
Abstract
Purpose
Liver regeneration is an orchestrated process that mainly comprises of the proliferation of the major liver cell types, that is, the hepatocytes and cholangiocytes, after liver injury (physical or chemical) in vivo. Although having a remarkable capacity to regenerate in vivo, hepatocytes are difficult to grow and maintain in culture as their viability and functions decline with time. The lack of a sufficient source of viable hepatocytes limits their clinical use for therapeutic applications.
Methods
In the current review, we have summarized the role of bile acids and their subsequent signaling pathways in liver regeneration in terms of both hepatocyte and cholangiocyte proliferation. We have also reviewed bile acid–based therapies in liver diseases.
Results
The expression of two major bile acid receptors, the farnesoid X receptor (FXR) and the Takeda G protein–coupled receptor (TGR5) in both the liver and the intestine immensely contribute to hepatocyte proliferation through varied mechanisms. Selective potent agonists of these two pathways are being synthesized for use as new therapies in several liver diseases.
Conclusion
FXR/TGR5 agonists hold immense potential to facilitate liver regeneration and ameliorate hepatic insufficiency in chronic liver diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The metabolic function of bile acids synthesized in the liver is to emulsify fats and fat-soluble vitamins in the intestine. Recent studies have now suggested that physiological concentrations of bile acids also modulate regenerative responses of the liver via the FXR and TGR5 signaling pathways in both the liver and intestinal cells. Agonists of these pathways are gaining attention as key stimulators of hepatocyte and cholangiocyte proliferation. They hold potential as therapeutic molecules in liver injury by affecting liver homeostasis, inflammation, and regeneration and should be investigated in future in the in vitro and in vivo studies.
Introduction
The liver is a vital organ that performs more than five hundred biological functions. It is the only visceral organ that has the capacity to regenerate in vivo. Liver regeneration can occur in both physiology and pathophysiology. Physiological liver regeneration comprises of the proliferation of healthy hepatocytes after surgical resection of some part of the liver, termed as partial hepatectomy (PHx) while pathophysiological regeneration means the proliferation of remaining hepatocytes/biliary epithelial cells (BECs) or cholangiocytes when some/most of the hepatocytes undergo replicative senescence [1]. The hepatocytes are among the major parenchymal cells that can re-enter the cell cycle and mount an efficient liver regeneration in response to partial ablation or liver injury. It is only during substantial hepatocyte loss, a population of BECs come into play and can act as facultative liver stem cells (LSCs) to repopulate the liver parenchyma [2, 3]. Liver regeneration is driven by a set of complex signals and factors released by the nonparenchymal liver cells like sinusoidal endothelial cells and Kupffer cells that activate tightly regulated signaling mechanisms in the parenchymal liver cells in a time-dependent fashion [4, 5]. The important signals released include metabolic signals, growth factors, cytokines, etc. that induce the expression of downstream target genes by activating the key transcription factors [1]. Some of the growth factors include hepatocyte growth factor (HGF), Wnt2, vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and IL-6 (Figure 1) [6]. Recently, bile acids (BAs) and their subsequent signaling pathways have been identified as one of the key endogenous metabolic signals affecting liver regeneration and homeostasis. In this review, we have summarized the BA signaling pathways in liver regeneration (majorly hepatocyte regeneration) and highlight how BA-based therapies could be developed for attaining an efficient hepatocyte proliferation and liver regeneration both in vitro and in vivo.

Physiological and pathophysiological liver regeneration: Physiological regeneration occurs after partial hepatectomy by the remaining healthy hepatocytes. Specific growth factors are released from the nonparenchymal liver cells including the sinusoidal endothelial cells, hepatic stellate cells, and Kupffer cells that drive the regeneration of hepatocytes. In case of liver injury, pathophysiological regeneration occurs, where the hepatocyte proliferation is impaired due to their massive loss and a subset of biliary epithelial cells, termed as the liver progenitor cells (oval cells in mice) get activated and govern the regeneration of the hepatic tissue. HGF, hepatocyte growth factor; WNT2, wingless-type MMTV integration site family, member 2; VEGF, vascular endothelial growth factor; EGF, epidermal growth factor; IL-6, interleukin 6; TNF-α, tumor necrosis factor-α; TWEAK, tumor necrosis factor (TNF)-related weak inducer of apoptosis; FGF-7, fibroblast growth factor 7, Wnt3a, wingless-type MMTV integration site family, member 3A
Bile Acids: Chemistry and Transport
BAs are steroids with 24 carbon atoms that contain cyclopentanoperhydrophenanthrene ring in their nucleus [7]. They are amphiphilic molecules synthesized from cholesterol degradation in the hepatocytes. BAs are classified into primary and secondary BAs and also conjugated and unconjugated. Primary unconjugated BAs such as cholic acid (CA) are synthesized in the liver by the classical pathway, which is initiated by the rate-limiting microsomal enzyme, cholesterol 7α-hydroxylase (CYP7A1), yielding 7α-hydroxycholesterol. The other pathway for BA synthesis is the alternative or the acidic pathway. This pathway is initiated by the mitochondrial sterol 27-hydroxylase (CYP27A1) enzyme and results in the formation of BAs such as chenodeoxycholic acid (CDCA)[8]. In humans, the classic pathway is thought to be the major BA biosynthesis pathway in physiological conditions. Once synthesized in the liver, they are transported across the canalicular membrane, modified in the cholangiocytes, and transported to the gallbladder, where they are stored. After ingestion of food, the release of BAs from the gall bladder into the digestive tract is provoked by the cholecystokinin hormone to aid in the absorption of food, nutrients, lipids, and fat-soluble vitamins [9, 10]. After their use in the intestine, BAs are efficiently reabsorbed via specific BA transporters in the ileum, and then carried back to the liver via the portal blood. This process is referred to as the enterohepatic circulation of the bile. In the intestine, primary BAs are converted into secondary BAs such as deoxycholic acid (DCA) and lithocholic acid (LCA) by the gut microbial enzymes [11]. The chemical structures, physiological functions, and other known effects of major BA species are described in Table 1.
In humans, about 0.2–0.6 g of BAs is being synthesized per day in the hepatocytes and is transported across the canalicular membrane of hepatocytes and pump out by bile salt export pump (BSEP) before being stored into the gallbladder and then into the small intestine. In the small intestine, about 95% of BAs are reabsorbed into enterocytes and sent back to the liver through the portal vein, and only 5% is excreted through faces, which is compensated by de novo BA synthesis in the liver. Most of the BAs are reabsorbed at the terminal ileum, where active uptake of conjugated BAs occurs through the apical sodium-dependent BA transporter (ASBT) [9]. BAs are taken up by the hepatocytes from portal blood through the two BA transporters, Na-dependent taurocholate transporter (NTCP), and organic anion transporters (OATPs) [10]. About 4–12 cycles of BA circulation take place between liver and intestine per day [9]. A summary of BA synthesis and transport in given in Figure 2.

Synthesis and transport of bile acids: Cholesterol breakdown results in the formation of primary bile acids, cholic acid, and chenodeoxycholic acid either by the classical pathway mediated by cytochrome p450 7A1(CYP7A1) or the alternative pathway accomplished by cytochrome p450 27A1(CYP27A1). Primary bile acids get conjugated by glycine or taurine (G/T) before they are secreted to the biliary ductules. Deconjugation and re-conjugation mediated by the gut microbiota in the distal ileum modify the primary bile acids forming the secondary bile acids, lithocholic acid (LCA), or deoxycholic acid (DCA). NTCP, sodium taurocholate cotransporting polypeptide; BSEP, bile salt exchange pump; ASBT, apical sodium-dependent bile acid transporter; OSTα-β, organic solute transporter α-β
Bile Acid Receptors and Signaling Pathways
In the last decade, several findings have revealed the role of BAs beyond emulsifiers and detergents of dietary fats and liposoluble vitamins. The discovery of specific BA cellular receptors such as the nuclear receptor, “farnesoid X receptor” (FXR), and the membrane receptors, including G protein–coupled receptor (TGR5) and pregnane X receptor (PXR) which respond to BAs, have led to the elucidation of BAs as crucial endocrine and paracrine signaling molecules affecting versatile functions [12]. Here we discuss the metabolic and physiological role of two important receptors, FXR and TGR5.
Physiological Functions of FXR and TGR5
The farnesoid X receptor (FXR) is the first identified nuclear receptor of BAs [12]. FXR is expressed in different cells of the liver, namely, hepatocyte, liver sinusoidal cells, hepatic stellate cells, and Kupffer cells, while in the intestine, it is highly expressed in the ileal enterocytes [13,14,15]. The FXR signaling pathway maintains the metabolic homeostasis affecting BA, cholesterol, and glucose metabolism[8]. The potency of FXR activation by different BAs, CDCA>DCA>LCA>CA [16]. In hepatocytes, FXR induces the expression of the small heterodimer partner (SHP) transcriptional repressor. SHP negatively interacts with other transcription factors, such as liver receptor homolog-1 and hepatocyte nuclear factor-4α (HNF-4α), that bind to the bile acid response elements located within the promoter region of the CYP7A1 and CYP8B1 genes, resulting in repression of BA synthesis. In the intestine, FXR regulates the absorption of BAs, vitamins, certain drugs, and xenobiotics by increasing the expression of the ASBT, organic solute transporters (OSTα and OSTβ), which are responsible for the enterohepatic circulation of BAs[17, 18].
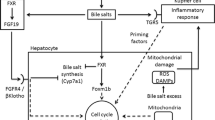
Takeda G protein–coupled receptor (TGR5) is a G protein–coupled receptor, also known as a G protein–coupled BA receptor 1 (GPBAR1). The receptor is widely distributed in tissues, such as the endocrine glands, gall bladder, adipocytes, muscles, intestine, and liver. In the liver, TGR5 is expressed in the cholangiocytes, sinusoidal endothelial cells, and Kupffer cells, but not in hepatocytes. BAs activate the TGR5 in a dose-dependent manner, in the order of potency: LCA>DCA>CDCA>CA. BAs bind with the TGR5 and activate adenyl cyclase to convert ATP to cAMP that induces protein kinase A to activate cAMP element response binding protein (CREBP), activating cAMP signaling pathways [19, 20]. Several studies have reported that TGR5 activation is associated with a transactivation of the EGFR [21, 22] and with the production of reactive oxygen species (ROS) [23], linking TGR5-dependent BA signaling to cell proliferation and apoptosis in different cancer cell lines and tumors [24]. As a crucial regulator of energy homeostasis, TGR5 has been reported as a potential target for the treatment of metabolic syndrome and its complications, including NASH [25, 26]. Given its high expression in the cholangiocytes and the gall bladder epithelial cells, TGR5 activation has been suggested to regulate CFTR-dependent Cl secretion via the regulation of chloride and bicarbonate transport in bile, reduce BA protonation, and protect bile duct cells and liver parenchyma from bile cytotoxicity [27,28,29]. TGR5 activation by BAs stimulates nitric oxide (NO) production in liver endothelial cells [30] and decreases LPS-induced cytokine gene induction in the Kupffer cells and macrophages [31, 32]. In the intestinal epithelial cells, BAs activate TGR5 to release glucagon-like peptide-1 (GLP-1) from enteroendocrine L cells, which stimulate pancreatic β cells to secrete insulin and decreases insulin tolerance [20]. TGR5 activation by BAs also increases the cAMP production in alveolar macrophages, which decreases the phagocytic activity and production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1α, IL-β, IL-6, and IL-8 [33].
Bile Acid Signaling and Liver Regeneration: The Role of FXR
The role of BAs in liver regeneration has been mostly highlighted in the PHx models, where hepatocytes are the major liver cells that undergo self-renewal. In 70% PHx models where more than 50% of the liver is resected, there is an acute overload of BAs in remaining liver tissue via enterohepatic circulation due to the immediate decrease in total liver functional mass. Studies have documented that both the pool size and composition of BAs are altered after PHx [34]. A relative decrease of CDCA and secondary BAs as compared with an increase in more hydrophilic CA has been observed in the first days after PHx in the rat.
The BA receptors act synergistically to facilitate both the regeneration process and also protecting the remnant liver and maintain biliary homeostasis. The remnant hepatocytes deal with excessive intracellular BA concentrations through the FXR-dependent adaptive pathways [34, 35]. Increased BA load activates FXR to increase the mRNA levels of a gene, SHP, which is a negative regulator of Cyp7a1 gene expression. A decreased expression of Cyp7a1 shuts down the BA synthesis in the liver. FXR also decreases the expression of basolateral BA transporter NTCP that allows BA uptake in the liver. Furthermore, the basolateral exit of BAs from the hepatocytes is strongly upregulated, through major induction of MRP3 and OSTa/b [34, 36]. These FXR-dependent adaptive responses are central to protect the regenerating liver in PHx. The inhibition of BA synthesis on its own (through both FXR-dependent and independent mechanisms) has also been reported to be a crucial step for the initiation of hepatocyte proliferation after CCl4 intoxication [37].
In addition to handling the BA overload, FXR stimulation in the hepatocytes by the BAs signals hepatocyte protection and division after PHx or even liver injury [38, 39]. Upon CCl4-induced toxic injury, the FXR null mice have been reported to exhibit a defective liver repair than the wild-type mice [39]. FXR stimulates hepatocyte cell cycle progression mainly by an activation of the major transcription factor, FOXM1B [38]. FOXM1B upregulates the expression of cell cycle proteins and cyclin-dependent kinase 2 (CDK2) and CDK1 cell cycle activity (Figure 3) [40, 41]. FXR activation is known to facilitate hepatocyte proliferation through the induction of pyruvate dehydrogenase kinase 4 (PDK4) gene as well as the subsequent metabolic reprogramming for rapid biomass generation, linking between liver regeneration and metabolic switch [42]. In addition to liver, intestinal FXR expression is also induced after PHx or liver injury. One critical FXR target gene in the intestine is FGF15. The induction of intestinal FXR increases the expression of its target gene, FGF15, a growth factor, in the ileum. FGF15 has also been identified as an important regulator of liver homeostasis and regeneration both after PHx and alcohol-related liver injury [43, 44]. In hepatectomized mice, genetic knockout of intestinal FXR reduces FGF-15 in the ileum and increases the expression of Cyp7a1 (Figure 3) [40, 45]. In FGF-15 knockout mice, the expression of protein levels JAK1 (Janus kinase 1), JAK2, and STAT3 (signal transducer and activation of transcriptional 3) is also downregulated in the liver tissues, indicating that FGF-15 modulates hepatocyte proliferation through JAK/STAT pathway [40]. The positive impact of BAs on liver regeneration has also been reinforced by BA supplementation and BA sequestering resin feeding experiments in different models of liver regeneration [38, 39, 46]. The feeding of 0.2% cholic acid, an agonist of FXR to mice, indeed favors liver regrowth after PHx, causing a 30% increase in the liver size. On the other hand, the use of BA resins that lead to sequestering of BAs inhibit regeneration and decrease the rate of liver growth and BrdU-positive nuclei in the hepatocytes [38]. In the acetaminophen-induced liver injury mouse model, feeding of cholic acid also enhances liver regeneration [46].

FXR-mediated liver regeneration: After a liver injury or partial hepatectomy, the increased bile acid flux activates the farnesoid X receptor (FXR) in the liver and the intestine. FXR suppresses liver CYP7A1 thus lowering the BA production and triggering the activation of FOXM1B which ultimately promotes liver regeneration. In the intestine, FXR activation induces the expression of FGF15 that binds to its receptor FGFR4 in the liver and activates the signaling pathway to promote liver regeneration
In mice, with an inhibited BA flux to the liver via the enterohepatic circuit, there is a deficient liver regeneration. For example, in rodents with a deficiency of intestinal Mrp3, expressed in the basolateral membrane, there was a reduced liver growth elicited by cholic acid feeding. These animals showed reduced FXR activation in the liver after CA administration and decreased portal serum levels of BAs [47]. In rats, knockout for ASBT mediates the reabsorption of BAs from the apical side of the ileum, hepatocyte proliferation is similarly attenuated after PHx as well as after CCl4 intoxication [48] in human patients undergoing hemihepatectomy with external bile drainage by cystic duct tube), and those having hemihepatectomy without drainage, the regenerated liver volumes on day 7 after hepatectomy were significantly greater in the latter indicating that external biliary drainage should be used judiciously after liver resection [49]. Although BAs and FXR agonists are known to trigger hepatocyte proliferation, their impact on stem cell-dependent models of liver regeneration, i.e., models in which hepatocytes are massively destroyed remain elusive [50].
Bile Acid Signaling and Liver Regeneration: The Role of TGR5
As compared to FXR, the role of TGR5 in liver regeneration and hepatocyte proliferation has been scarcely studied, precisely because TGR5 expression is absent in the hepatocytes. The role of TGR5 is mainly studied as a protector of BA overload after PHx. In systemic TGR5-KO mice, PHx is followed by massive cholestasis and hepatocyte necrosis, and also liver regeneration is markedly delayed as compared with wild-type mice [36].
The mechanisms through which TGR5 would efficiently protect the liver against this BA overload post-PHx are many. TGR5 contributes to fine-tuning of cytokine production, which is absent in TGR5 KO animals. TGR5 modulates the composition of BAs as TGR5-KO mice exhibit a more hydrophobic BA, suggesting that too much hydrophobic BAs accumulating in the liver of the systemic TGR5-KO mice immediately after PHx lead to liver injury [36]. It is well-known that a shift towards a more hydrophobic pool is associated with a diminished liver regeneration. The observed TGR5-dependent post-PHx increase in biliary HCO3– and Cl– output may be a part of an adaptive mechanism enhancing bile secretion and bile fluidity, thereby protecting the overloaded remnant liver from BA toxicity. TGR5 also enhances the expression of MRP2 and MRP4 in the kidneys and hence the BA efflux in the urine under conditions of BA overload in PHx, thus contributing to protect the liver from BA overload after PHx [36]. Post-PHx, BA-mediated effects via TGR5 might be occurring through the cholangiocytes or other nonparenchymal cells of the liver. For example, TGR5 might be allowing the liver to adapt to a change in ion composition in the bile after PHx, via cholangiocytes, in line with the evidence that TGR5 controls CFTR-dependent chloride secretion in different epithelial cells [51].
Although the direct effects of TGR5 in liver regeneration are less studied, it is to be appreciated that TGR5 agonists activate several signaling pathways pertaining to cell survival, development, and proliferation [52]. TGR5 agonists are known to enhance the AKT phosphorylation and increase NO production in vascular endothelial cells[53]. The activation of TGR5 has been documented to negatively regulate the hepatic inflammatory response through antagonizing NF-κB [25, 32]. The effects of BAs and TGR-5-mediated signaling on cholangiocytes have been discussed separately.
Bile Acid-Based Therapies for Liver Regeneration and Hepatocyte Proliferation
Despite the regenerative abilities of the liver in vivo, its major type of cells, hepatocytes, do not replicate and proliferate very efficiently in culture. Primary hepatocyte culture is the most challenging part of hepatic tissue engineering as they are short-lived in culture (lose their viability and functions with time) and also can dedifferentiate into other cell types. Thus, robust strategies for culturing primary hepatocytes are being explored since long time. Hepatocyte function is plastic and largely regulated by the surroundings, cells, molecules, and the extracellular matrix (ECM). In recent years, several studies have devised new approaches, including cellular, extracellular, and molecular to reproduce the exact in vivo environment and thus achieve a sustained and appreciable hepatocyte growth ex vivo [54]. Bile acid–based therapies offer considerable potential for both in vivo and in vitro hepatocyte regeneration. Taurocholate has shown to accelerate the bile canalicular network formation in rat hepatocyte sandwich cultures and concomitantly increase cAMP, prevented by adenyl cyclase inhibitor [55]. Tauroursodeoxycholic acid upregulates SIRT1-FXR activity, increasing FoxM1 expression, leading to decreased apoptosis and increased proliferative capacity in hepatocytes [56]. A study suggests that the physiological concentrations of BAs such as tauroursodeoxycholic acid also initiate hepatic differentiation of MSC via the farnesoid X receptor both in vitro and in vivo [57]. The hepatic differentiation of the cells has been shown to be accompanied by a decreased expression of mesodermal markers such as desmin and the transient acquisition of the expression profile of hepatic progenitor cells before they differentiate into hepatocyte-like cells. The study clearly shows the relevance of using naturally occurring or modified BAs that activate the FXR pathway as a novel tool to generate hepatocytes in vitro and also in vivo. This method could be used in conjugation with the growth factor treatment to facilitate the generation of patient-specific hepatocytes from stem cells for future therapeutic use. The surface modification of the biomaterials with versatile chemical functionalities offers enormous advantages in cell and tissue cultures. It would be intriguing to develop newer biomaterials functionalized with BAs for efficient hepatocyte cultures. Adding an appropriate BA to the biomaterial may help in creating a specific chemical and physical environment favorable for hepatocyte regeneration [58]. Studies have shown that the use of unconjugated BAs at higher concentrations is toxic to the primary hepatocyte cultures. Thus, an appropriate concentration range and specific BA species and composition which favor hepatocyte regeneration need to be identified. Also, functional hepatocytes are capable of biosynthesizing and excreting BAs in culture. Hence, endogenous levels of BAs in culture should be kept in mind before supplementing the hepatocytes with exogenous BA species.
Moreover, besides the known BAs, the contribution of agonists of the FXR and TGR5 pathways should be evaluated in vitro and in vivo. FXR agonists prevent cell death and promote hepatocyte proliferation and differentiation in vitro. Several potent and selective FXR agonists, GW4064, WAY-362450, etc., are being explored as potential therapeutic agents in hepatobiliary disease. GW4064 induces expressions of the cell survival gene and protein, p62/SQSTM1 in AML12 mouse liver cells [59]. The activation of FXR by GW4064 also has a pro-proliferative effect as it decreases the number of tetraploid (or binucleated) hepatocytes in vitro [60]. GW4064 also provides a significant protection to the cultured hepatocytes against cisplatin-induced toxicity [61]. Obeticholic acid (OCA), a 6α-ethyl derivative of the natural human BA chenodeoxycholic acid (CDCA) is the first-in-class selective FXR agonist that is ~100-fold more potent than CDCA. OCA is a potential therapeutic agent for nonalcoholic fatty liver diseases and also cholestatic diseases. Primary hepatocytes when treated with OCA display increased viability and secretion of FGF19 in cultures [62]. OCA has also been shown to suppress metabolic stress-induced p53 activation and cell death in hepatocytes [63]. Several new synthetic compounds have been synthesized that activate the TGR5 signaling. 6α-ethyl-23(S)-methyl-cholic acid (6-EMCA, INT-777) had been discovered as a selective, specific agonist for TGR5 with anti-inflammatory functions in the macrophages [64]. A small compound WB403 has been demonstrated to activate TGR5 and promote GLP-1 secretion [26]. TGR5-based therapies hold relevance for the proliferation of cholangiocytes or BECs.
Bile Acids and Biliary Epithelial Cells/Cholangiocytes
Cholangiocytes or biliary epithelial cells (BECs) are both morphologically and functionally heterogeneous along the network of bile ducts or the biliary tree. In the liver, small and large cholangiocytes are present according to the duct size. Small and large cholangiocytes differ in their nuclear-to-cytoplasmic ratio; this ratio is greater in small cholangiocytes, suggesting that they are less differentiated and have greater plasticity as compared to large cholangiocytes. The major physiological function of large cholangiocytes is the modification of hepatic bile along the biliary tree. The intrahepatic cholangiocytes lining the biliary tree secrete water and electrolytes [65]. In response to secretin binding with secretin receptor (SR) on these cells, there is an increase in cAMP synthesis, secretin-stimulated bile flow, HCO3− secretion. Cholangiocytes are normally in a quiescent state. In experimental models of ductal hyperplasia, such as bile duct ligation (BDL) or 70% hepatectomy, cholangiocytes proliferate markedly, leading to enlargement of intrahepatic ductal mass. Cholangiocyte proliferation is closely coupled with increased DNA synthesis, SR gene expression, and secretin-induced cAMP synthesis. Small cholangiocytes have also been postulated to represent a functional hepatobiliary progenitor cell population or liver progenitor cells (LPCs). In the face of massive hepatocyte loss, studies have shown that this population of BECs can repopulate the liver parenchyma [2]. During chronic liver injury, the normally quiescent resident LPCs are activated and expanded from the periportal to the pericentral zone of the liver, producing reactive ductules or ductular reaction in humans [65]. Reactive ductules or ductular reactions (DRs) refer to an increased number of ductules (the finest ramifications of the biliary tree), representing a proliferation of the BECs. The contribution of LPCs or BECs to hepatocytes during liver injury is dependent on impaired regenerative ability and enhanced senescence of the hepatocytes [66]
Since BECs are continuously exposed to endogenous BAs, many studies have reported that BAs alter the growth and functions of BECs both in vitro and in vivo. BAs such as taurocholate (TC) and taurolithocholate (TLC) have been shown to increase DNA synthesis, SR gene expression, and secretin-stimulated cAMP levels in the large cholangiocytes [67]. In vivo treatment of rats with BAs such as TC or TLC (1% for 1–4 weeks) has also been reported to cause a 2–3-fold increase in the total BA concentrations in the bile but not in the serum [68]. The study has demonstrated an increased number of PCNA-positive cholangiocytes, number of bile ducts, and [3H]thymidine incorporation in cholangiocytes in BA-treated rats. In BA-fed rats, SR gene expression and cAMP levels were also enhanced in cholangiocytes, which was associated with de novo secretin-stimulated bile flow and bicarbonate secretion. Importantly, there was no biochemical or histological evidence of hepatic damage or cholestasis in these animals after BA treatment [69]. TGR5 is required for BA-induced cholangiocyte proliferation in animal models as well as in isolated biliary epithelial cells [24]. Using isolated cholangiocytes, it has been demonstrated that TGR5 promotes cell proliferation through elevation of reactive oxygen species (ROS) and subsequent activation of Rous sarcoma oncogene (cSrc), epidermal growth factor receptor (EGFR), and Erk1/2. Under cholestatic conditions, TGR5-deficient mice showed reduced cholangiocyte hyperplasia and diminished hepatocyte proliferation and increased liver injury, suggesting that TGR5 exerts protective effects in models of BA overload [24].
Besides TGR5-cAMP and EGFR/Erk1/2 signaling, YAP/TAZ pathway is also activated in the BECs in response to BA treatment [70]. YAP or Yes-associated protein along with transcriptional coactivator with PDZ-binding motif (TAZ) is one of the downstream proteins in the Hippo signaling pathway, an evolutionarily conserved serine/threonine kinase signaling cascade involved in controlling organ size and development. In a healthy liver, YAP is expressed in adult BECs or cholangiocytes and endothelial cells but not in hepatocytes. Although YAP is essential for adult biliary cell survival and proliferation in health and disease, YAP and TAZ are not required for the proliferation of hepatocytes during liver development and regeneration. They are however required to achieve efficient regenerative responses and complete restoration of liver mass following PHx [76.] They are known to play an indirect role in liver regeneration by preserving bile duct integrity and securing immune cell recruitment and function [71]. A recent study examined the effect of chronically administering DCA-supplemented feed on the proliferation of BECs. The study showed that there was a dramatic increase in the number of BECs expressing the YAP transcriptional program after only 24h even after an intraperitoneal treatment with physiological levels of DCA. The study showed that upon chronic liver injury, a subset of BECs expressed Wnt-associated genes along with mature hepatocyte markers signifying the plasticity of BECs in response to nonphysiological levels of DCA treatment. The study also deduced that YAP activation in BECs required the intracellular presence of the DCA and hence a BA transporter, i.e., ASBT [72]. YAP activation is also known to drive tumorigenesis and proliferation of tumor cells in response to elevated pathological concentrations of BAs of more than 100 μM [73]. These studies indicate that at higher concentrations or during BA overload in the liver, BA may act as important activators of BECs and LPCs. It would be worthwhile to investigate if specific BA treatment would also promote hepatic differentiation of BECs in chronically injured livers with massive hepatocyte loss.
Conclusion
Liver transplantation is a well-established strategy for end-stage liver disease and liver failure, but it is largely restricted by the acute shortage of suitable donor organs. In the past few decades, extracorporeal bioartificial support and in vivo cell-based therapies for patients with liver failure have been explored. However, both bioartificial liver and hepatocyte cell therapies have not achieved much clinical success given the limited functional ability and viability of hepatocytes under ex vivo conditions. The use of decellularized liver scaffolds, 3D bio-printing of hepatocytes, and nano-based 3D scaffolds for in vitro and in vivo applications are being investigated as novel approaches for hepatocyte regeneration. Given a crucial role of BAs as endogenous signals for liver regeneration, using BAs and their derivatives along with other growth factors holds promise and immense potential as a lucrative approach for accelerating hepatocyte proliferation. More in-depth studies that focus on identifying the precise BA species and concentration, designing of bile acid derivatives, etc. would certainly lead to the development of BAs as a therapy for replacing ailing hepatocytes in liver diseases and failure. Pharmacological targeting of the FXR and TGR5 pathway would also provide novel approaches to accelerate liver regeneration after liver transplantation or surgery.
References
Ozaki M. Cellular and molecular mechanisms of liver regeneration: proliferation, growth, death and protection of hepatocytes. In Seminars in cell & developmental biology. 2020;100:62–73.
Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol. 2015;17:971–83.
Gadd VL, Aleksieva N, Forbes SJ. Epithelial plasticity during liver injury and regeneration: Cell Stem Cell. 2020;27:557–573.
Forbes SJ, Newsome PN. Liver regeneration—mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol. 2016;13:473–85.
Gilgenkrantz H, de l'Hortet AC. Understanding liver regeneration: from mechanisms to regenerative medicine. Am J Pathol. 2018;188:1316–27.
Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res. 1999;59:3954–60.
Mukhopadhyay S, Maitra U. Chemistry and biology of bile acids. Curr Sci. 2004 Dec;25:1666–83.
Chiang JY. Bile acid metabolism and signaling. Comprehensive Physiology. 2013;3:1191–212.
Di Ciaula A, Garruti G, Baccetto RL, Molina-Molina E, Bonfrate L, Wang DQ, et al. Bile acid physiology. Ann Hepatol. 2018;16:4–14.
Chiang JY, Ferrell JM. Bile acid biology, pathophysiology, and therapeutics. Clinical liver disease. 2020;15:91–4.
Li T, Chiang JY. Regulation of bile acid and cholesterol metabolism by PPARs. PPAR research. 2009;2009:501739.
Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67.
Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. 2018;9:1853.
Honke N, Shaabani N, Hardt C, Krings C, Häussinger D, Lang PA, et al. Farnesoid X receptor in mice prevents severe liver immunopathology during lymphocytic choriomeningitis virus infection. Cell Physiol Biochem. 2017;41:323–38.
Li J, Wilson A, Kuruba R, Zhang Q, Gao X, He F, et al. FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc Res. 2008;77:169–77.
Fiorucci S, Distrutti E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol Med. 2015;21:702–14.
Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2011;1812:893–908.
Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368:17–29.
Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis. 2014;46:302–12.
Chiang JY, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2020;318:G554–73.
Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, et al. The bile acid receptor TGR5 does not interact with β-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem. 2013;288:22942–60.
Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun. 2007;354:154–9.
Hong J, Behar J, Wands J, Resnick M, Wang LJ, DeLellis RA, et al. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut. 2010;59:170–80.
Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65:487–501.
Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54(6):1263–72.
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–77.
Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Häussinger D. The membrane-bound bilacid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009 Sep;50(3):861–70.
Hohenester S, Maillette de Buy Wenniger L, Paulusma CC, van Vliet SJ, Jefferson DM, Oude Elferink RP, et al. A biliary HCO3− umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology. 2012;55:173–83.
Baghdasaryan A, Claudel T, Gumhold J, Silbert D, Adorini L, Roda A, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2−/−(Abcb4−/−) mouse cholangiopathy model by promoting biliary HCO output. Hepatology. 2011;54:1303–12.
Keitel V, Reinehr R, Gatsios P, Rupprecht C, Görg B, Selbach O, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704.
Keitel V, Donner M, Winandy S, Kubitz R, Häussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84.
Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology. 2011;54:1421–32.
Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis. 2011;29:37–44.
Csanaky IL, Aleksunes LM, Tanaka Y, Klaassen CD. Role of hepatic transporters in prevention of bile acid toxicity after partial hepatectomy in mice. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2009;297:G419–33.
Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochimica et Biophysica Acta (BBA)-Molecular. Cell Res. 2007;1773:283–308.
Péan N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. 2013;58:1451–60.
Zhang L, Huang X, Meng Z, Dong B, Shiah S, Moore DD, et al. Significance and mechanism of CYP7a1 gene regulation during the acute phase of liver regeneration. Mol Endocrinol. 2009;23:137–45.
Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–6.
Meng Z, Wang Y, Wang L, Jin W, Liu N, Pan H, et al. FXR regulates liver repair after CCl4-induced toxic injury. Mol Endocrinol. 2010;24:886–97.
de Haan L, van der Lely SJ, Warps AL, Hofsink Q, Olthof PB, de Keijzer MJ, et al. Post-hepatectomy liver regeneration in the context of bile acid homeostasis and the gut-liver signaling axis. Journal of clinical and translational research. 2018;4:1.
García-Rodríguez JL, Barbier-Torres L, Fernández-Álvarez S, Gutiérrez-de Juan V, Monte MJ, Halilbasic E, et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology. 2014;59:1972–83.
Xie Y, Wang H, Cheng X, Wu Y, Cao L, Wu M, et al. Farnesoid X receptor activation promotes cell proliferation via PDK4-controlled metabolic reprogramming. Sci Rep. 2016;6:1–1.
Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut. 2013;62:899–910.
Kong B, Huang J, Zhu Y, Li G, Williams J, Shen S, et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2014;306:G893–902.
Fan M, Wang X, Xu G, Yan Q, Huang W. Bile acid signaling and liver regeneration. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2015;1849:196–200.
Bhushan B, Borude P, Edwards G, Walesky C, Cleveland J, Li F, et al. Role of bile acids in liver injury and regeneration following acetaminophen overdose. Am J Pathol. 2013;183:1518–26.
Fernández-Barrena MG, Monte MJ, Latasa MU, Uriarte I, Vicente E, Chang HC, et al. Lack of Abcc3 expression impairs bile-acid induced liver growth and delays hepatic regeneration after partial hepatectomy in mice. J Hepatol. 2012;56:367–73.
Naugler WE, Tarlow BD, Fedorov LM, Taylor M, Pelz C, Li B, et al. Fibroblast growth factor signaling controls liver size in mice with humanized livers. Gastroenterology. 2015;149:728–40.
Otao R, Beppu T, Isiko T, Mima K, Okabe H, Hayashi H, et al. External biliary drainage and liver regeneration after major hepatectomy. Br J Surg. 2012;99:1569–74.
Alison MR, Lin WR. Diverse routes to liver regeneration. J Pathol. 2016;238:371–4.
Hendrick SM, Mroz MS, Greene CM, Keely SJ, Harvey BJ. Bile acids stimulate chloride secretion through CFTR and calcium-activated Cl− channels in Calu-3 airway epithelial cells. Am J Phys Lung Cell Mol Phys. 2014;307:L407–18.
Guo C, Chen WD, Wang YD. TGR5, not only a metabolic regulator. Front Physiol. 2016;7:646.
Kida T, Tsubosaka Y, Hori M, Ozaki H, Murata T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1663–9.
Kaur S, Tripathi DM, Venugopal JR, Ramakrishna S. Advances in biomaterials for hepatic tissue engineering. Current Opinion in Biomedical Engineering. 2020;13:A1–6.
Martinez-Diez MC, Serrano MA, Monte MJ, Marin JJ. Comparison of the effects of bile acids on cell viability and DNA synthesis by rat hepatocytes in primary culture. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2000;1500:153–60.
Sun S, Zhao B, Qi M, Yao Y, Xu L, Ji R, et al. TUDCA Ameliorates liver Injury via activation of SIRT1-FXR Signaling in a Rat hemorrhagic shock model. Shock. 2020;53:217–22.
Sawitza I, Kordes C, Götze S, Herebian D, Häussinger D. Bile acids induce hepatic differentiation of mesenchymal stem cells. Sci Rep. 2015;5:1–5.
Bose S, Robertson SF, Bandyopadhyay A. Surface modification of biomaterials and biomedical devices using additive manufacturing. Acta Biomater. 2018;66:6–22.
Haga S, Yimin OM. Relevance of FXR-p62/SQSTM1 pathway for survival and protection of mouse hepatocytes and liver, especially with steatosis. BMC Gastroenterol. 2017;17:9.
Wigger L, Casals-Casas C, Baruchet M, Trang KB, Pradervand S, Naldi A, et al. System analysis of cross-talk between nuclear receptors reveals an opposite regulation of the cell cycle by LXR and FXR in human HepaRG liver cells. PLoS One. 2019;14:e0220894.
Vaquero J, Briz O, Herraez E, Muntané J, Marin JJ. Activation of the nuclear receptor FXR enhances hepatocyte chemoprotection and liver tumor chemoresistance against genotoxic compounds. Biochim Biophys Acta. 1833;2013:2212–9.
Dash A, Figler RA, Blackman BR, Marukian S, Collado MS, Lawson MJ, et al. Pharmacotoxicology of clinically-relevant concentrations of obeticholic acid in an organotypic human hepatocyte system. Toxicol in Vitro. 2017;39:93–103.
Goto T, Itoh M, Suganami T, Kanai S, Shirakawa I, Sakai T, et al. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci Rep. 2018;8:8157.
Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–57.
Tabibian JH, Masyuk AI, Masyuk TV, O'Hara SP, LaRusso NF. Physiology of cholangiocytes. Comprehensive Physiology. 2013 Jan;3(1):541–65.
Kaur S, Siddiqui H, Bhat MH. Hepatic progenitor cells in action: liver regeneration or fibrosis? Am J Pathol. 2015;185:2342–50.
Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547:350–4.
Alpini GI, Glaser SH, Robertson WI, Phinizy JL, Rodgers RE, Caligiuri AL, et al. Bile acids stimulate proliferative and secretory events in large but not small cholangiocytes. American Journal of Physiology-Gastrointestinal and Liver Physiology. 1997;273:G518–29.
Alpini G, Glaser SS, Ueno Y, Rodgers R, Phinizy JL, Francis H, et al. Bile acid feeding induces cholangiocyte proliferation and secretion: Evidence for bile acid–regulated ductal secretion. Gastroenterology. 1999;116:179–86.
Lu L, Finegold MJ, Johnson RL. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med. 2018;50:e423.
Verboven E, Moya IM, Sansores-Garcia L, Xie J, Hillen H, Kowalczyk W, et al. Regeneration defects in yap and taz mutant mouse livers are caused by bile duct disruption and cholestasis. Gastroenterology. 2021;160:847–62.
Pepe-Mooney BJ, Dill MT, Alemany A, Ordovas-Montanes J, Matsushita Y, Rao A, et al. Single-cell analysis of the liver epithelium reveals dynamic heterogeneity and an essential role for YAP in homeostasis and regeneration. Cell Stem Cell. 2019;25:23–38.
Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, Moore DD. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013;5:1060–9.
Funding
The work was supported by the SRF project (45/5/2020-PHY/BMS) by the Indian Council of Medical Research, India.
Author information
Authors and Affiliations
Contributions
IK and RT drafted the manuscript and the figures, DMT edited the figures, VGM and SR provided valuable inputs, and SK edited and finalized the manuscript and the figures. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaur, I., Tiwari, R., Naidu, V. et al. Bile Acids as Metabolic Inducers of Hepatocyte Proliferation and Liver Regeneration. Regen. Eng. Transl. Med. 8, 200–209 (2022). https://doi.org/10.1007/s40883-021-00221-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40883-021-00221-2