
Abstract
Six different Co(II) and Ni(II) complexes of a heterocyclic tridentate 2-acetyl-5-methylfuranthiosemicarbazone (AMFT) schiff’s base ligand were reported. Metal complexes were synthesized via conventional condensation and refluxing technique and characterized by different analytical techniques i.e., elemental analysis, molar conductance, magnetic moments and spectral i.e., IR, UV–Vis, EPR techniques. AMFT coordinated with the metal ion through three different heteroatoms i.e., oxygen, nitrogen and sulphur. Octahedral geometry was suggested by the experimental studies and also supported by theoretical studies. Chemical hardness and global softness value suggested high chemical reactivity of Co(II) complex. The charge transfer in the complexes were studied by MEP analysis. Antibacterial and antifungal activities of all the complexes were evaluated. The complexes showed better inhibition action than the parent ligand. Molecular docking was also performed to check the inhibition at atomic level and the result validated the experimental result. Ni(II) complexes showed better inhibition in comparison with Co(II) complexes.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Thiosemicarbazone is a well-known Schiff's base ligand that has been extensively investigated due to its exceptional capacity to produce potential coordination compounds that have been validated in a wide range of applications. (Khalid et al. 2021; Jain et al. 2019a). Condensation reaction of the thiosemicarbazide with different aldehyde or ketones results in the formation of thiosemicarbazones. The type of parent ketones/aldehydes used in coordination affect the bioactivity of synthesized thiosemicarbazones. Thiosemicarbazides can be monodentate, bidentate, tridentate, or multidentate depending on the parent ketone/aldehyde utilized for thiosemicarbazone production. Thiosemicarbazide has been a good precursor and is commonly used in the pharmacy and biomedical industries (Naveen et al. 2020; Jain et al. 2019b). Thiosemicarbazide containing ligands have been also reported to possess catalytical (Kostas et al. 2006), hydrogen evolution (Bakir et al. 2020), cholinesterase inhibition (Jawaria et al. 2020), analytical (Garg and Jain 1988), carbon releasing properties (Lawrence et al. 2020) and identification of different anions (Amuthakakala et al. 2020; Raicopol et al. 2020). The thiosemicarbazone transition metal complexes were studied for their coordination chemistry and structure functionality (Campbell 1975; Saswati et al. 2020; Casas et al. 2000). Metal complexes of thiosemicarbazones with diverse physical, chemical and structural features demonstrated antioxidant, (Dkhar et al. 2020) antimicrobial, (Almeida et al. 2020) antiviral, (Sevincli et al. 2020) antitumor/anticancer, (Alcaraz et al. 2020) antiproliferative, (Qi et al. 2018) antileulaemia, (Pahontu et al. 2015) antileshmanial, (Batista et al. 2019) anti-amoebic (Singh et al. 2006) and many more activities (Nehar et al. 2020; Amritha et al. 2020). This research paper focuses on the synthesis, characterization and computational studies of Co(II) and Ni(II) complexes along with antimicrobial potential assessment. The structures, charges and reactivity of the complexes were also confirmed by theoretical analysis. Molecular docking was performed to verify the experimental result as it is useful to provide more accurate interaction between the various proteins and synthesized complexes responsible for the infectious activity.
2 Materials and Methods
2.1 Starting Materials
All the metal salts i.e., Co(CH3COO)2.H2O (99%), Co(NO3)2.6H2O (98%), CoSO4.7H2O (99%), Ni(CH3COO)2.4H2O (98%), Ni(NO3)2.H2O (99%), NiSO4.6H2O (99%) and chemicals i.e., thiosemicarbazide used in the work were purchased from Sigma Aldrich and E. Merck, India and were of analytical grade. All the solutions were prepared via double distilled water. The solvents i.e., dimethyl sulfoxide (DMSO), dimethyl formamide (DMF) and absolute ethanol (EtOH) utilized in the research work, were of spectroscopic/standard grade. All the chemicals and solvents were used as such, without further purification.
2.2 Synthesis of Co(II) and Ni(II) Metal Complexes
The heterocyclic thiosemicarbazone ligand was synthesized using the reported method (Goel et al. 2018). Hot ethanolic solution of acetate and nitrate salts of Co(II) and Ni(II) i.e., Co(CH3COO)2.H2O (0.487 g), Co(NO3)2.6H2O (0.735 g), Ni(CH3COO)2.4H2O (0.62 g) and Ni(NO3)2.H2O (0.50 g), was added drop by drop to the hot ethanolic solution of the AMFT ligand (0.985 g) in 1:2 stoichiometry proportion. For the synthesis of sulphate complexes, aqueous solution of CoSO4.7H2O (0.70 g) and NiSO4.6H2O (0.655 g) were taken. With constant stirring, the mixture was refluxed for 9–10 h at 72–75 °C. The pH of the reaction mix was kept constant at around 6–7. The mixture was chilled for 4–6 h in an ice bath. Colored solids precipitated out after cooling, which were filtered, washed with EtOH and vacuum dried using anhydrous P2O5.
3 Physical Measurement
Carlo-Erbo 1106 analyzer was used to carry out the elemental analysis. The electrolytic and non-electrolytic natures were assessed using an ELICO Conductivity Bridge (model CM82T). At room temperature, the magnetic moment was measured using a Gouy magnetic susceptibility balance using CuSO4.5H2O as a calibrator. The IR spectrum of all the complexes were recorded on a Perkin-Elmer 577 (4000–400 cm−1) double beam FTIR spectrophotometer. KBr pellets were used for recording IR spectra. Electronic spectra of the complexes were recorded on a Shimadzu UV–Vis spectrophotometer in DMSO solution (HPLC grade) at room temperature. The EPR spectra of the complexes were recorded at room temperature on E4 EPR spectrometer and g-marker used was 2,2-diphenyl-1-picrylhydrazyl (DPPH). Computational analysis was carried out with the software Gaussian’09 (Frisch et al. 2009). Gauss View software version 5.0.9 was used to visualize (Frisch et al. 2001) the structures of all the synthesized complexes. The Well diffusion approach was used to test in vitro antifungal and antibacterial activities. The ligand and the complexes were evaluated for their antifungal activity for the inhibition of A. alternata, F. odum fungi. Chlorothalonil was employed as a standard drug. Whereas, Streptomycin was used as a standard drug for antibacterial inhibition against B. macerans and P. striata. For the ligand and synthesized metal complexes, all measurements were recorded in duplicate to assure accuracy (Gautam et al. 2018). The iGEMDOCK program was used to conduct the docking investigations (Vishvakarma et al. 2020). It was set in drug screening mode. The AMFT ligand, Co(II) complex, Ni(II) complex and protein were prepared from the Chimera in dock prep module. Blind docking was performed to check the inhibition of the bacteria and fungus.
4 Results and Discussions
Co(II) and Ni(II) complexes were synthesized by condensation and refluxing AMFT ligand and metal salts (Scheme 1). All the complexes were soluble in DMSO and DMF.

Synthesis of M(II) complexes where M = Co(II)/Ni(II)
As per analysis of analytical and spectral data, it was confirmed that complexes possessed [ML2]X2 formula where M = Co(II) and Ni(II), L = AMFT, X = CH3COO−, NO3−, and [ML2]X formula when X = SO42−. The acetate and nitrate group coordinated in monodentate manner whereas sulphate group coordinated in bidentate manner to the metal ion. Analytical and physical data of ligand and complexes are shown in Table 1. The complexation of metal ions with N, O and S donor atoms of the AMFT ligand was also confirmed by IR analysis. The molar conductance suggested the electrolytic nature of the complexes. UV–Visible spectral data and EPR data suggested octahedral geometry for all the complexes. The semiempirical (PM6) method was employed to carry out the computational analysis in the gaseous phase. The complete optimization of structures of Co(II) and Ni(II) complexes were performed. The bond lengths and bond angles of the ligand and complexes were calculated. Additionally, different other parameters like electron affinity (E), electronegativity (X), electrophilicity (ω), chemical hardness (η), chemical softness (S), global softness (σ), chemical potential (µ), ionization potential (I), EHOMO, ELUMO, energy gap (ΔE = EHOMO– ELUMO), ΔNmax were also reported. The transfer of the charge in the molecule was studied by MEP analysis. The antibacterial and antifungal activities were evaluated at four different concentrations against two different bacteria (B. macerans, P. striata) and two different fungi (A. alternata, F. odum). The complexes inhibited more effectively as compared to AMFT ligand. The findings were also backed up by a molecular docking study.
4.1 Spectroscopic Analysis
4.1.1 IR Spectra
A sharp band at 1619 cm–1 and a medium band at 800 cm–1 in the IR spectrum of ligand attributed to υ(C = N) and υ(C = S), respectively. The band due to υ(N − H) and υ(C − O − C) in the ligand were appeared at 3317 cm–1 and 1015 cm–1, respectively. The coordination of Co(II) and Ni(II) ions to the ligand resulted in the shifting of the three bands i.e., υ(C = N), υ(C = S), υ(C − O − C) to the lower frequency. The appearance of bands at lower frequency validated the association of N-atom of imine group, S-atom of thiocarbonyl group and O-atom of furan (C − O − C) ring in complex formation which concluded tridentate nature of the ligand (Goel et al. 2018). The bands between 514–551 cm–1 and 425–475 cm–1, suggested the existence of bonds owing to ν(M ← N) and ν(M ← O), respectively. The appearance of these bonds additionally confirmed the coordination between the ligand and metal(II) ions.
The stretching bands corresponding to νas(OAc) and νs(OAc) were absent in [CoL2](OAc)2 and [NiL2](OAc)2 complexes. Additionally, the existence of ν1 and ν2 bands at 1507 cm–1 and 1356 cm–1, respectively in [CoL2](OAc)2 (Fig. S1) confirmed the non-participation of acetate ions in complexation. Similarly, the non-participation of acetate ions in complexation were suggested by the presence of ν1 and ν2 bands at 1516 cm–1 and 1341 cm–1, respectively (Nakamoto 1970).
In case of nitrate complexes, the existence of a single broad band at 1384 cm–1 and the absence of ν1, ν2 and ν5 bands in the IR spectra of [CoL2](NO3)2 and [NiL2](NO3)2 reported the non-participation of nitrate group in complexation (Rao and Zacharias 1977). The splitting of the ν3 band into two in sulphate complexes indicates that sulphate coordinates in a unidentate manner, however if this splitting is expanded from two to three, sulphate coordinates in a bidentate manner. But in [CoL2]SO4 and [NiL2]SO4, such splitting is absent and a single broad band around 1105−1135 cm–1 was observed which confirmed the non-participation of sulphate group in coordination (Jain et al. 2020a). Table 2 shows some of the important IR peaks of the complexes.
4.1.2 Molar Conductance of the Complexes
The molar conductance of the Co(II) and Ni(II) complexes were evaluated and found to lie in the range of 113–125 Ω−1cm2mol−1 and 109–111 Ω−1cm2mol−1, respectively (Table 1). The non-participation of the anions in complex formation was concluded by molar conductance value and suggested the electrolyte nature of the synthesized complexes (Refat et al. 2009). Therefore, the complexes were assigned [ML2]X2 formula, where M = Co(II) and Ni(II), L = AMFT and X = CH3COO−, NO3−, and when X = SO42−, the formula was [ML2]X.
4.1.3 Magnetic Moment
The magnetic moment values (Table 3) for Co(II) complexes were observed in the range of 4.97−5.02 BM and corresponded to three unpaired electrons owing to orbital contribution (Tyagi et al. 2017), whereas the magnetic moment for Ni(II) complexes was observed in the range of 2.85−2.98 BM at room temperature, attributed to two unpaired electron. The magnetic moments were estimated using the Van Vleck formula as well, and the results were comparable. All of the compounds were thought to be paramagnetic, and octahedral geometry was predicted based on the findings.
4.1.4 Electronic Spectra Analysis
Four absorption bands i.e., 9597 − 9891 cm−1, 12484 − 12626 cm−1, 20040 − 20619 cm−1 and 35890 − 38418 cm−1 were exhibited by electronic spectra of Co(II) complexes (Fig. S2). The first three bands were attributed to 4T1g (F) → 4T2g (ν1), 4T1g (F) → 4A2g (ν2) and 4T1g (F) → 4T1g (P) (ν3) transitions, respectively and the occurrence of last band i.e., 35890 − 38418 cm−1 was due to charge transfer. The obtained transitions discovered that the complexes possess distorted octahedral geometry and possessed D4h symmetry (Jain et al. 2018). Similarly, the electronic spectra of Ni(II) complexes demonstrated four transition bands in the range of 11249 − 11455 cm−1, 15601 − 16051 cm−1, 25000 − 25707 cm−1 and 36231 − 36900 cm−1. The first three bands credited to three separate transitions i.e., 3A2g (F) → 3T2g (F) (ν1), 3A2g (F) → 3T1g (F) (ν2) and 3A2g (F) → 3T1g (F) (ν3), respectively. The positions of the absorption bands in electronic spectra of Ni(II) confirmed the octahedral geometry of the complexes. The bands in the range of 36231 − 36900 cm−1 correspond to charge transfer band (Chandra and Gupta 2004; Lever 1968).
4.1.5 Ligand Field Parameters of the Complexes
The energy associated with transitions is determined by the value of B (Racah parameter). The empirical values generated from free ion spectra are known as the Racah parameters. The first transition obtained in complex spectra is used to calculate the value of Dq. The nephelauxetic parameter (β) is evaluated by using β = B(complex)/B(free ion) expression, where B(free ion) for Co(II) = 1120 cm−1 and Ni(II) = 1041 cm−1. Table 3 illustrates values of Dq, B, β, and ligand field splitting energy (LFSE). The value of β found in the range of 0.89 − 0.92 for Co(II) complexes, suggesting some covalent nature between metal and ligand whereas for Ni(II) complexes the value of β was found to be in the range of 0.41 − 0.50, indicating strong covalent nature (Jain et al. 2020b).
4.1.6 Electronic Paramagnetic Resonance (EPR) Analysis
The polycrystalline form of Co(II) complexes were employed at room temperature (RT) to record EPR spectra. The EPR spectra were recorded on X band at 9.1 GHz frequency and 3000 G magnetic field in DMSO solution. A wide signal and the band width dependence on temperature were demonstrated by EPR spectra of Co(II) complexes. The signals are discovered due to rapid spin relaxation time span at low temperature. In comparison with, g-values do not show any dependency on temperature. The hyperfine splitting was absent in Co(II) complexes transitions. The values of giso were found in the range of 2.02–2.18, indicating covalent nature of metal ligand bond. The giso were determined by employing formula: giso = \(\frac{\mathrm{g}\parallel + 2\mathrm{g}\perp }{3}\). Table 4 depicts the g-values of Co(II) complexes, suggesting complexes to possess the octahedral geometry (Hathaway and Billing 1970). The various X-band EPR spectra of [CoL2](OAc)2 and [CoL2]SO4, are illustrated in Fig. S3(a) and S3(b), respectively.
4.2 Theoretical Analysis
In the gaseous phase, structural optimization of the Co(II) and Ni(II) complexes of the AMFT ligand was carried out at a semi-empirical level. Figure 1 depicts the optimized structures (with numbering) of the complexes. The bond lengths and bond angles of the complexes are given in Tables 5 and 6, respectively.

Optimized structures of complexes a [Co(L)2]X2, b [Ni(L)2]X2; Where, X = CH3COO−, NO3−, ½SO4.2− [Color Code: H-Light grey, C-Dark grey, N-Dark blue, O-Red, S-Yellow, Ni/Co-Light blue]
The tridentate nature of the AMFT ligand was confirmed by evaluating bond lengths and bond angles. The S-atom of the thiocarbonyl group and N-atom of imine group in AMFT occupied the equatorial position in the complexes [NiL2]+2 where L = AMFT. The equatorial bond length of two S-atoms and metal (S47-Mn and S48-Mn) were same i.e., 2.216 Å. Similarly, the bond length between N-atom and metal (N2-Ni and N10-Ni) in both the rings were also the same i.e., 1.879 Å. The axial position was occupied by O-atom of furan (C − O − C) ring and the bond length of O23-Ni and O38-Ni were also similar i.e., 2.986 Å. The bond lengths were also tabulated for [CoL2]+2 complexes. Similar to Ni(II) complexes, the S-atom of thiocarbonyl and N-atom of the imine occupied the equatorial positions and the axial positions were occupied by the O-atom of furan rings of the tridentate ligand. The bond length between S47-Co and S48-Co was 2.142 Å and 2.091 Å, respectively and between N2-Co and N10-Co, the bond length was 1.764 Å and 1.719 Å respectively, in both the rings of Co(II) complex. The axial bond length i.e., O23-Co and O38-Co was 2.088 Å and 2.479 Å respectively with some deviation. Most of the. Bond lengths of Co(II) and Ni(II) complexes are identical except few. The bond angles study revealed the difference in the structure of both the metal(II) complexes. The bond angles between O23-M-S47 and O38-M-S48 (128.892°) and O23-M-S48 and O38-M-S47 (45.747°) in the Ni(II) complexes confirmed the different orientation of the two ligands. The bond angles between the O23-M-S47 and O38-M-S48 were 111.610° and 123.597°, respectively and O23-M-S48 and O38-M-S47 were 69.444° and 55.101°, respectively in Co(II) complexes also confirmed the different orientation of the two ligands and reported a ‘χ’ like structure for all the complexes.
The bond angles between O23-M-O38, N2-M-N10 and S47-M-S48 were 107.431°, 170.987°, 173.221°, respectively in Ni(II) complexes and 90.549°, 141.358°, 178.069° in Co(II) complexes, respectively. In Ni(II) complexes, the angles reflected both the furan ring at an acute angle and both the N-atom and S-atom were in straight angle confirming the equatorial trans position. Whereas in Co(II) complexes, a little more distortion was observed. Both the furan ring were at right angle in Co(II) complexes and both the S-atom were in straight angle confirming the equatorial trans position but N atoms showed little distortion from straight angle. For Ni(II) complexes, the bond angles suggested octahedral geometry, but for Co(II) complexes, deformed octahedral geometry was suggested. The suggested structures of the Co(II) and Ni(II) complexes are shown in Fig. 2a and b, respectively.

Suggested structure of the complexes a [CoL2]X2, b [NiL2]X2; Where, X = CH3COO−, NO3−, ½SO4.2− [Color Code: H-Light grey, C-Dark grey, N-Dark blue, O-Red, S-Yellow, Ni/Co-Light blue]
4.2.1 Frontier Molecular Orbital (FMOs)
The FMOs hypothesis can help us to understand how complex systems interact. The ionization potential and electron affinity are closely connected to EHOMO and ELUMO. When the molecule gets energy, an electron is easily lost from EHOMO. Ionization potential is the negative value of EHOMO whereas ELUMO accepts an electron and electron affinity is its negative value. The energy difference between LUMO and HOMO is known as the energy gap, and it is always positive since LUMO energy is always more than HOMO energy. This is a highly essential chemical feature of the lowest possible energy electronic excitation in a compound. The small energy gap explains the high chemical reactivity and less kinetic stability of the complex (De and Geerlings 2001). In transition-metal complexes this gap has very important chemical implications, it explains the structure stability/reactivity of the complexes (Yan 2021). The FMOs values were evaluated for the complexes of AMFT ligand (Table 7).
The HOMO–LUMO gap was found to be 0.1472 eV, 0.2772 eV for Co(II) and Ni(II) complexes, respectively (Fig. 3) and suggested the possibility of charge transfer. Positive energy gap value suggested the stability of both the metal complexes (Yu and Pan 2021). The lower value of energy gap indicated the excitation energies for an electron from HOMO to LUMO, leading the high reactivity and less stability of the Co(II) complexes in comparison with Ni(II) complexes.

Frontier molecular orbitals of the synthesized complexes a [CoL2]X2, b [NiL2]X2, Where, X = CH3COO−, NO3−, ½SO42−
The different global reactivity descriptors were calculated with the value of EHOMO and ELUMO using the Koopman’s theorem (Koz et al. 2010). Global reactivity descriptors were used to describe the reactivity and stability of the complexes. Table 7 lists the values for all of the global reactivity characteristics. The Co(II) complexes have the highest chemical potential and softness, as well as the lowest hardness, which explained high chemical reactivity and lack of stability when compared to Ni(II) complexes. As electrophilicity is the measure of electrophilic power, therefore the high value of ω suggested Co(II) as the strongest electrophile (Drissi et al. 2015). The additional parameters like zero-point energy, enthalpy, entropy, energies and rotational constants are illustrated in Table 8.
4.2.2 Mulliken Population Analysis (MPA)
MPA is a method of calculating charges based on the population of linear combined atomic orbitals (LCAO) base (Pu and Pan 2022a); Pu and Pan 2022b). MPA was performed to obtain the charges on individual atoms (Jalilian et al. 2011). Partial atomic charge can be estimated by MPA. It describes the distribution of charges in the various sub-shells (core, valance, Rydberg) in the molecular orbital (Pan and Chen 2022). The charges on the individual atoms of the ligand are summarized in Fig. 4 and the values are listed in Table 9. Charge distribution in the ligand was analyzed on the optimized structure of the ligand obtained by DFT method in gaseous phase using a basis set 6-311 g + . The color, range and index are also depicted in Fig. 4. All C-atoms (red color), except C1, C14 and C19 were negatively charged whereas all the H-atoms and C1, C14 and C19 atoms (green color) were positively charged.

Mulliken charge distribution of AMFT ligand
The C-atoms of two methyl group i.e., C10 and C21 (deep red) possessed -0.692 and -0.535 values, respectively whereas, H8, H4, H7, C19, C14 (super green) reported 0.322, 0.285, 0.279, 0.281, 0.255 values, respectively. Sulphur atom (black) was the least negatively charged atom with -0.04 value (almost neutral). Oxygen of the furan ring (dark maroon) possessed -0.176 value.
4.2.3 Molecular Electrostatic Potential (MESP/MEP/ESP) Analysis
Molecular electrostatic potential (MESP) analysis calculation suggests the distribution of the negative and positive potential over the molecule. MESP can be used to find the electron-rich or electro-poor region in complex to determine best suited site for electrophilic or nucleophilic attack. It helps in prediction of the complex site that acts as a Lewis acid/base. Nuclear and electronic charge densities are used to carry out the MESP calculations. Mapping of the MESP on the molecular surface uses a color coding scheme. The potential energy order in the color coding scheme is red < orange < yellow < green < blue. Red to yellow color indicated a region of low electrostatic potential means the region of electrophilic reactivity sites whereas blue to green color indicated a region of high electrostatic potential means the region of nucleophilic reactivity sites (Djafri et al. 2020) The molecular electrostatic potential surface structures of the metal complexes are shown in Fig. 5.

Molecular electrostatic potential surfaces a [CoL2]X2, b [NiL2]X2
O-atom of the furan ring (red color) in the MESP indicated an electron rich site whereas the other site (yellow color) indicated the electrons deficient site. The charge clouds at S-atom, imine N-atom participating in the complex formation and specific locations of furan ring (red color) were characterized as donor whereas the other region in the complexes (green and yellow) were characterized as acceptor. The electron density isosurface was 0.02 a.u.
4.3 Biological Evaluation
The biological assessment of AMFT ligand and its complexes was conducted utilizing the Well diffusion technique (Nasiri and Zobi 2020). Different pathogens were tested on the prepared samples. The zone of inhibitions is given in Table S1. Compared to the ligand, the screening indicated a higher inhibition performance of complexes (Fig. 6).

Comparison of antibacterial and antifungal efficiency at a 250 ppm, b 500 ppm, c 750 ppm d 1000 ppm concentrations along with Standard deviations SD). P value for bacterial activity = 5.160. P value for fungal activity = 4.703
The improved activity has been widely recognized by chelation theory and principle of overtone (Tweedy 1964). The effect of enhanced concentration on the antimicrobial performance were also checked and revealed to be boosted with increasing concentration (Buldurun et al. 2019).
4.4 Molecular Docking
At the atomic level, determining the interaction between macromolecule and ligand is useful. The molecule is stabilized into active cavity by hydrogen bonding, electrostatic interactions, and van der Waals interactions (Vishvakarma et al. 2017). Molecular docking analysis of AMFT ligand, Co(II) complex and Ni(II) complex was performed against beta-glucanase of B. macerans and Allergen Alt a1 of A. alternate and results are given in Table 10. Co(II) complex possessed highest negative values in the case of beta-glucanase of B. macerans and Allergen Alt a 1 of A. alternate proteins i.e., -89.8461 kJ/mol & -78.7962 kJ/mol, respectively. Co(II) complex also showed the highest van der Waals interactions.
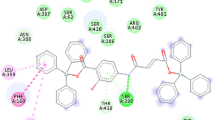
After molecular docking, pose analysis was done and the results were reviewed. H-bonds were produced in this study, and their bond length, hydrophobic interactions, and electrostatic interactions are depicted in Figs. 7 and 8. The five H-bonds with TRP-182, GLU-107, ASP-105, GLU-103 & TYR-92 were formed with ligand while four H-bonds were formed with SER-2, ILE-67 and TYR-209 amino acids in Co(II) complex. Similarly, four H-bonds were found in Ni(II) complex with ASP-58, THR-32, SER-23 & PHE-28 of beta-glucanase of B. macerans (Fig. 7).

Pose analysis of Endo-1,3–1,4-beta-glucanase of B. macerans (PDB ID-1MAC) a AMFT ligand b Co(II) complex and c Ni(II) complex

Pose analysis of Allergen Alt a 1 of A. alternate (PDB ID-3V0R) (PDB ID-1MAC) a AMFT ligand b Co(II) complex and c Ni(II) complex
In the case of Allergen Alt a 1 of A. alternate, H-bonding was absent in ligand whereas Co(II) complex demonstrated two H-bonds with VAL-39 and GLY-36. Similarly Ni(II) complex also showed two H-bonds with ASP-114 and ASP-115 (Fig. 8). Conventional, non-conventional and hydrophobic interactions with bond length are given in Table 11.
5 Conclusion
The coordination chemistry and biological efficiency of Co(II) and Ni(II) complexes were studied in the present work. Six different Co(II) and Ni(II) complexes were synthesized using the traditional condensation process. Different approaches were used to study the geometry and proposed octahedral structure of all the compounds. Theoretically hypothesized geometry and experimental geometry showed a strong connection in computational data. The ligand and complexes were also evaluated for their antibacterial and antifungal activity against two different bacteria and fungi strains and were found to better efficacy in comparison with AMFT ligand. Ni(II) complexes showed better inhibition activity than Co(II) complexes. Molecular docking result also demonstrated good agreement with the experimental result. As a result of this research, it may be inferred that thiosemicarbazone metal complexes can be used to treat a variety of diseases.
References
Alcaraz R, Muñiz P, Cavia M, Palacios O, Samper KG, Gil-García R, Jiménez-Pérez A, García-Tojal J, García-Girón C (2020) Thiosemicarbazone-metal complexes exhibiting cytotoxicity in colon cancer cell lines through oxidative stress. J Inorg Biochem 206:110993. https://doi.org/10.1016/j.jinorgbio.2020.110993
Almeida JCL, Amim RS, Pessoa C, Lourenço MCS, Mendes IC, Lessa JA (2020) Bismuth(III) complexes with pyrazine formamide thiosemicarbazones: Investigation on the antimicrobial and cytotoxic effects. Polyhedron 189:114709. https://doi.org/10.1016/j.poly.2020.114709
Amritha B, Manaf O, Nethaji M, Sujith A, Kurup P, Vasudevan S (2020) Mn(II) complex of a di-2-pyridyl ketone n(4)-substituted thiosemicarbazone: Versatile biological properties and naked-eye detection of Fe2+ and Ru2+ ions. Polyhedron 178:114333. https://doi.org/10.1016/j.poly.2019.114333
Amuthakala S, Bharathi S, Rahiman AK (2020) Thiosemicarbazone-based bifunctional chemosensors for simultaneous detection of inorganic cations and fluoride anion. J Mol Struct 1219:128640. https://doi.org/10.1016/j.molstruc.2020.128640
Bakir M, Lawrence MW, Yamin MB (2020) Novel κ2-Nim, S- and κ4-C, Nim,(μ-S),(μ-S)-coordination of di-2-thienyl ketone thiosemicarbazone (dtktsc). Hydrogen evolution and catalytic properties of palladacyclic [Pd(κ4-C, Nim,(μ-S),(μ-S)-dtktsc-2H)]4. InorganicaChim Acta 507:119592. https://doi.org/10.1016/j.ica.2020.119592
Batista SAA, Vandresen F, Falzirolli H, Britta E, de Oliveira DN, Catharino RR, Gonçalves A, Ramalho TC, La Porta FA, Nakamura CV, da Silva CC (2019) Synthesis and comparison of antileishmanial and cytotoxic activities of S-(−)-limonene benzaldehyde thiosemicarbazones with their R-(+)-analogues. J Mol Struct 1179:252–262. https://doi.org/10.1016/j.molstruc.2018.11.017
Buldurun K, Turan N, Savci A, Colak N (2019) Synthesis, structural characterization and biological activities of metal(II) complexes with Schiff bases derived from 5-bromosalicylaldehyde: Ru(II) complexes transfer hydrogenation. J Saudi Chem Soc 23:205–214. https://doi.org/10.1016/j.jscs.2018.06.002
Campbell MJM (1975) Transition metal complexes of thiosemicarbazide and thiosemicarbazones. Coord Chem Rev 15:279–319. https://doi.org/10.1016/S0010-8545(00)80276-3
Casas JS, Garcia-Tasende MS, Sordo J (2000) Main group metal complexes of semicarbazones and thiosemicarbazones. A Structural Review Coord Chem Rev 209:197–261. https://doi.org/10.1016/S0010-8545(00)00363-5
Chandra S, Gupta LK (2004) EPR, IR and electronic spectral studies on Mn(II), Co(II), Ni(II) and Cu(II) complexes with a new 22-membered azamacrocyclic [N4] ligand. Spectrochim Acta A 60:1751–1761. https://doi.org/10.1016/j.saa.2003.07.011
De Proft F, Geerlings P (2001) Conceptual and computational DFT in the study of aromaticity. Chem Rev 10:11451–11464. https://doi.org/10.1021/cr9903205
Djafri A, Perveen F, Benhalima N, Khelloul N, Rahmani R, Djafri A, Chouaih A, Kanoun MB, Goumri-Said S (2020) Experimental spectral characterization, Hirshfeld surface analysis, DFT/TD-DFT calculations and docking studies of (2Z,5Z)-5-(4-nitrobenzylidene)-3-N(2-methoxyphenyl)-2-N’(2-methoxyphenylimino) thiazolidin-4-one. Heliyon 6:e05754. https://doi.org/10.1016/j.heliyon.2020.e05754
Dkhar L, Banothu V, Poluri KM, Kaminsky W, Kollipara MR (2020) Platinum group complexes containing salicylaldehyde based thiosemicarbazone ligands: Their synthesis, characterization, bonding modes, antibacterial and antioxidant studies. J Organomet Chem 918:121298. https://doi.org/10.1016/j.jorganchem.2020.121298
Drissi M, Benhalima N, Megrouss Y, Rahmani R, Chouaih A, Hamzaoui F (2015) Theoretical and experimental electrostatic potential around the m-nitrophenol molecule. Molecules 20:4042–4054. https://doi.org/10.3390/molecules20034042
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersso GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J A, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth, GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.1, Gaussian Inc., Wallingford CT.
Frish A, Nielson AB, Holder AJ (2001) Gaussian view user manuals. Gaussian Inc, Pittsburg
Garg BS, Jain VK (1988) Analytical applications of thiosemicarbazones and semicarbazones. Microchem J 38:144–169. https://doi.org/10.1016/0026-265X(88)90017-3
Gautam S, Chandra S, Rajor H, Agrawal S, Tomar PK (2018) Structural designing, spectral and computational studies of bioactive Schiff’s base ligand and its transition metal complexes. Appl Organometal Chem 32:e3915. https://doi.org/10.1002/aoc.3915
Goel P, Kumar D, Chandra S, Kumar A (2018) Synthesis and spectroscopic study of biologically active tridendtate Schiff’s base ligand 2-Acetyl-5-methyl-furanthiosemicarbazone and its Mn(II), Co(II), Ni(II) and Cu(II) complexes. Iran J Sci Technol A 42:557–565. https://doi.org/10.1007/s40995-016-0140-6
Hathaway BJ, Billing DE (1970) The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion. Coord Chem Rev 5:143–207. https://doi.org/10.1016/S0010-8545(00)80135-6
Jain P, Kumar D, Chandra S (2018) Synthesis, spectroscopic approach, biological screening and geometrical optimization of bidentate Schiff’s base ligand and their Mn(II) and Co(II) complexes. Asian J Chem 30:1664–1670. https://doi.org/10.14233/ajchem.2018.21347
Jain P, Pandey G, Kumar D, Chandra S (2019a) Prospects of biologically active schiff’s base ligand and metal complexes in drug discovery. Adv Sci Eng Med 11:144–154. https://doi.org/10.1166/asem.2019.2302
Jain P, Kumar D, Chandra S (2019b) Biological applications of Co(II) and Ni(II) complexes of semicarbazones and thiosemicarbazones. Asian J Chem 31:1–8. https://doi.org/10.14233/ajchem.2019.21475
Jain P, Kumar D, Chandra S, Misra N (2020a) Experimental and theoretical studies of Mn(II) and Co(II) metal complexes of a tridentate Schiff’s base ligand and their biological activities. ApplOrganomet Chem 34:e5371. https://doi.org/10.1002/aoc.5371
Jain P, Sharma S, Kumar N, Misra N (2020b) Ni(II) and Cu(II) complexes of bidentate thiosemicarbazone ligand: Synthesis, structural, theoretical, biological studies and molecular modelling. Appl Organomet Chem 34:e5736. https://doi.org/10.1002/aoc.5736
Jalilian R, Jauregui LA, Lopez G, Tian J, Roecker C, Yazdanpanah MM, Cohn RW, Jovanovic I, Chen YP (2011) Scanning gate microscopy on graphene: charge inhomogeneity and extrinsic doping. Nanotechnology 22:295705. https://doi.org/10.1088/0957-4484/22/29/295705
Jawaria R, Hussain M, Ahmad HB, Ashraf M, Hussain S, Naseer MM, Khalid M, Hussain MA, Al-Rashida M, Tahir MN, Asim S, Shafiq Z (2020) Probing ferrocene-based thiosemicarbazones and their transition metal complexes as cholinesterase inhibitors. InorganicaChim Acta 508:119658. https://doi.org/10.1016/j.ica.2020.119658
Khalid M, Jawaria R, Khan MU, Braga AAC, Shafiq Z, Imran M, Zafar HMA, Irfan A (2021) An efficient synthesis, spectroscopic characterization, and optical nonlinearity response of novel salicylaldehyde thiosemicarbazone derivatives. ACS Omega 6:16058–16065. https://doi.org/10.1021/acsomega.1c01938
Kostas ID, Heropoulos GA, Kovala-Demertzi D, Yadav PN, Jasinski JP, Demertzis MA, Andreadaki FJ, Vo-Thanh G, Petit A, Loupy A (2006) Microwave-promoted Suzukie-Miyaura cross-coupling of aryl halides with phenylboronic acid under aerobic conditions catalyzed by a new palladium complex with a thiosemicarbazone ligand. Tetrahedron Lett 47:4403–4407. https://doi.org/10.1016/j.tetlet.2006.04.088
Koz G, Özdemir N, Astley D, Dinçer M, Astley ST (2010) Synthesis, spectroscopic and structural characterization of cobalt(II) complex with uracil-containing 2,6-diformylpyridine ligand: Theoretical studies on the ligand and pentagonal-bipyramidal [Co(L)(H2O)2]2+ and [Zn(L)(H2O)2]2+ cations. J Mol Struct 966:39–47. https://doi.org/10.1016/j.molstruc.2009.12.001
Lawrence ML, Shell SM, Beckford FA (2020) Binuclear manganese-iron complexes containing ferrocenyl thiosemicarbazones: Biological activity and carbon monoxide-releasing properties. InorganicaChim Acta 507:119548–119557. https://doi.org/10.1016/j.ica.2020.119548
Lever ABP (1968) Inorganic electronic spectroscopy, 1st edn. Elsevier, Amsterdam, p 249
Nakamoto K (1970) Infrared spectra of inorganic coordination compounds, 2nd edn. Wiley-Interscience, New York, p 17
NasiriSovari S, Zobi F (2020) Recent studies on the antimicrobial activity of transition metal complexes of groups 6–12. Chemistry 2:418–452. https://doi.org/10.3390/chemistry2020026
Naveen TRK, Ghule VD, Kumar N, Kumar L, Lal K, Kumar A (2020) Design, synthesis, biological activity, molecular docking and computational studies on novel 1,4-disubstituted-1,2,3-triazole-thiosemicarbazone hybrid molecules. J Mol Struct 1209:127951. https://doi.org/10.1016/j.molstruc.2020.127951
Nehar OK, Mahboub R, Louhibi S, Roisnel T, Aissaoui M (2020) New thiosemicarbazone Schiff base ligands: Synthesis, characterization, catecholase study and hemolytic activity. J Mol Struct 1204:127566. https://doi.org/10.1016/j.molstruc.2019.127566
Pahontu E, Julea F, Rosu T, Purcarea V, Chumakov Y, Petrenco P, Gulea A (2015) Antibacterial, antifungal and in vitro antileukaemia activity of metal complexes with thiosemicarbazones. J Cell Mol Med 119:865–878. https://doi.org/10.1111/jcmm.12508
Pan Y (2021) Role of hydrogen on the structural stability, mechanical and thermodynamic properties of the cubic TM3Si silicides. Int J Hydrogen Energ 46:28338–28345. https://doi.org/10.1016/j.ijhydene.2021.06.038
Pan Y, Chen S (2022) Influence of alloying elements on the mechanical and thermodynamic properties of ZrB2 boride. Vacuum 198:110898. https://doi.org/10.1016/j.vacuum.2022.110898
Pu D, Pan Y (2022a) First-principles investigation of solution mechanism of C in TM-Si-C matrix as the potential high-temperature ceramics. J Am Ceram Soc 105:2858–2868. https://doi.org/10.1111/jace.18254
Pu D, Pan Y (2022b) New insight into the structural stability, ductility and melting point of Mo5SiB2 under high-pressure environment. Vacuum 196:110727. https://doi.org/10.1016/j.vacuum.2021.110727
Qi J, Yao Q, Qian K, Tian L, Cheng Z, Yang D, Wang Y (2018) Synthesis, antiproliferative activity and mechanism of gallium(III)-thiosemicarbazone complexes as potential anti-breast cancer agents. Eur J Med Chem 154:91–100. https://doi.org/10.1016/j.ejmech.2018.05.016
Raicopol MD, Chira NA, Pandele AM, Hanganu A, Ivanov AA, Tecuceanu V, Bugeana IG, BuicaG O (2020) Electrodes modified with clickable thiosemicarbazone ligands for sensitive voltammetric detection of Hg(II) ions. Sens Actuators B Chem 313:128030
Rao CRK, Zacharias PS (1977) Studies on dinuclear metal complexes of macrocyclic ligands with varying chelate ring size. Polyhedron 16:1201–1209. https://doi.org/10.1016/S0277-5387(96)00364-6
Refat MS, Chandra S, Tyagi M (2009) Spectroscopic, thermal and biocidal studies on Mn(II), Co(II), Ni(II) and Cu(II) complexes of tridentate ligand having semicarbazone moieties. J Therm Anal Calorim 100:261–267. https://doi.org/10.1007/s10973-009-0397-5
Saswati MM, Banerjee A, Biswal S, Horn A, Schenk G, Brzezinski K, Sinn E, Reuter H, Dinda R (2020) Polynuclear zinc(II) complexes of thiosemicarbazone: Synthesis, X-ray structure and biological evaluation. J Inorg Bio Chem 203:110908. https://doi.org/10.1016/j.jinorgbio.2019.110908
Sevinçli ZS, Duran GN, ÖzbilM KN (2020) Synthesis, molecular modeling and antiviral activity of novel 5-fluoro-1H-indole-2,3-dione 3-thiosemicarbazones. Bioorg Chem 104:104202. https://doi.org/10.1016/j.bioorg.2020.104202
Singh S, Athar F, Mauryab MR, Azama A (2006) Cyclooctadiene Ru(II) complexes of thiophene-2-carboxaldehyde-derived thiosemicarbazones: Synthesis, characterization and antiamoebic activity. Eur J Med Chem 41:592–598. https://doi.org/10.1016/j.ejmech.2006.01.014
Tweedy BG (1964) Plant extracts with metal ions as potential antimicrobial agents. Phytopathology 55:910–918
Tyagi P, Tyagi M, Agrawal S, Chandra S, Ojha H, Pathak M (2017) Synthesis, characterization of 1,2,4-triazole Schiff base derived 3d- metal complexes: Induces cytotoxicity in HepG2, MCF-7 cell line, BSA binding fluorescence and DFT study. Spectrochim Acta A 171:246–257. https://doi.org/10.1016/j.saa.2016.08.008
Vishvakarma VK, Singh P, Kumari K, Chandra R (2017) Rational design of threo as well erythro noscapines, ananticancer drug: a molecular docking and molecular, dynamic approach. Biochem Pharmacol 6:1–7. https://doi.org/10.4172/2167-0501.1000229
Vishvakarma VK, Kumari K, Singh P (2020) A model to study the inhibition of arginase II with noscapine & its derivatives. J pro Res Bioinf 2:1–14. https://doi.org/10.24966/PRB-1545/100008
Yu E, Pan Y (2021) Influence of noble metals on the electronic and optical properties of LiH hydride: first-principles calculations. Int J Hydrogen Energ 46:35342–35350. https://doi.org/10.1016/j.ijhydene.2021.08.095
Acknowledgements
IIT (Delhi), IIT (Jodhpur) and IIT (Mumbai), SAIF is acknowledged for providing IR and UV report, computational data and EPR data, respectively.
Funding
No funding received.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors state that they have no known competing financial interests or personal ties that might have influenced the research presented in this study.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jain, P., Vishvakarma, V.K., Singh, P. et al. Co(II) and Ni(II) Complexes of a Heterocyclic Ligand: Synthesis, Characterization, Docking and Biological Activity. Iran J Sci Technol Trans Sci 46, 793–805 (2022). https://doi.org/10.1007/s40995-022-01312-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40995-022-01312-7