
Abstract
Dried blood spots (DBS) are the best sampling for large and population-based genetic studies. The study aimed to develop a DNA extraction protocol for DBS samples in a 96-well microtiter plate format using DNA salting-out principle. The developed protocol was named High-Throughput Salting-Out (HTSO). The DBS samples were collected from 20 suspected Glutaric Acidemia Type I (GA-I) patients from unrelated families and 50 controls without any metabolic disorders. The DNA was isolated from DBS samples using HTSO protocol in a 96-well microtiter plate. The assessment of yield and quality of DNA after isolation by HTSO method was done using NanoDrop-8000. The DNA isolated by the HTSO was used in the genotyping of multiple mutations, R402W and W225X in the Glutaryl-CoA Dehydrogenase gene in GA-I patients by multiplex PCR and RFLP. The HTSO showed at least a 2- to 34-fold increased DNA yield against manual in-house, commercial, and automation-based protocols. The per sample DNA extraction cost of the HTSO method was four times lesser than that of the commercial kits. Without sophisticated instruments or automation, HTSO method considerably reduced the turnaround time of DNA extraction compared to manual in-house and commercial kit protocols. The HTSO method is efficient, inexpensive, and time-saving for large-scale and potential high-throughput DNA extraction for DBS samples. This method is adequate for high-end diagnostic and institution facilities, in addition to institutions having financial constraints or lacks sophisticated instruments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Dried blood spots (DBS) have been used since the 1960s for newborn heel prick tests to identify neonates with phenylketonuria (Guthrie and Susi 1963). Simple, non-invasive sampling, reduced risk of infection, inexpensive transportation, and easy storage make them a preferred choice of specimen in newborn screening (NBS) programs (Guthrie and Susi 1963; Hue et al. 2011; Mössner et al. 2016; Wilhelm et al. 2014; Chaisomchit et al. 2005; Saavedra-Matiz et al. 2013). The DBS are extensively used in drug monitoring (Edelbroek et al. 2009), genetic analysis (Hollegaard et al. 2011; St Julien et al. 2013), epidemiological studies (Parker et al. 1999), and in biobanks (Choi et al. 2014). Presently, there is an urge for molecular genetic testing in NBS programs. The DNA is robust and stable for many decades on a filter paper matrix (Choi et al. 2014). Subsequently, it makes an ideal choice of sampling for genotyping studies in a high-throughput manner for diagnostic and research laboratories (Hollegaard et al. 2011; Hollegaard et al. 2009).
The significant step in molecular biology is the isolation of DNA from biological samples, which is difficult, time-consuming, and involves multiple steps (Kalendar et al. 2021). Due to less blood on the DBS matrix, the genetic material obtained will also be less (Kumar et al. 2019). It is essential to have improved techniques at a cheaper cost to acquire a good quantity and quality DNA from DBS samples for large-scale or high-throughput genetic studies. There are numerous high-throughput protocols for extracting DNA from DBS samples like manual in-house, commercial kits (truXTRAC® DBS DNA Kit, sbeadex™), and automation-based high-throughput methods. However, the manual in-house methods are usually crude or have pre-treatment or multiple washing steps or provide low DNA yield (Saavedra-Matiz et al. 2013; St Julien et al. 2013; De Vries et al. 2009; Zhili et al. 2005). The commercial kits and automation-based high-throughput protocols are costly and may not be accessible for laboratories with limited resources or have financial limits (Saavedra-Matiz et al. 2013; De Vries et al. 2009; Zhili et al. 2005; Sirdah 2014; Ellen et al. 1999). The present study focuses on developing a large-scale and potential high-throughput DNA extraction protocol for DBS samples that provides good quality DNA at a lesser cost. The anticipated method should be adequate for high-end facilities and institutions with financial constraints or lack sophisticated instruments.
The newly proposed DNA extraction method for DBS was called High-Throughput Salting-Out or HTSO, formulated based on the DNA salting-out principle (Miller et al. 1988). The DNA salting-out protocol is a simple, affordable, and non-toxic. This protocol is commonly used by research laboratories worldwide. Unlike conventional DNA salting-out method, HTSO process DBS samples in a microtiter plate and provides quality DNA usable for genetic studies. The DNA extracted by the HTSO method was used in genotyping of multiple mutations such as R402W (c. 1204C > T) and W225X (c. 675G > A), in Glutaryl-CoA Dehydrogenase gene (GCDH) in Indian patients having Glutaric Aciduria Type I (GA-I), a neurometabolic disorder caused due to defective GCDH enzyme (EC 1.3. 99.7) (Hedlund et al. 2006).
2 Materials and Methods
2.1 Sample Collection
The Institutional Human Ethics Committee approved this work. The study followed the ethical standards laid down in the 1964 Declaration of Helsinki. All participants of this study gave informed consent. The blood spots were collected on S&S 903 filter paper, air-dried, and stored at − 20 °C. Fifty children without any clinical history of metabolic disorders presented at the Department of Pediatrics, Victoria Multispecialty Hospital, Bengaluru, India, were recruited as controls for this study. The DBS collected from control subjects were used for standardizing the HTSO method.
Twenty suspected GA-I patients were recruited based on the following inclusive criteria 1. Patients having clinical symptoms like macrocephaly, acute encephalitis-like crisis, spasticity, hypotonia, dystonia, choreoathetosis, ataxia, dyskinesia, and seizures. 2. Neuroimaging studies suggestive of frontal cerebral atrophy, subdural hematomas, atrophy in the basal ganglia or caudate and putamen, and hypoattenuation or atrophy of lentiform nuclei or 3. Biochemical report suggestive of elevated levels of Glutaric Acid and 3-Hydroxy Glutaric Acid in urine.
3 Extraction of DNA from DBS by HTSO Method
Three DBS spots of 3 mm diameter were punched into the wells of a sterile 96-well microtiter plate (Cat No: P-DW-11-C, Axygen) using a handheld manual DBS card puncher or a DBS Puncher® (Product No: 1296–071 by PerkinElmer). The 50 µl of methanol was added to each well and incubated for 1 min at room temperature (RT). The plate was then air or vacuum dried to remove methanol. The extraction buffer, 300 µl (30 mM Tris.HCl; 5 mM MgCl2; Triton X-100 1% (v/v); SDS 3% (w/v); 20 mM EDTA, pH 8) was added to each well. Subsequently, the plate was sealed and incubated at 80 °C for 15 min. The 10 µl of Proteinase K (20 mg/ml) was added to each well and further incubated at 60 °C for 45 min. Saturated NaCl (6 M) of 200 µl was added to each well and mixed using a multichannel pipette. The plate was then sealed and centrifuged at 4500 rpm for 10 min at RT. The upper clear aqueous phase was dispensed into the corresponding wells of a fresh 96-deep well microtiter plate using a multichannel pipette. To each well, 10% (v/v) sodium acetate (3 M; pH 5) and two volumes (v/v) of chilled absolute ethanol was added. The plate wassealed and centrifuged at 4700 rpm using a plate rotor (Sorvall™ Legend™ XT Centrifuge) for 10 min at 4 °C. The pellets were washed using 70% ethanol and centrifuged again at 4700 rpm using a plate rotor for 10 min at 4 °C. The pellets were air-dried and suspended in 50 µl of autoclaved Milli Q water. The 96-deep well microtiter plate was then covered with aluminum foil and stored at -20 °C or -80 °C for the long-term.
4 Extraction of DNA from DBS by Slso Method (Shaik et al. 2016)
Three DBS of 3 mm diameter were punched into sterile 1.5 ml microcentrifuge tubes. The protocol was followed as described in our previous study (Shaik et al. 2016). The DNA obtained was suspended in 50 µl of autoclaved Milli Q water and was stored at -20 °C or -80 °C for the long-term.
5 Extraction of DNA from DBS by Phenol–chloroform Method (Gold Standard) (Hue et al. 2012)
Three DBS of 3 mm diameter were punched into a sterile 1.5 ml microcentrifuge tube. To the spots, added 200 µl of lysis buffer (10 mM Tris–HCl; 5 mM MgCl2; Triton X100 1% (v/v); SDS 1% (w/v); 10 mM EDTA, pH 8) and incubated at 85 °C for 20 min with gentle mixing. To it added 0.03 mg of proteinase K and continue to incubate at 60 °C for 1 h. The second lysis was done by adding 200 µl of lysis buffer (Tris HCl 30 mM; EDTA 20 mM; SDS 3%, pH 8) and further incubating the spots for 20 min at 85 °C. To these spots, an equal volume of phenol: chloroform: iso-amyl alcohol (25:24:1 v/v) was added, mixed well, and centrifuged at 12,000 rpm for 4 min at RT. After centrifugation, the upper aqueous phase was removed and dispensed into a fresh microcentrifuge tube. To it added 10% (v/v) 3 M sodium acetate (pH 5) and two volumes (v/v) of chilled absolute ethanol for the precipitation of DNA from the aqueous solution. The precipitated DNA was centrifuged at 12,000 rpm at 4 °C for 15 min. The pellets were washed with 70% ethanol, air-dried and suspended in 50 µl of autoclaved Milli Q water. The isolated DNA was stored at − 20 °C or − 80 °C for the long-term.
6 Quantity and Quality Assessment of DNA
The measurement at A260 nm using NanoDrop-8000 spectrophotometer provided the measure of quantity of DNA obtained by the HTSO method. The measure of A260/A280 ratio (indicators of contaminants), and the ability of DNA to amplify exon 7 (639 bp) and exon 10 (481 bp) of the GCDH gene in a PCR reaction (indicates any inhibitory material interfering with the PCR amplification), provided the quality of DNA obtained by the HTSO method. The assessment of DNA degradation during purification from DBS samples was analyzed by loading 3 μl of DNA isolated by the HTSO method onto 0.8% agarose gel and carrying out horizontal gel electrophoresis.
7 Genotyping of R402W and W225X Mutations
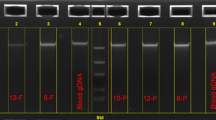
The DNA of 10 µl from 96-deep well microtiter plate was transferred to the corresponding well of the 96-well PCR plate. To each well of 96-well PCR plate, added 15 µl of PCR reaction mixture, which had 2.5 µl of 10X Taq DNA polymerase assay buffer (Merck, GeneiTM), 0.5 µl of Taq polymerase of 5 U/µl (Merck, GeneiTM), 6 µl de-ionized water, 2 µl of dNTPs of 2.5 mM each (Merck, GeneiTM) and 2 µl (20 µM) forward and reverse primers of R402W (c. 1204C > T) mutation (FP: 5'-TGTTACCCTCATGTGCCAC-3 and RP: 5'-TGCCTTCGGAGCTTACCTGT-3') and W225X (c. 675G > A) mutation (FP: 5'-CCGGCTAGTAAGAATCACCA-3' and RP: 5'-CCGGCTGAGTAAGAATCACCA-3'). The thermocycler amplification program consisted of following steps: initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 60 °C for 1 min 30 s, extension at 72 °C for 2 min and final extension of 72 °C for 5 min, to obtain the PCR products of 639 bp and 481 bp. After amplification, 10 µl of the PCR product from each well, transferred to the corresponding well of fresh 96-well deep well plate. Double restriction digestion using Msp I and Sac I restriction enzymes were performed by incubating the reaction mixture at 37 °C for 2 h. Depending on the gain or loss of the restriction site, specific band patterns on the agarose gel indicated the presence or absence of mutations (Figs. 1, 2). The mutants identified by RFLP were confirmed by sequencing the PCR products (Chromous Biotech Pvt. Ltd, Bengaluru, India).

a Horizontal agarose gel electrophoresis using 0.8% agarose for the DNA isolated by HTSO method. b Restriction fragment length polymorphism of R402W and W225X mutations Lane L represents 100 bp ladder (Genei TM, Merck, Bangalore); Lane 1 represents W225X (AA) and R402W (CC) genotype with bands at 335 bp, 305 bp, 243 bp, 146 bp and 91 bp; Lane 2 represents W225X (GG) and R402W (TT) genotype with bands at 548 bp, 481 bp, and 91 bp; Lane 3 represents W225X (GG) and R402W (CT) genotype with bands at 548 bp, 481 bp, and 335 bp

Diagrammatic representation of the restriction pattern of W225X and R402W mutations
8 Statistical Analysis
The statistical analysis was carried out by using SPSS 17.0. The mean, standard deviation, and Student's t-test statistical methods were used for the analysis of demographic data. The "p" value of < 0.05 was considered significant. The Hardy–Weinberg formula was used to calculate genotypic and allelic frequencies.
9 Results
The mean DNA yield obtained using the HTSO method from control DBS samples was 20.17 ± 4.4 ng/µl (ranging from 3 to 29 ng/µl) (Table 1), and mean purity was 2.0 ± 0.3 (ranging from 1.6 to 2.7). The average DNA yield from DBS samples of GA-I patients using the HTSO method was 16.3 ± 6.8 mg/µl (ranging from 7 to 27 mg/µl), and the average purity found to be 1.94 ± 0.18 (ranging from 1.65 to 2.3).
The average DNA yield from SLSO and phenol–chloroform methods was 23.8 ng/µl and 51.9 ng/µl, respectively. The mean purity of DNA obtained by SLSO was 1.91 and with the phenol–chloroform was 1.55. The yield and purity of DNA of the HTSO method, when compared with SLSO, showed no statistical significance (p = 0.642 and p = 0.840, respectively). Though the phenol–chloroform method gave good DNA yield compared to HTSO (p = 0.0001), the average purity of DNA obtained was low (1.5 ± 0.3; p = 0.001).
The DNA extracted using the HTSO method gave a non-degraded and good intense band on 0.8% agarose gel (Fig. 1a). The co-amplified PCR products of exon 7 and 10 amplified by using multiplex PCR also showed two distinct, bright bands corresponding to 639 bp (exon 7) and 481 bp (exon 10), respectively on 1.5% agarose gel (figure not shown). The double restriction PCR products also showed clear bands on 1.5% agarose gel (Fig. 1b). In addition, the chromatograms of all sequenced specimens showed clear nucleotide peaks.
The reliability and reproducibility of the HTSO method were assessed by intra and interassays which were performed using the DBS samples of control subjects. The intra assay done on the same day in duplicate sets of DBS samples showed no statistical difference between the isolates (p = 0.29; r2 = 0.933) (Fig. 3). Similarly, the interassay on two consecutive days also showed no statistical difference (p = 0.15; r2 = 0.924) (Fig. 4). However, the DNA yield varied from 3 to 25 mg/µl between different DBS samples.

Intra reproducibility assay of HTSO method: The DNA yield obtained using the HTSO method after two consecutive extracts performed on the same day using the DBS samples of 50 control subjects showed no statistical difference between the assays

Inter reproducibility assay of HTSO method: The DNA yield obtained using the HTSO method after two consecutive days of DNA extraction with the same DBS samples of 50 control subjects showed no statistical difference between the assays
The genotyping of 20 GA-I patients for W225X and R402W mutations (Table 2) showed 14 (70%) GA-I patients had W225X (GG) and R402W (CC) genotype. Two (10%) GA-I patients had W225X (AA) and R402W (CC) genotype. One (5%) patient had W225X (GG) and R402W (CT) and three (15%) GA-I patient had W225X (GG) and R402W (TT) genotype. None of the patients had W225X (GA) and R402W (CC) or W225X (AA) and R402W (TT) or W225X (GA) and R402W (CT) genotypes. The mutant allele frequencies for R402W and W225X mutations were 17.5% and 10%, respectively.
10 Discussion
In this study, a novel large-scale and potential high-throughput DNA extraction protocol for DBS samples called High-throughput Salting-Out or HTSO was formulated by exploiting the DNA salting-out principle. The initial step in the HTSO protocol is to treat DBS punches with methanol to prevent diffusion of PCR inhibitors from the filter paper matrix (McCabe et al. 1991; Panteleeff et al. 1999), followed by lysis step using concentrated lysis buffer that assists in the degradation of cellular contents. The degraded cellular proteins were then precipitated by adding saturated sodium chloride. To precipitate DNA from the supernatant cold absolute ethanol was added. The HTSO method showed a considerable reduction in per sample DNA extraction cost and processing time against single-vial protocols like SLSO and phenol–chloroform (Table 1). By replacing the microcentrifuge vials with a microtiter plate, the HTSO protocol cost less than 1$ per sample and took less than 2 h to extract DNA from 96 DBS samples. The yield and purity of DNA obtained by HTSO were similar to SLSO method, whose DNA quality was equivalent to the DNA obtained through commercial kits (silica-based column) (Shaik M et al. 2016). The DNA extracted by HTSO did not give any problems while carrying out PCR or during genotyping studies suggesting that the HTSO method provides better quality DNA compared to the phenol–chloroform method. Previous studies have shown that the low quality of DNA during the extraction process is due to impurity or cellular debris, which results in inhibition of PCR reactions and degradation of DNA on long-term storage (Singh et al. 2018).
The HTSO also gave more yield and better DNA purity compared to 96-well-based protocols such as the manual in-house (Saavedra-Matiz et al. 2013; St Julien et al. 2013), commercial kits (Sirdah 2014), and automation-based high-throughput methods (Zhili et al. 2005; Ellen et al. 1999). Perhaps single-step lysis of cellular components using concentrated extraction buffer and fewer washing steps helped in minimal sample loss and better DNA yield, as opposed to multiple lysis and washing steps resulting in loss of DNA. The HTSO method has proven cost-effective and time-saving compared to commercial kits (sbeadex™ Kit by LGC Biosearch Technologies). Though automation-based high-throughput methods are faster, they require exclusive robotic systems, which are less affordable for low- to mid-scale laboratories or institutions (Table 1).
The archived DBS samples of GA-I patients also gave good DNA yield and purity when extracted by the HTSO method suggesting that this method can efficiently extract DNA from stored samples. The consistency in intra and inter assays suggest that the HTSO is a reliable and reproducible method. The variation in the DNA yield between DBS samples is due to the difference in the number of blood cells on the filter paper matrix (Caboux et al. 2012).
The DNA extracted from GA-I patients by HTSO method did not cause any difficulties during genotyping multiple mutations, R402W and W225X in the GCDH gene. Overall, the HTSO method gave a twofold increased yield of DNA, help to detect two mutations in a single run, and grossly reduced the genotyping time compared to other high-throughput methods. We were unable to test the HTSO protocol on an automated high-throughput instrument due to financial constraints and the unavailability of the robotic system. Nonetheless, the HTSO method could be well-designed for the automated high-throughput system as well.
The genotyping study revealed that the frequency of mutant “T” allele of R402W mutation is 17.5% (7/40), which is similar to the frequency reported among Europeans (10–20%) (Heringer et al. 2015). Previous studies with GA-I patients have shown mutant “T” allele frequency of 15%, 23.5%, and 18.8% for R402W mutation (Shaik M et al. 2016; Tp KV et al. 2017; Gupta et al. 2015). A study from India suggested the absence of R402W mutation in GA-I patients from South India (Gupta et al. 2015). In our study, 78% of GA-I patients from South India had R402W mutation, indicate the prevalence of this mutation in South India as well. For the first time, in a study with 12 GA-I patients, Radha Rama Devi et al. (2016) reported W225X mutation in the Indian population with mutant “A” allele frequency of 16% (4/24). In our study, 10% (2/20) of the patients were homozygous for W225X mutation, making mutant “A’ allele frequency of 10% (4/40). The difference in allelic frequency for R402W and W225X mutations in different studies is due to the difference in the cohort size. In the present study, only GA-I patients from South India had W225X mutation. None of the GA-I patients from North India had this mutation, suggesting that this mutation is probably prevalent only in GA-I patients of Southern origin.
In conclusion, the HTSO is a successful large-scale and potential high-throughput DNA extraction protocol for DBS samples formulated based on the DNA salting-out principle. The method is proven to be simple, low-cost, and time-efficient. As HTSO is an affordable method, it can be easily incorporated into institutions and corporates with limited resources. Further, the HTSO is considered non-toxic as no hazardous chemicals such as phenol or chloroform was used during the DNA extraction process. A pilot genotyping study reveals that the yield and quality of DNA isolated from DBS samples by HTSO method was suitable for genetic studies. For the first time, two mutations in the GCDH gene of GA-I patients were screened simultaneously in a single reaction mixture, which implies that multiple genotyping is strategically possible and proves economical in the genetic diagnosis of GA-I disorder by incorporating methods like the HTSO. Overall, the HTSO is efficient, reliable, and superior to other 96-well-based methods with similar output.
References
Caboux E, Lallemand C, Ferro G et al (2012) Sources of pre-analytical variations in yield of DNA extracted from blood samples: analysis of 50,000 DNA samples in EPIC. PLoS One 7: e39821
Chaisomchit S, Wichajarn R, Janejai N (2005) Stability of genomic DNA in dried blood spots stored on filter paper. Southeast Asian J Trop Med Public Health 36:270–273
Choi EH, Lee SK, Ihm C, Sohn Y-H (2014) Rapid DNA extraction from dried blood spots on filter paper: potential applications in biobanking. Osong Public Health Res Perspect 5:351–357
De Vries JJ, Claas EC, Kroes AC, Vossen AC (2009) Evaluation of DNA extraction methods for dried blood spots in the diagnosis of congenital cytomegalovirus infection. J Clin Virol 46:37–42
Edelbroek PM, van der Heijden J, Stolk LM (2009) Dried blood spot methods in therapeutic drug monitoring: methods, assays, and pitfalls. Ther Drug Monit 31:327–336
Gupta N, Singh PK, Kumar M et al (2015) Glutaric acidemia type 1-clinico-molecular profile and novel mutations in GCDH gene in Indian patients. JIMD Rep 21:45–55
Guthrie R, Susi A (1963) A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32:338–343
Heath EM, O’Brien DP, Banas R et al (1999) Optimization of an automated DNA purification protocol for neonatal screening. Arch Pathol Lab Med 123:1154–1160
Hedlund GL, Longo N, Pasquali M (2006) Glutaric acidemia type 1. Am J Med Genet C Semin Med Genet 142C:86–94
Heringer J, Boy N, Burgard P et al (2015) Newborn screening for glutaric aciduria type I: benefits and limitations. Int J Neonat Screen 1:57
Hollegaard MV, Grove J, Grauholm J et al (2011) Robustness of genome-wide scanning using archived dried blood spot samples as a DNA source. BMC Genet 12:58
Hollegaard MV, Grove J, Thorsen P et al (2009) High-throughput genotyping on archived dried blood spot samples. Genet Test Mol Biomarkers 3:173–179
Hue NT, Phong PT, Chan NDH et al (2011) An efficiency human genomic DNA extraction from dried blood spots. Procedia Environ Sci 8:179–185
Hue NT, Chan NDH, Phong PT et al (2012) Extraction of human genomic DNA from dried blood spots and hair roots. Int J Biosci Biochem Bioinform 2:21–26
Kalendar R, Boronnikova S, Seppänen M (2021) Isolation and Purification of DNA from Complicated Biological Samples. Methods Mol Biol 2222:57–67
Kumar A, Mhatre S, Godbole S, Jha P et al (2019) Optimization of extraction of genomic DNA from archived dried blood spot (DBS): potential application in epidemiological research & bio banking. Gates Open Res 14:57
Lin Z, Suzow JG, Fontaine JM, Naylor EW (2005) A simple automated dna extraction method for dried blood specimens collected on filter paper. JALA 10:310–314
McCabe ER (1991) Utility of PCR for DNA analysis from dried blood spots on filter paper blotters. PCR Methods Appl 1:99–106
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Mössner BK, Staugaard B, Jensen J et al (2016) Dried blood spots, valid screening for viral hepatitis and human immunodeficiency virus in real-life. World J Gastroenterol 22:7604–7612
Panteleeff DD, John G, Nduati R et al (1999) Rapid method for screening dried blood samples on filter paper for human immunodeficiency virus type 1 DNA. J Clin Microbiol 37:350–353
Parker SP, Cubitt WD (1999) The use of the dried blood spot sample in epidemiological studies. J Clin Pathol 52:633–639
Radha Rama Devi A, Ramesh VA, Nagarajaram HA et al (2016) Spectrum of mutations in glutaryl-CoA dehydrogenase gene in glutaric aciduria type I–study from South India. Brain Dev 38:54–60
Saavedra-Matiz CA, Isabelle JT, Biski CK et al (2013) Cost-effective and scalable DNA extraction method from dried blood spots. Clin Chem 59:1045–1051
Shaik M, Shivanna DK, Kamate M et al (2016) Single lysis-salting out method of genomic DNA extraction from dried blood spots. J Clin Lab Anal 30:1009–1012
Singh UA, Kumari M, Iyengar S (2018) Method for improving the quality of genomic DNA obtained from minute quantities of tissue and blood samples using Chelex 100 resin. Biol Proced Online 1:12
Sirdah MM (2014) Superparamagnetic-bead based method: an effective DNA extraction from dried blood spots (DBS) for diagnostic PCR. J Clin Diagn Res 8:FC01–FC04
St Julien KR, Jelliffe-Pawlowski LL, Shaw GM et al (2013) High Quality Genome-Wide Genotyping from Archived Dried Blood Spots without DNA Amplification. PLoS One 8:e64710
Tp KV, Muntaj S, Devaraju KS et al (2017) Genetic screening of selected disease-causing mutations in Glutaryl-CoA Dehydrogenase gene among Indian patients with Glutaric Aciduria type I. J Pediatr Genet 6:142–148
Wilhelm AJ, den Burger JC, Swart EL (2014) Therapeutic drug monitoring by dried blood spot: progress to date and future directions. Clin Pharmacokinet 53:961–973
Acknowledgements
We acknowledge the Science and Engineering Research Board (SERB) under the Department of Science and Technology (DST), Government of India for funding this work. Registration No: SERB/LS-771/2012. We also acknowledge all the clinicians for providing patients samples, GA-I patients, and their families who participated in this study.
Funding
The Science and Engineering Research Board (SERB) under the Department of Science and Technology (DST), Government of India funded this work (Registration no: SERB/LS-771/2012).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest was declared by the authors.
Rights and permissions
About this article

Cite this article
Shaik, M., Alladi, A., Vedamurthy, A. et al. Large-scale Extraction of DNA by Using Salting-out Principle for Dried Blood Spots to Screen Multiple Mutations in GCDH Gene. Iran J Sci Technol Trans Sci 46, 33–40 (2022). https://doi.org/10.1007/s40995-021-01225-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40995-021-01225-x