilines were inapplicable.

Cooperative iron and CPA-catalyzed asymmetric reductive amination of ketones
In addition to ketones, Beller’s group also used commercially available alkynes 30 in reductive hydroamination in a relay cascade approach with primary amines 28 and molecular hydrogen (Scheme 9) [92]. This enantioselective reductive hydroamination was carried out by a key three-component catalytic system, comprising a gold(I) complex-catalyzed hydroamination of alkynes 30 for synthesizing imines, and sequential asymmetric hydrogenation to give 25 (via cooperative 26/(R)-L5 catalysis).

Enantioselective reductive hydroamination of alkynes with primary amines
Afterwards, Beller’s group extended the dual iron-phosphoric acid catalytic system in an enantioselective hydrogenation of substituted quinoxalines and benzoxazines 32, producing chiral tetrahydroquinoxalines and dihydro-2H-1,4-benzoxazines 33 in excellent yields and good-to-excellent enantioselectivities (up to 94% ee) (Scheme 10a) [93]. Quinoxalines 32 with aromatic, heteroaromatic, cyclic and aliphatic substituents at the heteroaromatic core were all applicable under standard conditions. Moreover, treating 1,2-phenylenediamine 34 and phenylglyoxal 35 with the ligand (R)-L6 and several iron(II)-based hydrogenation catalysts also led to the chiral tetrahydroquinoxaline efficiently and selectively; the best catalyst 26 could give the desired tetrahydroquinoxaline 33a in 75% yield and 90% ee (Scheme 10b).

Enantioselective hydrogenation of quinoxalines and 2H-1,4-benzoxazines
According to density functional theory (DFT) calculations and experimental observations, Hopmann [94] proposed a concerted imine hydrogenation mechanism with synergistic effect of Knölker’s complex 26 and CPA catalyst (Scheme 11). For such cyclic imine benzoxazine substrates 32, the reaction occurred with an involvement of noncovalent interactions (including electrostatic and dispersion interactions, as shown in 36), that is, the hydroxycyclopentadienyl ligand and the iron complex do not change oxidation state during the catalytic cycle, rather than the redox mechanism. The H2 splitting assisted by phosphate promotion is the rate-limiting step. The subsequent stepwise hydrogenation of imine, where phosphoric acid acts as a proton donor, afforded the final product 33.

Hopmann’s mechanism of iron complex-catalyzed asymmetric hydrogenation
In 2015, Beller’s group reported an asymmetric hydrogenation of benzoxazinones 37 by a relay iron/chiral Brønsted acid catalysis (Scheme 12) [95]. Both chiral 3-aryl and, more challenging, 3-alkyl substituted dihydrobenzoxazinones 38 were obtained in good yields and uniformly high enantioselectivities (84–96% ee). Rather than the possible participation of chiral iron phosphate species, the authors proposed a relay catalytic mechanism involving Fe3(CO)12-catalyzed reduction of phenantridine 40 to dihydrophenantridine 41 in a molecular hydrogen atmosphere, and sequential asymmetric transfer hydrogenation of benzoxazinone 37. The latter step, as depicted in Scheme 12, using a CPA catalyst (S)-L7 provides a high level of enantioselectivity through a possible hydride transfer process. The achiral phosphine ligand tris(4-methoxyphenyl)phosphine (TMP) 39 effectively modulates the reactivity of Fe3(CO)12 to decrease unselective background hydrogenations.

Relay iron/chiral Brønsted acid-catalyzed hydrogenation of benzoxazinones 37
3.3 Enantioselective Additions
In 2009, Huang and co-workers reported an enantioselective Friedel–Crafts alkylation of indoles 43 with enones 42 by using iron(III) as Lewis acid and CPA as Brønsted acid to establish a binary catalyst (Scheme 13) [96]. Enones 42, especially those having an electron-withdrawing group at the para position of the phenyl ring delivered chiral indoles (R)-44 in good to excellent yields and enantioselectivities (up to 90 % yield and 91 % ee). In this catalytic system, the key catalytic species iron(III) phosphate salt (45 and 46) formed in situ was confirmed by electrospray ionization mass spectrometry (ESI–MS) studies, which seems to cause high activity and good enantioselectivity. The hydrogen-bonding interaction (45 and 46) between the basic site of CPA and indole 43 is important for the catalytic process.

Chiral iron(III) phosphate catalyzed Friedel–Crafts alkylation of indoles
Instead of proton transfer, an alternative protocol via selective β-proton elimination of the resulting carbocationic intermediate was developed by Luo and co-workers in 2014 [97]. The authors combined FeCl3 and sodium phosphate Na[(S)-L4] to promote a direct conjugate nucleophilic addition of alkenes 47 to α,β-enones 48 (Scheme 14) [97]. A remarkably selective β-proton elimination of carbocationic intermediate 50 was revealed, wherein the anionic phosphate ligand was vital to inhibit cationic olefin polymerization and nucleophilic interception. The use of phosphate (S)-L4 provides better results than the corresponding acid. Further, an initial catalytic asymmetric version with the 3,3′-substituted phosphate ligand was conducted and gave 35% ee [97].

FeCl3-catalyzed direct conjugate addition of alkenes to α,β-enones
In 2017, Tan and colleagues developed the first asymmetric Paal–Knorr reaction under a simple binary iron/CPA catalysis (Scheme 15) [98]. 1,4-Diones 51 and substituted anilines 28 underwent the reaction smoothly in the presence of 10 mol% of Fe(OTf)3 and 10 mol% of (S)-L9 at 0 °C. This Paal–Knorr reaction allowed rapid access to a wide range of axially chiral arylpyrroles (R)-52 in good yields and enantioselectivities (85–98% ee). When using CCl4/EtOH as co-solvent instead, (S)-52 was obtained.

Asymmetric iron-catalyzed Paal–Knorr reaction of 1,4-diones and anilines
4 Combination of Cu with CPAs
4.1 Cyclizations
Cascade cycloisomerization reactions between carbonyl, imine, and alkenyl group or alkynyl group have provided a versatile tool for synthesis of heterocyclic scaffolds. In 2011, Toste's group developed a tandem cycloisomerization reaction for synthesis of furan derivatives 54 by using a chiral anionic copper(II) phosphonate catalyst (Scheme 16) [54]. Heterocyclization of 2-(1-alkynyl)-2-alkene-1-ones 53 followed by nucleophilic attack by indoles 43 yields products 54 with a high level of enantioselectivity (72–93% ee).

Cu-catalyzed enantioselective cycloisomerization–indole addition reactions
Lalli and van de Weghe developed the first catalytic enantioselective Prins cyclization in 2014 (Scheme 17) [99]. A superior binary-acid catalysis derived from CuCl and chiral BINOL-derived bis-phosphoric acid (R)-L10 was used to activate the key oxonium ion intermediate 58 (through a hemiacetal pathway) and acted as a chiral counterion for enantioselective control [100, 101]. Treatment of aldehydes 55 and homoallylic alcohol 56 provides tetrahydropyrans 57 containing three contiguous stereogenic centers in high yields, good enantio- and excellent diastereoselectivities. The presence of a phenyl group as an internal nucleophile in homoallylic alcohol urges the tandem Prins/Friedel–Crafts process. The absolute configuration of 57 was confirmed as 4S,4aR,10bR by single crystal X-ray analysis.

First enantioselective Prins cyclization
In addition to the sequential addition with nucleophiles, Akiyama and co-workers reported a chiral copper(II) phosphate-catalyzed enantioselective cycloisomerization/hydrogenation of o-alkynyl(oxo)benzenes 59 with Hantzsch esters 60 as a hydrogen source (Scheme 18) [102]. The reaction is believed to proceed by sequential intramolecular cyclization and asymmetric transfer hydrogenation. This strategy furnishes an enantioselective synthesis of multisubstituted isochromenes 61 containing various substituents in high yields with good-to-excellent enantioselectivities.

Enantioselective synthesis of isochromene o-alkynylacetophenones
4.2 Conjugate Additions
By using copper catalysis, Kumagai and Shibasaki described a direct catalytic asymmetric conjugate addition of alkynes to α, β-unsaturated thioamides in 2010 (Scheme 19) [103, 104]. The combined use of a soft copper(I) Lewis acid and a hard Brønsted base [e.g., Li(OC6H4-p-OMe)] resulted in simultaneous activation of alkynes 30 and thioamides 62, giving the β-alkynylthioamide 63 with excellent enantioselectivity. The presence of (S)-L5 enhances the basicity of Li(OC6H4-p-OMe), enabling promotion of reaction rate and enantiomeric control, especially for the success of aliphatic terminal alkyne 30. While the (R)-L5 was ineffective for the conversions, bisphosphine oxide was applied for aryl alkynes. Crystallography analysis showed Cu/(R)-64/(S)-L5 association can be possible at the transition state. Based on the experiment results, the thioamide functional group was vital for the asymmetric alkynylation via coordination with copper alkynylide intermediates. Furthermore, the thioamide is readily converted into carboxylic acid, and thus the protocol has found useful synthetic application in a concise synthesis of a potent GPR40 agonist AMG 837 66 via the formation of chiral alkyne intermediate 65 [105].

Asymmetric conjugate addition of terminal alkynes to α, β-unsaturated thioamides
4.3 Radical-Involved Transformations
Recently, Shi and co-workers established a simple protocol for assembling vicinal diamines from conjugated dienes 67 with 1,3-di-tert-butyldiaziridinone 68 as nitrogen source [106]. The copper salt CuX-PPh3 (1:2) was applied to the diamination reaction, and provided the diaminated products 69 in excellent yields and terminal regioselectivities (Scheme 20) [107]. It was found that the chiral copper(I) diphenyl phosphate produced from mesitylcopper(I) and the corresponding phosphoric acid (R)-L4 achieve an asymmetric diamination process in moderate enantiomeric excesses (49–61% ee), where the catalyst provides an anionic counterion effect. As illustrated in Scheme 20, two distinct mechanistic pathways for Cu(I)-catalyzed diamination were proposed to account for the different regioselectivity at either the terminal or internal double bond affected by the reaction conditions. First, in the presence of copper(I) catalyst Cu[(R)-L4], the N–N bond of 68 is activated and forms a Cu(II) nitrogen radical 71′ or a four-membered Cu(III) species 71. The Cu(II) nitrogen radical 71′ further reacts with the conjugated diene 67 to form a new Cu(II) allyl radical species 72 or a Cu(III) complex 72′, which ultimately results in the terminal diaminated product. While under simple CuBr catalysis, the four-membered Cu(III) species 71 reacts with dienes 67, providing the internal diamination target 70 through π-allyl species 73 [108, 109].

Diamination of conjugated dienes with 1,3-di-tert-butyldiaziridinone
In contrast to the well-documented enantioselective nucleophilic and electrophilic transformations, asymmetric radical chemistry still remains a formidable challenge owing to the high reactivity of such free radical species [71, 72, 110,111,112]. To address this problem, Liu and co-workers developed a dual-catalytic method that combines copper and CPA to handle these intrinsically reactive species and further capture different nucleophiles to create new bonds [113]. In 2014, they reported the first example of highly enantioselective radical trifluoromethylation of alkenes which uses a dual copper(I)/CPA catalytic system for simultaneous installation of two new C–CF3 and C–O bonds (Scheme 21) [53]. This multicomponent protocol produces a wide range of valuable enantioenriched trifluoromethylated N,O-aminals 77 with good to excellent yields and with excellent regio-, chemo-, and enantioselectivities. The reaction is initiated by addition of the in situ generated trifluoromethyl radical to unactivated alkenes 74 to generate 78 under Cu(I)/CPA catalysis with Togni’s reagent 75b, followed by a 1,5-hydrgoen atom transfer to give a new C-centered radical 79 adjacent to nitrogen atoms. A further single-electron oxidation step produces the imine intermediate 80. The attack of an alcohol to imine through a two-point hydrogen bonding interaction in the presence of CPA via a zwitterionic transition state yields the final product. Functionalized primary and secondary alkyl and benzyl alcohols 76 were applicable for the reaction.

Enantioselective remote C–H bond functionalization of unactivated alkenes
Similar to the enantioselective attack of alcohols with N-(2-allylbenzyl)amides 74, the use of N-benzyl or aryl substituted indoles 9 instead of alcohols as a nucleophile efficiently leads to two new C–CF3 and C–C(sp2) bonds in a one-pot manner (Scheme 22) [114]. Under the synergistic catalytic system of CuSCN and (R)-L7, the reaction provides a simple and straightforward method to access various trifluoromethylated indole compounds 81 in moderate to good yields and enantioselectivities (up to 86% ee).

Enantioselective α-C–H functionalization of amides with indoles
In 2016, Liu and co-workers disclosed a novel asymmetric radical aminotrifluoromethylation of alkenes 82 for the first time, providing straightforward access to densely functionalized CF3-containing pyrrolidines 85 bearing an α-tertiary stereocenter with excellent enantioselectivity (Scheme 23) [115]. The key to success is not only the utilization of a Cu(I)/CPA dual-catalytic system but also the use of urea (82) bearing two acidic N–H as both the nucleophile and directing group. The in situ generated trifluoromethyl radical from 75b would attack the alkene to provide a benzylic radical intermediate 83. The benzylic radical and urea could be trapped by Cu(II) phosphate to generate a Cu(III) species 84 (path 1) followed by reductive elimination to forge the C–N bond formation. An alternative SET process between benzylic radical and Cu(II) phosphate to yield the carbocation intermediate 84′ and subsequent C–N bond formation is also possible (path 2). The stereoselectivity of this process was probably controlled by chiral phosphate (S)-L12 via both hydrogen-bonding interactions with N–H bond adjacent to aryl group and ion-pairing interaction in a concerted transition state.

Asymmetric radical aminotrifluoromethylation of alkenes
Besides the radical trifluoromethylation with Togni’s reagent, commercially available fluoroalkylsulfonyl chlorides 86, such as CF3SO2Cl, n-C4F9SO2Cl, CH3O2CCF2SO2Cl, etc. were also suitable as precursors for aminoperfluoroalkylation and aminodifluoromethylation of alkenes 82 (Scheme 24) [116]. The method provides a sustainable, widely applicable and remarkable enantioselective platform for efficiently obtaining four types of enantioenriched functionalized α-tertiary pyrrolidines 87 bearing different β-fluoroalkyl groups. It is worth noting that the use of silver carbonate can absorb in situ generated HCl and inhibit background and side hydroamination reactions.

Asymmetric radical aminoperfluoroalkylation and aminodifluoromethylation of alkenes
Based on the cooperative Cu(I)/CPA catalysis, Liu’s group further extended the strategy to diamination and azidoamination of unactivated alkenes by using O-acylhydroxylamines 88 and azidoiodinane 90 as N-radical precursors, respectively [117]. As shown in Scheme 25, these transformations enable a facile and selective route to enantioenriched α-tertiary pyrrolidines 89 bearing a β-alkylamine and azido moiety with good yields, giving two C–N bonds in one-pot manner. The results indicated that urea 82 bearing electron-withdrawing N-aryl group is key to the success of conversions. The current program provides a complementary and efficient method for the synthesis of various chiral vicinal diamines 89 and 91, such as β-primary, secondary, or tertiary amine-containing pyrrolidines, and bicyclic amines. Further, an asymmetric copper-catalyzed aminoarylation of urea-derived alkenes was also established, and provided the expected chiral pyrrolidines with good yields and enantioselectivities [118]. Aryldiazonium salts with methylsulfonyl and nitro group at phenyl moiety were used as the aryl radical precursor. Very recently, a variant of enantioselective radical aminosilylation with alkene 82 with (TMS)3SiH was developed by Liu’s group using Cu(I)/CPA cooperative catalysis [119].

Copper-catalyzed asymmetric radical diamination of alkenes
Liu’s group revealed that alcohols are also appropriate for asymmetric radical reactions. They treated alkenes 92 with a pendant intramolecular alcohol moiety with 75b under copper(I)/phosphoric acid and realized asymmetric oxytrifluoromethylation (Scheme 26) [120]. Conversion with cooperative CuBH4(PPh3)2/chiral VAPOL-based acid catalysis (R)-L14 provides various trifluoromethyl-substituted tetrahydrofurans 93 bearing an α-tertiary stereocenter with excellent enantioselectivity. Remarkably, the presence of achiral pyridine (e.g., N,N-diethylnicotinamide 95) was vital to the radical oxytrifluoromethylation to improve the enantiotopic selectivity, which was considered as a coordinative ligand on copper metal (94) to stabilize the high-valent copper species in the asymmetric control process.

Asymmetric radical oxytrifluoromethylation of alkenes with alcohols
Very recently, the authors completed the asymmetric intermolecular dicarbofunctionalization of 1,1-diarylalkenes 96 catalyzed by a dual Cu(I) and sterically bulky SPINOL phosphoric acid (S)-L15 with diverse carbon-centered radical precursors 97 and electron-rich heteroaromatics 9 (Scheme 27) [121]. This three-component radical reaction provides direct access to chiral triarylmethanes 98 bearing quaternary all-carbon stereocenters with high efficiency as well as excellent chemo- and enantioselectivities. Various sulfonyl chlorides 97 and Togni’s reagent were applied to C-centered radicals. DFT calculations elucidated that hydrogen-bonding and ion-pair interactions between CPA and substrates (N–H and O–H moieties, as shown in 99 and resonance 99′) creates a chiral environment for enantiodiscrimination. The incorporation of a hydroxy group as the directing group, and introduction of a sterically demanding CPA will favor the desired radical difunctionalization over the otherwise remarkable side reactions.

Asymmetric radical 1,2-dicarbofunctionalization of alkenes with heterocycles
The effect of chiral copper(I) phosphates generated from BINOL-derived phosphoric acids on Kharasch–Sosnovsky of acyclic alkenes 100 were evaluated in 2015 (Scheme 28) [122]. It is proposed that peroxybenzoate 101 converts Cu(I) to copper(II) benzoate and OtBu radical. The latter abstracts the allylic hydrogen atom to product a key allylic radical species. The combination of copper(II) benzoate and allylic radical species provides the allyl ester 102 in good regioselectivity but low enantioselectivity.

Cu-catalyzed asymmetric allylic oxidation of linear alkenes
5 Combination of Zn with CPA
Charette and colleagues [123,124,125] first introduced iodine methyl zinc phosphate 105 as a new available cyclopropanation reagent in 2005, providing a method for asymmetrical Simmons–Smith cyclopropanation of alkenes (Scheme 29). A strong background reaction without the phosphate catalyst always occurs, which prevents high enantiopurity. Initial attempts at the racemic reaction were conducted with diphenylphosphate 104 and diethylzinc 103 (Scheme 29a). The results showed that 105 was obtained in excellent yield, and was quite stable and crystallized as a dimeric structure possessing tetrahedral geometry with respect to the Zn atom (Scheme 29a). Afterwards, the iodomethylzinc phosphate 105 was extended for the cyclopropanation reaction with alkenes and the reaction furnished the expected products in good yields. Upon treatment of the reaction with chiral zinc phosphate derived from (R)-L16 with cinnamyl alcohol and homoallylic ether 106, the desired cyclopropanes 107 were obtained in very good yields and excellent enantioselectivities. Different additives were optimized to prevent the use of a stoichiometric amount of phosphoric acid (R)-L16 (1.2 equivalents). They finally identified that the addition of 0.5 equivalent of dimethyl ether (DME) and 0.9 equivalent of Zn(CH2I)2 could reduce the (R)-L16 to catalytic amount with a slight loss of enantioselectivity (up to 88% ee, Scheme 29c).

Cyclopropanation of alkenes with iodomethylzinc phosphates
Analogously, Charette’s group applied the chiral zinc TADDOL phosphate species to the cyclopropanation of allylic ethers and 1-phenyl-3,4-dihydronaphthylene [126]. Allylic ethers showed more reactivity than unfunctionalized olefins, resulting in high yields and selectivity (up to 75% ee). They also identified (n-BuO)2P(O)OZnCH2I derived from dibutyl phosphate as a very stable reagent and extended this to the cyclopropanation reactions [127]. The substrate scope was very broad, including allylic alcohols and their derivatives, styrenic substrates with electron-donating or electron-withdrawing substituents on the aromatic ring, and even unfunctionalized alkenes were applicable to afford the desired cyclopropanes in excellent yields (72–99%).
Recently, Orthaber and Faber [128] developed an asymmetric allylation of (hetero)aromatic aldehydes 108 with the in situ generated zinc(II)-allylbutyrolactone species from 119 (Scheme 30). Catalyzed by CPA (S)-L5, the Barbier-type allylation results in β-substituted α-methylenebutyrolactones 110 in good yields and enantioselectivities. NH4Cl is a key additive to activate Zn surface. Based on the experimental observations and DFT studies, the proton of CPA has an important influence on the success of allylation of activated aldehydes 108, and chiral induction occurs by forming key zinc complexes (111 and 112) bearing a six-membered ring in the transition state, as shown in Scheme 30. Unfortunately, aliphatic aldehydes provided the products only in low enantioselectivities. This allylation strategy has witnessed synthetic applications, such as in the concise synthesis of natural product (S)-(–)-hydroxymatairesinol 113 in 46% overall yield and 98% ee.

Asymmetric allylation reaction for synthesis of α-methylenebutyrolactones
Enantioselective control of tertiary α-carbon in the Nazarov cyclization of enones is challenging because the reaction involves an enantioselective proton transfer process. In 2017, Zhou and Zhu [129] developed a scalable, highly enantioselective Nazarov cyclization of indole enone substrates 114 bearing one coordinating site. The reaction was cooperatively catalyzed Lewis acid (ZnCl2) and a chiral Brønsted acid (R)-L17 (Scheme 31). The mechanism studies by DFT calculation and experimental results showed that both (R)-L17 and Zn(II) ionic activate the enone by coordination to promote cyclization and produce carbocation species 116. Then, ZnCl2 dissociation and CPA-catalyzed [1,3]-proton transfer through the eight-membered transition state 117 to selectively provide a Nazarov cyclized target 115. The proton transfer of the enol intermediate is the stereochemistry-determining step of the process.

Enantioselective zinc(II)-catalyzed Nazarov cyclization of indole enones
6 Combination of Sc with CPA
As early as in 1998, Inanaga and co-workers [130] first reported chiral rare-earth-metal phosphate catalysts in the enantioselective hetero-Diels–Alder reaction. In this platform, scandium organophosphate complexes, Sc[(R)-L4]3, Sc[(R)-L18]3 could act as homogeneous Lewis acid catalysts for the enantioselective hetero-Diels–Alder reactions [131, 132]. As shown in Scheme 32, treating carbonyl compounds 118 with Danishefsky’s diene 119 afforded the corresponding cycloadducts 120 with excellent ee values.

Scandium phosphate-catalyzed hetero-Diels–Alder reaction and conjugate addition
Inanaga et al. [133,134,135] further introduced chiral scandium complex Sc[(R)-L4]3 into the aziridination of chalcone 121 with O-substituted hydroxylamines 122 to provide optically active α-keto aziridines 124 with good enantioselectivity (Scheme 32). The reaction occurred by an enantioselective Michael addition to generate 123, followed by treatment with La(OiPr)3 or sodium tert-butoxide to give the target aziridine-ring (αR, βS)-124 in quantitative yields and without loss of ee values.
In 2006, Inanaga’s group identified the perfluorinated phosphate complex Sc[(R)-L19]3 as a chiral catalyst in the fluorination reaction of β-ketoesters 125 with electrophilic 1-fluoropyridinium triflate 126 (Scheme 33) [136]. The presence of fluorine atom in Sc[(R)-L19]3 is critical and causes a stronger Lewis acidity than its analogous Sc[(R)-L4]3. Under simple conditions, both cyclic and acyclic dicarbonyl substrates 125 were applicable for the asymmetric fluorination to provide 127 with high enantioselectivities (up to 88% ee).

Asymmetric fluorination of β-keto esters with 1-fluoropyridinium triflate
In 2013, Luo and co-workers [61] published a highly selective [4 + 2] cycloaddition of olefins 47 with β, γ-unsaturated α-ketoesters 10 (Scheme 34). Significant binary catalysis with Sc(BArF)3 [BArF = [3,5-(CF3)2C6H3]4B] and chiral calcium phosphate proved to be effective for the synthesis of endo-cycloaddition products 128 in excellent diastereoselectivity (Scheme 34). Based on the literature and experimental observations, a bidentate activation (129) between ketoester 10 and the cationic scandium metal center was proposed to promote the reaction. The TRIP group of ligands (S)-L5 in complex with Ca/Sc constitute a well-defined chiral environment that tunes the orbital-favored and less-space-demanding endo transition state (Scheme 34).

Asymmetric binary acid-catalyzed [4 + 2] cycloadditions with simple olefins
In 2014, Luo and co-workers developed a new intermolecular azo-transfer of aryl triazenes 132 with 1,3-dicarbonyl compounds under Sc(OTf)3 catalysis (Scheme 35) [137]. The combination of Sc(III)-activated triazenes 133 and enol ether species 132 completed a conjugate addition and a N–N bond cleavage providing aliphatic azo compounds 131 with good yields and wide substrate scopes. However, an initial catalytic asymmetric variant with (R)-L20 showed a disappointing ee value (24% ee).

Sc(OTf)3-catalyzed transfer diazenylation of dicarbonyl compounds with triazenes
7 Miscellaneous
Very recently, Kim and co-worker [138] developed a gram-scale asymmetric synthesis of 2,4-diaryl-1-benzopyrans 137 via a one-pot two-step transformation (Scheme 36). With the aid of (R)-L6, the β-keto acid 135 is efficiently subjected to a decarboxylative process to provide an enol intermediate 139 at room temperature, which then reacts with ortho-quinone methide 138 in situ generated from o-hydroxy benzylic alcohol 134 to produce the chiral adduct 136. Subsequently, Sc(OTf)3 promotes the cyclization and dehydration sequence to give synthetically useful chiral benzopyrans 137 in good yields and enantioselectivities (up to 94% ee).

Enantioselective decarboxylative alkylation of β-keto acids to form chiral benzopyrans
Compared to the enantioselective nucleophilic substitution reactions at the prochiral sp2-hybridized carbon atom, the involvement of sp3-hybridized carbon atom has been underdeveloped [139]. In 2016, Terada and co-workers developed a novel nucleophilic addition of thiols 141 to the racemic alkyne–dicobalt complex 140 derived from the corresponding secondary propargylic alcohols (Scheme 37) [140]. A key planar cobalt-based carbenium ion 143 was probably formed by phosphoric acid-catalyzed rearrangement and stabilized by the conjugate base phosphate anion (R)-L6 or L21. The racemization process is markedly quick over nucleophilic addition with thiols 141, enabling better results for addition conversion. Increasing temperature would advance the enantioconvergent of (S)-substrate 140 to intermediate 144 for forming (R)-configured product 142.

Nucleophilic substitution reaction of racemic alkyne–dicobalt complexes with thiols
Further, the authors developed an intramolecular enantioconvergent Nicholas reaction version to synthesize seven-membered cyclic ethers 147 in high yields with good enantioselectivities (Scheme 38) [141]. In the presence of CPAs, racemic diols 145 containing an (hetero)aryl substituent at the propargylic position reacted efficiently via key intermediates 148. The resulting enantioenriched cyclic ethers (R)-146 were further treated in one-pot manner with 1,3-dibromo-5,5-dimethylhydantoin 149 for de-complexation, to afford densely functionalized cyclic ethers 147 bearing an unsaturated diester moiety without loss of enantiomeric excess.

Enantioselective intramolecular Nicholas reaction of racemic diols
In 2009, Terada’s group reported a highly enantioselective synthesis of optically active anti β-amino alcohols 154 (Scheme 39) [142, 143]. Under optimal conditions, various racemic hemiaminal allyl ethers 150 bearing alkyl-, benzyl- and phenyl-substituents at the β-position were well applicable. A Ni(II) complex catalyzed the olefin isomerization of starting material 150 to racemic hemiaminal vinyl ethers 151, followed by a CPA catalyzed aza-Petasis–Ferrier reaction (e.g., (R)-L5) to afford the β-amino aldehydes 153 via key intermediate 152. The racemic vinyl ethers 151 underwent a sequential C–O bond cleavage and C–C bond formation to form β-amino aldehydes 153 with high level of anti-, diastereo- and enantioselectivities (up to 99% ee). Finally, a formyl reduction by NaBH4/methyl alcohol yielded the β-amino alcohol compounds 154.

Alkene isomerization/enantioselective aza-Petasis–Ferrier rearrangement reaction of hemiamial vinyl ethers
8 Conclusions and Prospects
In this chapter, we have provided an overview of the development and applications of organic phosphoric acids in combination with different first-row transition metals in asymmetric catalysis, providing a mechanistic discussion to better understand the interaction of the metal/phosphate and the factors for asymmetric control. The combination of metal cations/complexes with phosphates provides a variety of catalytic modes, including relay catalysis, counterion-pairing catalysis, and binary-acid catalysis, thus enabling a range of enantioselective transformations. This progress represents an essential part of the implement of asymmetric counteranion-directed catalysis reported so far. In addition, concerning the property of metals, e.g., copper, iron, and manganese can act as one or two-electron transfer catalysts to permit redox reactions. Significant efforts in enantioselective oxidations and hydrogenations, C–H bond functionalizations and radical-initiated difunctionalizations of olefins have been made recently. Consequently, the complexity and diversity of the catalytic systems between first-row transition metals and phosphate derivatives potentially provide a powerful, viable and accessible route for asymmetric synthesis, although it is still less developed and limited to some transition metals. Due to the versatility of metal Lewis acids and chiral Brønsted acids, the dual metal-CPA catalysis is bound to stimulate more evolutionary research in the future, especially in the field of radical-involved enantioselective chemistry, which remains largely underdeveloped.
References
Mlynarski J (ed) (2017) Chiral Lewis acids in organic synthesis. Wiley, Weinheim
Akiyama T (2007) Chem Rev 107:5744–5758
Parmar D, Sugiono E, Raja S, Rueping M (2014) Chem Rev 114:9047–9153
Parmar D, Sugiono E, Raja S, Rueping M (2017) Chem Rev 117:10608–10620
Maji R, Mallojjala SC, Wheeler SE (2018) Chem Soc Rev 47:1142–1158
Akiyama T, Itoh J, Yokota K, Fuchibe K (2004) Angew Chem Int Ed 43:1566–1568
Uraguchi D, Terada M (2004) J Am Chem Soc 126:5356–5357
Terada M (2008) Chem Commun 35:4097–4112
Terada M (2010) Synthesis 12:1929–1982
Masahiro T (2010) Bull Chem Soc Jpn 83:101–119
Schenker S, Zamfir A, Freund M, Tsogoeva SB (2011) Eur J Org Chem 12:2209–2222
Stemper J, Isaac K, Duret V, Retailleau P, Voituriez A, Betzer J-F, Marinetti A (2013) Chem Commun 49:6084–6086
Stemper J, Isaac K, Ghosh N, Lauwick H, Le Duc G, Retailleau P, Voituriez A, Betzer J-F, Marinetti A (2017) Adv Synth Catal 359:519–526
Stemper J, Isaac K, Pastor J, Frison G, Retailleau P, Voituriez A, Betzer J-F, Marinetti A (2013) Adv Synth Catal 355:3613–3624
Zhu J-C, Cui D-X, Li Y-D, Jiang R, Chen W-P, Wang P-A (2018) ChemCatChem 10:907–919
Isaac K, Stemper J, Servajean V, Retailleau P, Pastor J, Frison G, Kaupmees K, Leito I, Betzer J-F, Marinetti A (2014) J Org Chem 79:9639–9646
Yang C, Xue X-S, Jin J-L, Li X, Cheng J-P (2013) J Org Chem 78:7076–7085
Rueping M, Kuenkel A, Atodiresei I (2011) Chem Soc Rev 40:4539–4549
Chen D-F, Han Z-Y, Zhou X-L, Gong L-Z (2014) Acc Chem Res 47:2365–2377
Allen AE, MacMillan DWC (2012) Chem Sci 3:633–658
Du Z, Shao Z (2013) Chem Soc Rev 42:1337–1378
Shao Z, Zhang H (2009) Chem Soc Rev 38:2745–2755
Zhong C, Shi X (2010) Eur J Org Chem 16:2999–3025
Inamdar SM, Shinde VS, Patil NT (2015) Org Biomol Chem 13:8116–8162
Yang Z-P, Zhang W, You S-L (2014) J Org Chem 79:7785–7798
Rueping M, Koenigs RM, Atodiresei I (2010) Chem Eur J 16:9350–9365
Phipps RJ, Hamilton GL, Toste FD (2012) Nat Chem 4:603–614
Brak K, Jacobsen EN (2013) Angew Chem Int Ed 52:534–561
Mayer S, List B (2006) Angew Chem Int Ed 45:4193–4195
Parra A, Reboredo S, Martín Castro AM, Alemán J (2012) Org Biomol Chem 10:5001–5020
Mahlau M, List B (2013) Angew Chem Int Ed 52:518–533
Lv J, Luo S (2013) Chem Commun 49:847–858
Mukherjee S, List B (2007) J Am Chem Soc 129:11336–11337
Yan S-Y, Han Y-Q, Yao Q-J, Nie X-L, Liu L, Shi B-F (2018) Angew Chem Int Ed 57:9093–9097
Lin H-C, Wang P-S, Tao Z-L, Chen Y-G, Han Z-Y, Gong L-Z (2016) J Am Chem Soc 138:14354–14361
Rueping M, Antonchick AP, Brinkmann C (2007) Angew Chem Int Ed 46:6903–6906
Terada M, Li F, Toda Y (2014) Angew Chem Int Ed 53:235–239
Hu W, Xu X, Zhou J, Liu W-J, Huang H, Hu J, Yang L, Gong L-Z (2008) J Am Chem Soc 130:7782–7783
Jiang J, Xu H-D, Xi J-B, Ren B-Y, Lv F-P, Guo X, Jiang L-Q, Zhang Z-Y, Hu W-H (2011) J Am Chem Soc 133:8428–8431
Alamsetti SK, Spanka M, Schneider C (2016) Angew Chem Int Ed 55:2392–2396
Li C, Wang C, Villa-Marcos B, Xiao J (2008) J Am Chem Soc 130:14450–14451
Miura T, Nishida Y, Morimoto M, Murakami M (2013) J Am Chem Soc 135:11497–11500
Rong Z-Q, Zhang Y, Chua RHB, Pan H-J, Zhao Y (2015) J Am Chem Soc 137:4944–4947
Han Z-Y, Xiao H, Chen X-H, Gong L-Z (2009) J Am Chem Soc 131:9182–9183
Liu X-Y, Che C-M (2009) Org Lett 11:4204–4207
Muratore ME, Holloway CA, Pilling AW, Storer RI, Trevitt G, Dixon DJ (2009) J Am Chem Soc 131:10796–10797
Hamilton GL, Kang EJ, Mba M, Toste FD (2007) Science 317:496
Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ (2012) Science 336:324–327
Cai Q, Zhao Z-A, You S-L (2009) Angew Chem Int Ed 48:7428–7431
Sorimachi K, Terada M (2008) J Am Chem Soc 130:14452–14453
Komanduri V, Krische MJ (2006) J Am Chem Soc 128:16448–16449
Pellissier H (2019) Coord Chem Rev 386:1–31
Yu P, Lin J-S, Li L, Zheng S-C, Xiong Y-P, Zhao L-J, Tan B, Liu X-Y (2014) Angew Chem Int Ed 53:11890–11894
Rauniyar V, Wang ZJ, Burks HE, Toste FD (2011) J Am Chem Soc 133:8486–8489
Ren L, Lei T, Ye J-X, Gong L-Z (2012) Angew Chem Int Ed 51:771–774
Chen L, Zhang L, Lv J, Cheng J-P, Luo S (2012) Chem Eur J 18:8891–8895
Mori K, Isogai R, Kamei Y, Yamanaka M, Akiyama T (2018) J Am Chem Soc 140:6203–6207
Hatano M, Moriyama K, Maki T, Ishihara K (2010) Angew Chem Int Ed 49:3823–3826
Alix A, Lalli C, Retailleau P, Masson G (2012) J Am Chem Soc 134:10389–10392
Domżalska A, Ulikowski A, Furman B (2017) Alkaline earth metal based chiral lewis acids. In: Mlynarski J (ed) Chiral Lewis acids in organic synthesis. Wiley-VCH, Weinheim, pp 1–23 https://doi.org/10.1002/9783527802142.ch1
Lv J, Zhang L, Luo S, Cheng J-P (2013) Angew Chem Int Ed 52:9786–9790
Wang L, Lv J, Zhang L, Luo S (2017) Angew Chem Int Ed 56:10867–10871
Chirik P, Morris R (eds) (2015) Special Issue: Earth abundant metals in homogeneous catalysis. Acc Chem Res 48:(whole issue)
Egorova KS, Ananikov VP (2016) Angew Chem Int Ed 55:12150–12162
Alig L, Fritz M, Schneider S (2019) Chem Rev 119:2681–2751
Crossley SWM, Obradors C, Martinez RM, Shenvi RA (2016) Chem Rev 116:8912–9000
Pellissier H, Clavier H (2014) Chem Rev 114:2775–2823
Bauer I, Knölker H-J (2015) Chem Rev 115:3170–3387
Tzouras NV, Stamatopoulos IK, Papastavrou AT, Liori AA, Vougioukalakis GC (2017) Coord Chem Rev 343:25–138
Chirik P, Morris R (2015) Acc Chem Res 48:2495
Choi J, Fu GC (2017) Science 356:7230–7239
Wang F, Chen P, Liu G (2018) Acc Chem Res 51:2036–2046
Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC (2016) Science 351:681
Fischer C, Fu GC (2005) J Am Chem Soc 127:4594–4595
Son S, Fu GC (2008) J Am Chem Soc 130:2756–2757
Schley ND, Fu GC (2014) J Am Chem Soc 136:16588–16593
Tasker SZ, Standley EA, Jamison TF (2014) Nature 509:299
Liao S, List B (2010) Angew Chem Int Ed 49:628–631
McGarrigle EM, Gilheany DG (2005) Chem Rev 105:1563–1602
Merten C, Pollok CH, Liao S, List B (2015) Angew Chem Int Ed 54:8841–8845
Gebauer K, Reuß F, Spanka M, Schneider C (2017) Org Lett 19:4588–4591
El-Sepelgy O, Haseloff S, Alamsetti SK, Schneider C (2014) Angew Chem Int Ed 53:7923–7927
Chen X, Jiang H, Hou B, Gong W, Liu Y, Cui Y (2017) J Am Chem Soc 139:13476–13482
Liao S, List B (2012) Adv Synth Catal 354:2363–2367
Narute S, Parnes R, Toste FD, Pappo D (2016) J Am Chem Soc 138:16553–16560
Narute S, Pappo D (2017) Org Lett 19:2917–2920
Morris RH (2016) Chem Rev 116:8588–8654
Zhang Z, Butt NA, Zhou M, Liu D, Zhang W (2018) Chin J Chem 36:443–454
Quintard A, Rodriguez J (2014) Angew Chem Int Ed 53:4044–4055
Zhou S, Fleischer S, Junge K, Beller M (2011) Angew Chem Int Ed 50:5120–5124
Zhou S, Fleischer S, Jiao H, Junge K, Beller M (2014) Adv Synth Catal 356:3451–3455
Fleischer S, Werkmeister S, Zhou S, Junge K, Beller M (2012) Chem Eur J 18:9005–9010
Fleischer S, Zhou S, Werkmeister S, Junge K, Beller M (2013) Chem Eur J 19:4997–5003
Hopmann KH (2015) Chem Eur J 21:10020–10030
Lu L-Q, Li Y, Junge K, Beller M (2015) J Am Chem Soc 137:2763–2768
Yang L, Zhu Q, Guo S, Qian B, Xia C, Huang H (2010) Chem Eur J 16:1638–1645
Lv J, Zhong X, Luo S (2014) Chem Eur J 20:8293–8296
Zhang L, Zhang J, Ma J, Cheng D-J, Tan B (2017) J Am Chem Soc 139:1714–1717
Lalli C, van de Weghe P (2014) Chem Commun 50:7495–7498
Breugst M, Grée R, Houk KN (2013) J Org Chem 78:9892–9897
Tsui GC, Liu L, List B (2015) Angew Chem Int Ed 54:7703–7706
Saito K, Kajiwara Y, Akiyama T (2013) Angew Chem Int Ed 52:13284–13288
Yazaki R, Kumagai N, Shibasaki M (2010) J Am Chem Soc 132:10275–10277
Yazaki R, Kumagai N, Shibasaki M (2011) Chem Asian J 6:1778–1790
Yazaki R, Kumagai N, Shibasaki M (2011) Org Lett 13:952–955
Zhu Y, Cornwall RG, Du H, Zhao B, Shi Y (2014) Acc Chem Res 47:3665–3678
Zhao B, Du H, Shi Y (2009) J Org Chem 74:8392–8395
Zhao B, Peng X, Cui S, Shi Y (2010) J Am Chem Soc 132:11009–11011
Zhao B, Peng X, Zhu Y, Ramirez TA, Cornwall RG, Shi Y (2011) J Am Chem Soc 133:20890–20900
Plesniak MP, Huang H-M, Procter DJ (2017) Nat Rev Chem 1:77–92
Miyabe H, Kawashima A, Yoshioka E, Kohtani S (2017) Chem Eur J 23:6225–6236
Sibi MP, Manyem S, Zimmerman J (2003) Chem Rev 103:3263–3296
Tian Y, Chen S, Gu Q-S, Lin J-S, Liu X-Y (2018) Tetrahedron Lett 59:203–215
Li T, Yu P, Du Y-M, Lin J-S, Zhi Y, Liu X-Y (2017) J Fluor Chem 203:210–214
Lin J-S, Dong X-Y, Li T-T, Jiang N-C, Tan B, Liu X-Y (2016) J Am Chem Soc 138:9357–9360
Lin J-S, Wang F-L, Dong X-Y, He W-W, Yuan Y, Chen S, Liu X-Y (2017) Nat Commun 8:14841–14851
Wang F-L, Dong X-Y, Lin J-S, Zeng Y, Jiao G-Y, Gu Q-S, Guo X-Q, Ma C-L, Liu X-Y (2017) Chem 3:979–990
Li X-F, Lin J-S, Wang J, Li Z-L, Gu Q-S, Liu X-Y (2018) Acta Chim Sin 76:878–882
Zeng Y, Liu X-D, Guo X-Q, Gu Q-S, Li Z-L, Chang X-Y, Liu X-Y (2019) Sci China Chem. https://doi.org/10.1007/s11426-019-9528-2
Cheng Y-F, Dong X-Y, Gu Q-S, Yu Z-L, Liu X-Y (2017) Angew Chem Int Ed 56:8883–8886
Lin J-S, Li T-T, Liu J-R, Jiao G-Y, Gu Q-S, Cheng J-T, Guo Y-L, Hong X, Liu X-Y (2019) J Am Chem Soc 141:1074–1083
Incerti-Pradillos CA, Hudson D, Malkov AV (2015) Asymmetric Catal 2:45–50
Lacasse M-C, Poulard C, Charette AB (2005) J Am Chem Soc 127:12440–12441
Cornwall RG, Wong OA, Du H, Ramirez TA, Shi Y (2012) Org Biomol Chem 10:5498–5513
Zheng Y, Zhang J (2010) Adv Synth Catal 352:1810–1817
Voituriez A, Charette AB (2006) Adv Synth Catal 348:2363–2370
Voituriez A, Zimmer LE, Charette AB (2010) J Org Chem 75:1244–1250
Fuchs M, Schober M, Orthaber A, Faber K (2013) Adv Synth Catal 355:2499–2505
Wang G-P, Chen M-Q, Zhu S-F, Zhou Q-L (2017) Chem Sci 8:7197–7202
Furuno H, Hanamoto T, Sugimoto Y, Inanaga J (2000) Org Lett 2:49–52
Furuno H, Kambara T, Tanaka Y, Hanamoto T, Kagawa T, Inanaga J (2003) Tetrahedron Lett 44:6129–6132
Furuno H, Hayano T, Kambara T, Sugimoto Y, Hanamoto T, Tanaka Y, Jin YZ, Kagawa T, Inanaga J (2003) Tetrahedron 59:10509–10523
Jin XL, Sugihara H, Daikai K, Tateishi H, Jin YZ, Furuno H, Inanaga J (2002) Tetrahedron 58:8321–8329
Jin XL, Sugihara H, Daikai K, Tateishi H, Jin YZ, Furuno H, Inanaga J (2003) Tetrahedron 59:877
Sugihara H, Daikai K, Jin XL, Furuno H, Inanaga J (2002) Tetrahedron Lett 43:2735–2739
Suzuki S, Furuno H, Yokoyama Y, Inanaga J (2006) Tetrahedron Asymmetry 17:504–507
Liu C, Lv J, Luo S, Cheng J-P (2014) Org Lett 16:5458–5461
Jeong HJ, Kim DY (2018) Org Lett 20:2944–2947
Gualandi A, Rodeghiero G, Cozzi PG (2018) Asian J Org Chem 7:1957–1981
Terada M, Ota Y, Li F, Toda Y, Kondoh A (2016) J Am Chem Soc 138:11038–11043
Ota Y, Kondoh A, Terada M (2018) Angew Chem Int Ed 57:13917–13921
Terada M, Toda Y (2009) J Am Chem Soc 131:6354–6355
Terada M, Komuro T, Toda Y, Korenaga T (2014) J Am Chem Soc 136:7044–7057
Acknowledgements
Financial support from the National Natural Science Foundation of China (Nos 21722203, 21831002, and 21801116) and Shenzhen Nobel Prize Scientists Laboratory Project (C17783101) is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection “Asymmetric Organocatalysis Combined with Metal Catalysis”, edited by Bruce A. Arndtsen, Liu-Zhu Gong.
Rights and permissions
About this article

Cite this article
Fang, GC., Cheng, YF., Yu, ZL. et al. Recent Advances in First-Row Transition Metal/Chiral Phosphoric Acid Combined Catalysis. Top Curr Chem (Z) 377, 23 (2019). https://doi.org/10.1007/s41061-019-0249-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-019-0249-0