
Abstract
Phosphate rocks mainly contain Ca3(PO4)2, SiO2, and MgO. The vacuum carbothermal reduction of Ca3(PO4)2 with different MgO contents was investigated to determine the maximum volatilization rate of P2 and the transformation of phases in a SiO2–C–Ca3(PO4)2–MgO-based system. The application of reduced slag in the dephosphorization of a converter was discussed. The carbon excess coefficient (CEC), reduction temperature, and equilibrium phase were calculated using FactSage 8.1. The quinoline phosphomolybdate weight method, scanning electron microscopy-energy dispersive X-ray spectroscopy, and X-ray diffraction were used to characterize the volatilization rate of P2, phases, and micromorphology of the reduced slag. The volatilization rate of P2 first increased and then decreased, with the highest volatilization rate of 93.77% obtained at n(MgO/CaO) = 0.50. The reduced slag phase changed from CaSiO3 to 2CaO·SiO2·MgO·SiO2, CaO·SiO2·MgO·SiO2, and 3CaO·P2O5·2CaO·SiO2 solid solutions with increasing MgO content. The diffusion of graphite was hindered by an excessive MgO solid phase when n(MgO/CaO) > 0.50, resulting in the reaction of unreacted Ca3(PO4)2 with 2CaO·SiO2·MgO·SiO2 to form 2CaO·SiO2·3CaO·P2O5 (C2S–C3P2) during the holding process, thus reducing the reduction rate of Ca3(PO4)2. Reduced slag with a small amount of phosphorus can be used in converter smelting to achieve low-temperature dephosphorization.
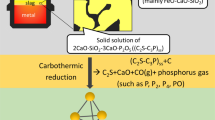
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phosphorus, a non-renewable resource, is commonly used in battery production, military operations, agriculture, phosphoric acid, fine chemicals, food, and medicine. Phosphate rock can be classified as high-grade (26–35% P2O5), medium-grade (17–25% P2O5), or low-grade (12–16% P2O5) [1]. The total phosphate reserves worldwide are 70 billion tons. Reserves of 50 billion tons in Morocco account for 71.43% of the total reserves, and these reserves are mainly high-grade phosphate rock (P2O5 > 30%) [2, 3]. Although China’s phosphate rock reserves rank second in the world, they only account for 4.7% of the total reserves, with only approximately 132 million tons of high-grade phosphate rock [4, 5]. The average grade of phosphate rock in China reaches approximately 17% mainly with low- and medium-grade phosphate rock [6]. China is a large producer, consumer, and exporter of phosphorus, providing approximately 53% of the phosphorus consumed by the rest of the world [7].
Natural phosphate rocks mainly contain carbonate fluorapatite, calcite, dolomite, natural gypsum, quartz, calcite-Mg, apatite, pyrite, fluorite, and sphalerite-Cd [8]. The main components of phosphate rock are CaO, SiO2, P2O5, MgO, CaF2, and a few heavy metal elements (U and Cd) after beneficiation [9,10,11]. The phosphorus in phosphate rock is mainly in the form of fluorapatite (9CaO·3P2O5·CaF2), hydroxyapatite [9CaO·3P2O5·Ca(OH)2], colloidal apatite (9CaO·3P2O5·CaCO3·H2O + H2O), carbonate fluorapatite [9CaO·3P2O5·Ca(F, CO3)·H2O], carbonate apatite (9CaO·3P2O5·CaCO3‚ H2O), and particularly Ca3(PO4)2 under high-temperature conditions [12].
Ninety-five percent of the world’s phosphate rock is used to prepare phosphoric acid for fertilizer production, with the remainder being smelted in electric furnaces for other applications [13]. Currently, two mature phosphoric acid production processes exist, namely, wet and thermal phosphoric acid production processes [14]. In the wet phosphoric acid production process, sulfuric acid is used to leach phosphate from phosphate ore to obtain phosphoric acid [15]. However, in the thermal phosphoric acid production process, the phosphate is first reduced to P2, and then the P2 is used as a raw material to obtain phosphoric acid by oxidation–hydration [16]. The thermal phosphoric acid production process is a reduction–oxidation–hydration process, which produces phosphoric acid with higher purity than that produced by the wet phosphoric acid process but uses more operating procedures and a higher reduction temperature (1400–1600 °C). In addition, the thermal phosphoric acid production process is more complex and has a higher energy consumption than the wet phosphoric acid production process [17]. Therefore, the wet phosphoric acid production process is used on a much larger scale than the thermal phosphoric acid production process owing to its lower economic cost. However, 5–5.5 tons of phosphogypsum are produced from 1 ton of H3PO4, and the worldwide production of phosphogypsum is 200–300 million tons per year, resulting in serious environmental pollution [18]. Furthermore, phosphogypsum production in China is 80 million tons per year, but the utilization rate is only approximately 27% [19].
Hence, some scholars have reported a thermal (kiln) phosphoric acid process to overcome the shortcomings of the wet phosphoric acid process. Cao et al. used NiSO4, K2CO3, and NiSO4 as additives to promote phosphate ore reduction and found that these additives could enhance the reduction rate of phosphate ore [20]. However, the reduction time was as long as 4 h, and the negative environmental impact of the harmful gases CO2 and SO2 produced by the decomposition of additives at high temperatures was not considered. Jacob et al. determined the suitability of SiO2 over Al2O3 as a co-solvent for the thermal phosphoric acid production process [21]. Although the reduction of the thermal phosphoric acid production process has been improved, the production process consumes more energy and causes CO gas pollution.
Therefore, the vacuum carbothermal reduction of phosphate ore, which is a cleaner process than previous reduction processes, has been proposed. Wu et al. studied the reduction of low-grade phosphate ore by pulverized coal under isothermal conditions at a pressure of 1 Pa and found that the phosphate ore had a faster reduction rate at temperatures greater than 1250 °C [9, 22]. He et al. investigated the effects of different carbon sources on the vacuum carbothermal reduction process of low-grade phosphate ore at 1 Pa [14]. The results demonstrated that the reduction rate of graphite was higher than that of pulverized coal and coke, and SiO2, Al2O3, and MgO formed a low-temperature eutectic to promote the reduction reaction.
The aforementioned studies primarily focused on the reduction process of phosphate ore as a whole. However, the phosphorus in phosphate ore is mainly present in the form of Ca3(PO4)2 under high-temperature conditions, and mechanistic studies on the vacuum carbothermal reduction process of Ca3(PO4)2 with a MgO additive have not been thoroughly investigated. Therefore, the vacuum carbothermal reduction of Ca3(PO4)2 in a SiO2–C–Ca3(PO4)2–MgO-based system was studied. The vacuum carbothermal reduction mechanism of Ca3(PO4)2 in a SiO2–C–Ca3(PO4)2–MgO-based system with the addition of MgO was elucidated, and the application of reduced slag in the converter smelting process was analyzed according to the reduced slag physical phase composition for the clean production of thermal phosphoric acid.
The carbothermal reduction temperature and carbon excess coefficient (CEC: mass ratio of actual carbon and theoretical carbon) of Ca3(PO4)2 under vacuum (P = 1 Pa) were calculated using thermodynamic software FactSage 8.1. The reduced slag was characterized by X-ray diffraction (XRD) and scanning electron microscopy–energy dispersive X-ray spectroscopy (SEM–EDS). The phosphorus content in the reduced slag was measured using the quinoline phosphomolybdate weight method, and the volatilization level of P2 in the SiO2–C–Ca3(PO4)2–MgO-based system was calculated. The P2 volatilization rate in the SiO2–C–Ca3(PO4)2–MgO-based system at T = 1300 °C and P = 1 Pa first increased and then decreased, reaching a maximum of 93.77% at n(MgO/CaO) = 0.50. When n(MgO/CaO) > 0.50, the unreduced Ca3(PO4)2 in the slag is endowed with a 2CaO·SiO2·3CaO·P2O5 solid solution. The fugacity of this solid solution proves that the low-melting-point slag remaining after vacuum carbothermal reduction can be used for converter smelting dephosphorization and reducing lime consumption, which is of great significance for the harmless production of phosphoric acid and the low-carbon development of converter smelting.
Experimental
Raw Materials
The samples were prepared from Ca3(PO4)2, SiO2, MgO, and graphite based on the main components of phosphate rock. The quantities of Ca3(PO4)2 and graphite and the CEC were 5.00 g, 0.50 g, and 1.03, respectively, according to the thermodynamic calculations.
The SiO2 content was determined to be 1.21 g [n(SiO2/CaO) = 1.25] based on preliminary experiments. To study the effect of MgO on the vacuum carbothermal reduction of Ca3(PO4)2, MgO quantities were determined to be 0.00, 0.97, 1.45, 1.93, 2.42, and 2.90 g, respectively. Methylcellulose (M), a binder, was added (0.020 g), which started to decompose at 250 °C and had a small effect on the vacuum carbothermal reduction of Ca3(PO4)2. The experimental samples were identified as S0, S1, S2, S3, S4, and S5 based on the different MgO contents of the raw materials, as listed in Table 1.
Experimental Procedure
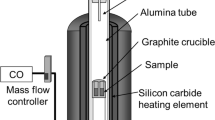
Each analytically pure chemical reagent was weighed according to the raw material composition listed in Table 1 using an electronic balance (AL204) and poured into the mixing tank. A dropper was used to add five drops of deionized water to the mixing tank, followed by stirring for 40 min with a glass stir bar. After mixing, the experimental samples were formed into cylindrical samples with a diameter of 30 mm and a height of 13 mm using an electric tablet press (P = 10 MPa). The pressed samples were dried for 120 min in an electric thermostatic oven (DHG924013) at a temperature of 110 °C, and after the water was removed from the samples, the dehydrated samples were weighed. The weighed and dried samples were placed in a graphite crucible, which was placed in a constant temperature zone inside a vacuum carbon-tube furnace (MTLQ-TG-40). The pressure inside the vacuum carbon tube furnace was pumped to 1 Pa, and the temperature program was set as follows: for T ≤ 500 °C, the heating rate was 10 °C/min; for 500 ≤ T ≤ 1000 °C, the heating rate was 8 °C/min; and for 1000 ≤ T ≤ 1500 °C, the heating rate was 6 °C/min.
The reaction temperatures of the experimental samples in the vacuum carbon tube furnace were determined to be 1250 °C and 1300 °C, and the holding time of the experimental samples was 60 min based on the thermodynamic calculations and the results of the previous experiments. After the samples were held at the set temperature for 60 min, the procedure was stopped. The reduced slag was cooled in the furnace and removed for weighing, followed by a calculation of the volatilization rate of P2 in Ca3(PO4)2. The volatilization process and P2 volatilization rate calculation formula are shown in Fig. 1 and Eq. (1), respectively.
where η, m0, m1, w0, and w1 are volatilization rate, original masses, masses of the reduced sample, phosphorus content in the original sample, and phosphorus content in the reduced sample, respectively.

Experimental procedure: T1 = 500 °C and T2 = 1000 °C
Characterization
The Equilib module of the thermodynamic software FactSage 8.1 and the Ftoxid and FTSalt databases were selected to calculate the masses of P2 volatilized and the composition of the reduced slag at a system pressure of 1 Pa. An electronic balance (AL204) was used to weigh the raw material and reduced slag, and the phosphorus content of the reduced slag was tested using the quinoline phosphomolybdate weight method. The phases and microscopic morphology of the reduced slag were characterized by XRD and SEM–EDS, respectively.
Results
Thermodynamic Calculation of the Masses of P2 Volatilized in Vacuum Carbothermal Reduction
Using the Equilib module in FactSage 8.1, the thermodynamic calculations of the masses of P2 volatilized and CEC were carried out for the vacuum carbothermal reduction of 100 g of Ca3(PO4)2, as shown in Fig. 2 At 1300 °C, the masses of P2 produced by the vacuum carbothermal reduction of Ca3(PO4)2 gradually increased with increasing CEC. The masses of P2 produced by the vacuum carbothermal reduction of Ca3(PO4)2 reached a maximum when the CEC ≥ 1.03, and the masses of P2 volatilized demonstrated little change, which showed that 1.03 was the optimal CEC for the vacuum carbothermal reduction of Ca3(PO4)2 at constant temperature.

Thermodynamic calculations of the masses of P2 volatilized and CEC
The changes in the thermodynamic volatilization rates of P2 and PO with temperature in the process of vacuum carbothermal reduction at P = 1 Pa, n(SiO2/CaO) = 1.25, and CEC = 1.03 were calculated using FactSage 8.1, as shown in Fig. 3.

Effect of temperature on the masses of P2 volatilized in the Ca3(PO4)2 reduced system by thermodynamic calculations [P = 1 Pa, n(SiO2/CaO) = 1.25, CEC = 1.03]
The masses of P2 volatilized reached a maximum value when the temperature was 840 °C. The PO in the reaction system did not change with increasing temperature between 840 °C and 1300 °C. The masses of P2 decreased while that of PO increased gradually in the reduced system when the temperature > 1300 °C because the vacuum carbothermal reduction of Ca3(PO4)2 was progressive. The reaction between PO and graphite is an exothermic reaction, which would be hindered at higher temperatures.
Furthermore, the thermodynamic transformation of phases in the reduced slag samples with different MgO contents was simulated at 1250 °C and 1300 °C when the CEC = 1.03 by FactSage 8.1 according to the volatilization rate of P2 at P = 1 Pa, as shown in Fig. 4, the slag samples reduced at 1250 °C and 1300 °C exhibited identical phases, mainly CaSiO3, 2CaO·SiO2·MgO·SiO2, CaO·SiO2·MgO·SiO2, and 2MgO·SiO2. When n(MgO/CaO) = 0.50, the main phases of reduced slag were CaSiO3, 2CaO·SiO2·MgO·SiO2, and CaO·SiO2·MgO·SiO2. At 0.50 ˂ n(MgO/CaO) ˂ 1.00, the content of 2CaO·SiO2·MgO·SiO2 increased, but the contents of CaSiO3 and CaO·SiO2·MgO·SiO2 decreased gradually with increasing n(MgO/CaO). When n(MgO/CaO) = 0.75, the CaSiO3 phase disappeared completely, and the 2MgO·SiO2 phase began to appear in the reduced slag. When n(MgO/CaO) ≥ 1, the CaO·SiO2·MgO·SiO2 phase disappeared, and the reduced slag contained only 2CaO·SiO2·MgO·SiO2 and 2MgO·SiO2. The content of 2CaO·SiO2·MgO·SiO2 decreased gradually with increasing n(MgO/CaO), while the content of 2MgO·SiO2 gradually increased.

Thermodynamic calculations of the phase contents with different MgO contents (CEC = 1.03)
Effect of MgO on Volatilization Rate of P2
According to previous thermodynamic calculations, Ca3(PO4)2 samples with different MgO contents were reduced at 1250 °C and 1300 °C with P = 1 Pa, and the volatilization rates of P2 are shown in Fig. 5 The volatilization rate of P2 at 1300 °C was higher than that at 1250 °C. At a constant temperature, the volatilization rate of P2 in the SiO2–C–Ca3(PO4)2–MgO-based system gradually increased with increasing n(MgO/CaO) when 0 ≤ n(MgO/CaO) < 0.50, and the maximum volatilization rate of P2 in the system occurred when n(MgO/CaO) = 0.50. At 1250 °C, the volatilization rate of P2 decreased to 29.40% at n(MgO/CaO) = 0.75 due to the uneven mixing of components. The volatilization rate of P2 at 1300 °C was similar to that at 1250 °C, with both showing an increase followed by a gradual decrease. The volatilization rate of P2 reached a maximum of 93.77% when n(MgO/CaO) = 0.50; however, for n(MgO/CaO) > 0.50, the volatilization rate of P2 decreased with an increase in n(MgO/CaO), indicating that the excessive MgO was not conducive to the reduction of Ca3(PO4)2.

Volatilization rate of P2 with different n(MgO/CaO) values
Effect of MgO on the Phase Transformation of Reduced Slag
The main phases of reduced slag without the addition of MgO were characterized using XRD, as shown in Fig. 6. The reduced slag mainly contained SiO2 and CaSiO3 phases. The relative peak intensity of SiO2 at 2θ = 26.84° in the reduced slag was higher than that of the CaSiO3 peak at 1250 °C. However, the peak intensity of SiO2 at 2θ = 26.84° in the reduced slag was weaker than that of the CaSiO3 peak at 1300 °C. As shown in Fig. 6, a higher content of SiO2 could be transformed into CaSiO3 at the higher temperature of 1300 °C, promoting the reaction between graphite and Ca3(PO4)2 during the vacuum carbothermal reduction without the addition of MgO.

Main phases of reduced slag without MgO additive
Furthermore, the phases in the reduced slag with different amounts of MgO added were analyzed using XRD, as shown in Fig. 7. The strong characteristic peaks at 2θ = 31.32°, 26.42°, 34.26°, 47.42° and 62.57° were attributed to 2CaO·SiO2·MgO·SiO2, SiO2, MgO, CaO·SiO2·MgO·SiO2, and 2CaO·SiO2·3CaO·P2O5, respectively.

Phase analysis of reduced slag with the addition of different amounts of MgO by XRD
When the reduction temperature was 1250 °C, the intensity of the characteristic peak of 2CaO·SiO2·MgO·SiO2 at 2θ = 26.42° decreased with increasing n(MgO/CaO), and the intensity of the characteristic peak of MgO at 2θ = 26.42° increased gradually. The addition of MgO could promote the conversion of SiO2 into other phases, such as 2CaO·SiO2·MgO·SiO2 and CaO·SiO2·MgO·SiO2 in the reduced slag, resulting in a decrease in the peak intensity of SiO2, but the strongest characteristic peaks of MgO and SiO2 overlap; therefore, the change in the strongest characteristic peak of SiO2 is not found in Fig. 7. At n(MgO/CaO) = 0.75, the characteristic peaks of the CaO·SiO2·MgO·SiO2 phase began to appear in the reduced slag, and the intensity of the characteristic peaks transformed slightly with increasing n(MgO/CaO). In addition, the characteristic peaks of 2CaO·SiO2·3CaO·P2O5 phase began to appear, and the intensity of the characteristic peak of 2CaO·SiO2·3CaO·P2O5 at 2θ = 62.57° increased gradually in the reduced slag.
When the reduction temperature was 1300 °C, the characteristic peak intensity of 2CaO·SiO2·MgO·SiO2 at 2θ = 31.32° in the reduced slag first increased and then decreased, and the phases were similar to those at 1250 °C. However, the reduction of Ca3(PO4)2 at 1300 °C was more complete, and the characteristic peaks of 2CaO·SiO2·3CaO·P2O5 in the reduced slag were smaller than those of the phase in the slag reduced at 1250 °C. The reaction of MgO with other components was more complete at 1300 °C than at 1250 °C. When n(MgO/CaO) ≥ 0.75, the relative intensity of the characteristic diffraction peak of MgO at 2θ = 42.41° increased with increasing MgO addition, and the residual MgO content of the reduced slag increased gradually.
As shown in Fig. 7, 2CaO·SiO2·3CaO·P2O5 solid solution was found to be the primary constituent of the unreduced Ca3(PO4)2 in the reduced slag. When n(MgO/CaO) < 1.00, the intensity of the characteristic diffraction peaks of 2CaO·SiO2·MgO·SiO2 in the reduced slag increased gradually, and when 1.00 ≤ n(MgO/CaO) ≤ 1.50, the intensity of the characteristic diffraction peaks of 2CaO·SiO2·MgO·SiO2 in the reduced slag decreased gradually.
Effect of MgO on the Micromorphology of Reduced Slag
To analyze the micromorphology transformation with different amounts of MgO added, S0, S1, and S3 reduced at 1250 °C and 1300 °C were characterized by SEM, as shown in Fig. 8. At 1250 °C, the morphology of the reduced slag without MgO added was primarily a fish scale structure. A large number of liquid phases and cubic crystals began to appear in the reduced slag when n(MgO/CaO) = 0.50. Moreover, the cubic crystals in the slag disappeared, and the slag formed a viscous planar structure when n(MgO/CaO) = 1.00.

Micromorphology of reduced slag
At 1300 °C, the reduced slag S0 without MgO added formed a rough surface structure, and the slag contained many micropores. Furthermore, when n(MgO/CaO) = 0.50, the slag morphology was dense and flat, with only a few cubic grains remaining unmelted. When n(MgO/CaO) = 1.00, the reduced slag almost completely formed a viscous liquid during the holding process because the increase in MgO increased the Ca and Mg feldspar contents, decreasing the melting point of the minerals and promoting the melting of the reduced slag. When n(MgO/CaO) > 0.50, the volatilization rate of P2 decreased with an increase in the liquid phase in the system owing to the increase in residual MgO particles enriched in the liquid phase, which hindered contact between graphite and Ca3(PO4)2, according to the XRD analysis.
Additionally, the phosphorus and Mg in the reduced slag were analyzed by EDS surface scanning to determine the distribution of residual Ca3(PO4)2, as shown in Fig. 9. When n(MgO/CaO) = 0.50, the phosphorus content of the reduced slag was the lowest at n (MgO/CaO) = 0.50. Aggregated Mg is found in Fig. 9-S3 at 1250 °C, and no aggregated Mg is found in Fig. 9-S3 when T = 1300 °C. These results indicated that MgO could react more completely at 1300 °C and promote slag solubilization. Comparing the distribution of phosphorus in the reduced slag at a constant temperature (T = 1250 °C and 1300 °C), the reduced slag contained the least amount of phosphorus when n(MgO/CaO) = 0.50, indicating that the effect of MgO on the solubilization of the vacuum carbothermal reduction of Ca3(PO4)2 was most evident when n(MgO/CaO) = 0.50.

Surface scanning of reduced slag. S0-T = 1250 °C, n(MgO/CaO) = 0.00; S1-T = 1250 °C, n(MgO/CaO) = 0.50; S3-T = 1250 °C, n(MgO/CaO) = 1.00; S0-T = 1300 °C, n(MgO/CaO) = 0.00; S1-T = 1300 °C, n(MgO/CaO) = 0.50; S3-T = 1300 °C, n(MgO/CaO) = 1.00
Discussion
Effect of MgO on the Vacuum Carbothermal Reduction Mechanism of Ca3(PO4)2
Thermodynamic calculations in Fig. 2 revealed that the masses of P2 volatilized in the SiO2–C–Ca3(PO4)2–MgO-based system gradually decreased with increasing temperature. However, Fig. 5 shows that the actual volatilization rate of P2 was higher at 1300 °C than at 1250 °C because the vacuum carbothermal reduction of Ca3(PO4)2 is exothermic, as shown in Eq. (2). The enthalpy change of Eq. (2) calculated by FactSage 8.1 was \(\Delta H\) > 0 at P = 1 Pa and T = 1250 °C and 1300 °C, indicating that temperature enhancement is favorable for the vacuum carbothermal reduction of Ca3(PO4)2. Furthermore, a higher temperature of 1300 °C would improve the kinetics of the vacuum carbothermal reduction of Ca3(PO4)2 according to the Arrhenius formula, as shown in Eq. (3).
where D, ED, D0, and T are the graphite diffusion coefficient at a certain temperature, the activation energy of atomic diffusion, the diffusion coefficient in the standard state, and temperature (K), respectively.
Moreover, to analyze the reduction behavior of Ca3(PO4)2 at a constant temperature, a physical model of the Ca3(PO4)2 vacuum carbothermal reduction in a SiO2–C–Ca3(PO4)2–MgO-based system with different MgO contents was established, as shown in Fig. 10. The kinetics of the vacuum carbothermal reduction of Ca3(PO4)2 in a SiO2–C–Ca3(PO4)2–MgO-based system are discussed using the contact point model of Dalvi and Suresh [23,24,25]. The vacuum carbothermal reduction between Ca3(PO4)2 and graphite in the original samples mainly proceeded at the Ca3(PO4)2 interface. The Dalvi and Suresh model reported that the solid-phase reaction was mainly influenced by the diffusion rate of the solid phase in the product layer and the interfacial contact area of the reactant particles. Therefore, the vacuum carbothermal reduction of Ca3(PO4)2 was divided into three stages, to analyze the effect of the Ca3(PO4)2 particle interface area on the Ca3(PO4)2 conversion at different stages.

Vacuum carbothermal reduction mechanism of Ca3(PO4)2 in SiO2–C–Ca3(PO4)2–MgO-based systems
According to the contact and geometric conditions of Ca3(PO4)2 and graphite in the original samples, the equation relating the contact area to the reaction time for the vacuum carbothermal reduction of Ca3(PO4)2 was obtained, as shown in Eq. (4) [26, 27].
where \(S(r)\) is the surface area of any unreacted kernel surface of radius r measured at the center of the contact point and \(K = \frac{3{C}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}}}{{C}_{\mathrm{graphite}}}\).
According to Eq. (4), the curvature radius at the contact point before the reduction of Ca3(PO4)2 is ξ1. The curvature radius of the surface at which the reduced products of each contact point come into contact with each other is ξc. The conversion rate and the number of contact points between graphite and Ca3(PO4)2 are α and \({N}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}-\mathrm{C}}\), respectively. Cgraphite and \({C}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}}\) are the Ca3(PO4)2 and graphite contents of the original sample, respectively. Based on the above parameters, the mathematical relationship between the conversion rate α1 of Ca3(PO4)2 and \({N}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}-\mathrm{C}}\) can be derived in the first stage, as shown in Eq. (5). The vacuum carbothermal reduction of Ca3(PO4)2 is proportional to \({N}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}-\mathrm{C}}\) in the first stage if the radius change is neglected.
In the second stage, the product layers begin to overlap, but the product layers do not completely encapsulate Ca3(PO4)2 (ξc ˂ ξ ≤ \(\xi^{\prime}_{{\text{c}}}\)). According to the geometrical relationship of particle contact, the mathematical relationship between the conversion rate of Ca3(PO4)2 and graphite is shown in Eq. (6). The third stage is the gradual reduction and contraction of Ca3(PO4)2 to a smaller bulk mass (\(\xi^{\prime}_{{\text{c}}}\) ≤ ξ ≤ \({r}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}}\)), as shown in Eq. (7).
where ξf and Vs are spherical radii of the inner unreacted Ca3(PO4)2 when tangent to the product zone and volume of unreduced Ca3(PO4)2, respectively. The total conversion rate (α) of Ca3(PO4)2 can be characterized using Eq. (8).
Based on Eqs. (5)–(8), the P2 volatilization rate gradually increased for 0 < n(MgO/CaO) ≤ 0.50 because the increase in MgO content promoted the formation of low melting point phases. These phases facilitated the diffusion of graphite into the unreacted Ca3(PO4)2 interface to increase \({N}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}-\mathrm{C}}\), promoting the full reduction reaction. However, the P2 volatilization rate gradually decreased for 0.50 ˂ n(MgO/CaO) ≤ 1.50 because the excess MgO resulted in the enrichment of solid particles around the unreacted Ca3(PO4)2 particles decreasing \({N}_{{\mathrm{Ca}}_{3}{({\mathrm{PO}}_{4})}_{2}-\mathrm{C}}\), thus hindering the diffusion channels of graphite. In addition, when 0.50 < n(MgO/CaO) ≤ 1.25, the 2CaO·SiO2·3CaO·P2O5 solid solution was characterized by XRD, and the results indicated a decrease in the diffusion rate of graphite.
Effect of MgO on the Phase Transformation Mechanism of Reduced Slag
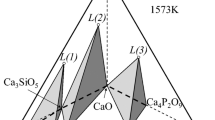
Figure 11, a ternary phase diagram of MgO–SiO2–CaO at 1 Pa, shows the transformation of the reduction products from CaSiO3 to 2CaO·SiO2·MgO·2SiO2 as the amount of MgO added gradually increased. This result, along with the evolution of phases in the reduced slag with increasing MgO addition in Fig. 11, indicated that the 2CaO·SiO2·MgO·SiO2 content decreased in the reduced slag when n(MgO/CaO) > 0.50 because the evolution of the product zone from the 2CaO·SiO2·MgO·SiO2 to the MgO region was promoted by the increase in the MgO content.

Ternary phase diagram of MgO–SiO2–CaO at 1 Pa
Moreover, Table 2 shows the thermodynamics of the reduced slag phases at 1 Pa and temperatures of 1250 °C and 1300 °C, revealing that the formation of all phases in the reduced slag can proceed spontaneously. As shown in Table 2, the ∆G of Eq. (11) was more negative than that of Eq. (12), indicating a greater thermodynamic tendency to form 2CaO·SiO2 in the reduced slag, but when n(MgO/CaO) = 0, the phase analysis of the reduced slag indicated that it was dominated by CaSiO3 and SiO2. This result was attributed to the reaction between the Ca3(PO4)2 surface intermediate CaO and SiO2 in the absence of MgO, which was influenced by the production rate of the interfacial intermediate CaO.
A low CaO content formed at the Ca3(PO4)2 interface owing to the kinetic limitations of the vacuum carbothermal reduction of Ca3(PO4)2. Therefore, the reaction of CaO with SiO2 primarily followed the pathway depicted in Eq. (12). At 1300 °C, the intensity of the characteristic peak of the SiO2 in the reduced slag was weaker than that at 1250 °C, which can be attributed to the higher energy of the reducing system increasing the activity of SiO2 molecules and promoting the full diffusion of SiO2 from the slag to the CaO interface of the Ca3(PO4)2 intermediate product layer. This allows the CaO at the Ca3(PO4)2 interface to react with SiO2 to form CaSiO3 and promotes the rapid vacuum carbothermal reduction of Ca3(PO4)2.
Furthermore, phases in the reduced slag gradually evolved from CaSiO3 to 2CaO·SiO2·MgO·SiO2 and CaO·SiO2·MgO·SiO2. The relative intensity of the characteristic peaks of 2CaO·SiO2·MgO·SiO2 in the reduced slag is greater than that of CaO·SiO2·MgO·SiO2 because the \(\Delta G\) of the direct reaction of MgO with the intermediate products CaO and SiO2 to form CaO·SiO2·MgO·SiO2 during the vacuum carbothermal reduction of Ca3(PO4)2 is much lower than those of other equations, as shown in Eqs. (15)–(17). Therefore, the intermediate product CaO produced from the slag during the vacuum carbothermal reduction of Ca3(PO4)2 reacts preferentially with MgO and SiO2 to form 2CaO·SiO2·MgO·SiO2. When n(MgO/CaO) = 0.75, the characteristic peaks of 2CaO·SiO2·3CaO·P2O5 solid solution began to appear in the reduced slag, and the intensity of the characteristic peaks gradually increased, while the relative intensity of the characteristic peaks of 2CaO·SiO2·MgO·SiO2 gradually decreased, which can be attributed to the unreacted Ca3(PO4)2 reacting with the 2CaO·SiO2 of 2CaO·SiO2·MgO·SiO2 at the interface to form a 2CaO·SiO2·3CaO·P2O5 solid solution. The phase diagram of the reaction between unreduced Ca3(PO4)2 and 2CaO·SiO2 in the 2CaO·SiO2·MgO·SiO2 solid solution is shown in Fig. 12.

Ternary phase diagram of SiO2–Ca3(PO4)2–CaO
In addition, the thermodynamic calculation of the reduced slag in Fig. 4 shows that 2MgO·SiO2 began to appear in the reduced slag at n(MgO/CaO) = 0.75. However, the main phases in the reduced slag were 2CaO·SiO2·MgO·SiO2, CaO·SiO2·MgO·SiO2, 2CaO·SiO2·3CaO·P2O5, SiO2, and MgO, and 2MgO·SiO2 was not found in the reduced slag because of the more negative Gibbs free energy changes in Eqs. (14)–(17) than that of MgO and SiO2 reacting to form 2MgO·SiO2.
Application of Reduced Slag in a Basic Oxygen Furnace
The above analysis indicated that the volatilization rate of P2 reached a maximum of 93.77% at n(MgO/CaO) = 0.50. When n(MgO/CaO) > 0.50, the volatilization rate of P2 in the SiO2–C–Ca3(PO4)2–MgO-based system decreased gradually, and the unreduced Ca3(PO4)2 in the reduced slag primarily comprised 2CaO·SiO2·3CaO·P2O5 solid solution. Therefore, the 2CaO·SiO2·MgO·SiO2 produced by the reduced slag had a fixing effect on the Ca3(PO4)2.
Phosphorus in the converter smelting process is mainly enriched in the converter slag as 2CaO·SiO2·3CaO·P2O5 solid solution [28]. In the converter smelting process, the beginning of smelting occurs at approximately 1400 °C, and a lower temperature is more conducive to initial phosphorus removal by converter smelting. Pahlevani et al. reported that dephosphorization in the converter was mainly influenced by CaO dissolution behavior in the liquid phase of converter slag and the 2CaO·SiO2·3CaO·P2O5 solid solution [29]. The conventional converter dephosphorization process mainly increases the phosphorus capacity of the converter slag by increasing the CaO content. However, the high melting point of CaO (2580 °C) and slow dissolution rate are not kinetically conducive to generating Ca3(PO4)2 at the steel–slag interface to diffuse rapidly into the slag and react to form 2CaO·SiO2 solid solution. During the smelting process, increasing the temperature is necessary to promote slagging and improve the kinetic conditions for the diffusion of Ca3(PO4)2 in the molten slag. However, a higher temperature is thermodynamically unfavorable for the oxidation of phosphorus and leads to phosphorus reversion. Sun et al. studied the behavior of converter slag dephosphorization by industrial tests at 1350–1450 °C [28]. The results indicated that the effect of molten slag dephosphorization first increased and then decreased with increasing temperature, and the dephosphorization rate was above 55% in the range of 1385–1410 °C. Zhu et al. studied the distribution of phosphorus in the solid solution and liquid phases of 2CaO·SiO2·3CaO·P2O5 and reported that at an alkalinity of 2.00–3.50, reducing the alkalinity could promote phosphorus enrichment in the solid solution 2CaO·SiO2·3CaO·P2O5 phase [30]. In addition, Wang et al. reported that the low dephosphorization rate at the beginning of converter smelting owing to the high melting point of CaO resulted in a lower slagging rate [31].
The process of dephosphorization in the converter is shown in Fig. 13. Phosphorus and silicon diffuse from the molten steel to the molten slag interface, where they react with oxygen to form oxides. In the next step, the oxides produced at the molten slag interface diffuse into the molten slag to form 3FeO·P2O5 and Ca3(PO4)2. Then, the 2CaO·SiO2 phase produced at the interface of CaO in the molten steel slag reacts with Ca3(PO4)2 to form a stable 2CaO·SiO2·3CaO·P2O5 solid solution [32].

Phosphorus removal process in converter smelting
Based on the analysis of dephosphorization in the converter, the 2CaO·SiO2·MgO·SiO2 of the reduced slag applied in dephosphorization by converter smelting can supply 2CaO·SiO2 directly to the molten slag, which will decrease the energy consumption of CaO dissolution to achieve low-temperature dephosphorization in the early stages of converter smelting.
Conclusion
In this study, the effects of MgO on the volatility of Ca3(PO4)2 reduction and the evolution mechanism of the phases and micromorphology in reduced slag were analyzed thermodynamically and kinetically. The feasibility of the comprehensive utilization of reduced slag for phosphorus removal in converter smelting was also discussed. The following conclusions can be drawn from this study.
-
(1)
With increasing n(MgO/CaO), the P2 volatilization rate in the SiO2–C–Ca3(PO4)2–MgO-based system first increased and then gradually decreased. At n(MgO/CaO) = 0.50, the P2 volatilization rate reached 93.77% after 60 min, which was much more efficient than the traditional fire process.
-
(2)
With increasing MgO content, the phases in the reduced slag gradually evolved from the CaSiO3 phase to 2CaO·SiO2·MgO·SiO2, 2CaO·SiO2·3CaO·P2O5, and other phases.
-
(3)
When n(MgO/CaO) > 0.50, the excessive solid-phase MgO hindered the contact between graphite and unreduced Ca3(PO4)2, resulting in the reaction of residual Ca3(PO4)2 with interfacial 2CaO·SiO2·MgO·SiO2 to form 2CaO·SiO2·3CaO·P2O5 solid solution.
-
(4)
Using the reduced slag as premelting slag for converter smelting can achieve low-temperature dephosphorization at the beginning of converter smelting and reduce converter smelting costs and carbon emissions in phosphorus chemical and steel enterprises.
References
Sengul H, Ozer AK, Gulaboglu MS (2006) Beneficiation of Mardin-Mazıdaği (Turkey) calcareous phosphate rock using dilute acetic acid solutions. Chem Eng J 122:135–140. https://doi.org/10.1016/j.cej.2006.06.005
Ait-Ouakrim EIH, Chakhchar A, Modafar CEI, Douira A, Amir S, Ibnsouda-Koraichi S, Belkadi B, Filali-Maltouf A (2023) Valorization of Moroccan phosphate sludge through isolation and characterization of phosphate solubilizing bacteria and assessment of their growth promotion effect on Phaseolus vulgaris. Waste Biomass Valoriz 14:2673–2690. https://doi.org/10.1007/s12649-023-02054-2
Liu WB, Huang WX, Ma H, Chi XP, Rao F (2020) China’s phosphate ore resources distribution and the progress of phosphate ore dressing technology. Ind Miner Process 49:19–25. https://doi.org/10.16283/j.cnki.hgkwyjg.2020.12.005. (in Chinese)
Cui RG, Zhang FY, Guo J, Guo ZH, Xiao YP (2019) Development strategy of phosphate rock in China under global allocation of resources. Strateg Study CAE 12:128–132. https://doi.org/10.15302/J-SSCAE-2019.01.018. (in Chinese)
Jia H, Liu JX, Jiao S, Shang PQ, Wang CG, Chi HX (2021) Situation analysis and countermeasures of phosphate rock resources exploitation and ecological protection in Yangtze River Economic Belt, China. Min Mag 30:67–72. https://doi.org/10.12075/j.issn.1004-4051.2021.02.028. (in Chinese)
Wang B, Zhou ZX, Xu DH, Wu JH, Yang XS, Zhang ZY, Yan ZJ (2022) A new enrichment method of medium-low grade phosphate ore with high silicon content. Miner Eng 181:1–10. https://doi.org/10.1016/j.mineng.2022.107548
Wu FF, Wang JX, Liu JT, Zeng GP, Xiang P, Hu P, Xiang WS (2021) Distribution, geology and development status of phosphate resources. Geol China 48:67–72. https://doi.org/10.12029/gc20210106. (in Chinese)
Zrelli RE, Rabaoui L, Daghbouj N, Abda H, Castet S, Josse C, Beek PV, Souhaut M, Michel S, Bejaoui N, Courjault-Radé P (2018) Characterization of phosphate rock and phosphogypsum from Gabes phosphate fertilizer factories (SE Tunisia): high mining potential and implications for environmental protection. Environ Sci Pollut Res 25:14690–14702. https://doi.org/10.1007/s11356-018-1648-4
Wu QH, Huang R, Lv XD, Qian X, Liu L, Zhang JZ (2019) Effect of temperature on the vacuum carbothermal reduction of low-grade phosphate ore. Mater Res Express 6:1–11. https://doi.org/10.1088/2053-1591/ab17a6
Shang DL, Geissler B, Mew M, Satalkina L, Zenk L, Tulsidas H, Barker L, El-Yahyaoui A, El-Yahyaoui A, Tahhusseina M, Zheng YH, Wang ML, Yao Y, Liu XD, Deng HD, Zhong J, Li ZY, Steiner G, Bertau M, Haneklaus N (2021) Unconventional uranium in China’s phosphate rock: review and outlook. Renew Sustain Energy Rev 140:1–9. https://doi.org/10.1016/j.rser.2021.110740
Haneklaus N, Sun YJ, Bol R, Lottermoser B, Schnug E (2017) To extract, or not to extract uranium from phosphate rock, that is the question. Environ Sci Technol 51:753–754. https://doi.org/10.1021/acs.est.6b05506
Abouzeid AZM (2008) Physical and thermal treatment of phosphate ores—an overview. Int J Miner Process 85:59–84. https://doi.org/10.1016/j.minpro.2007.09.001
Aydin I, Imamoglu S, Aydin F, Saydut A, Hamamci C (2009) Determination of mineral phosphate species in sedimentary phosphate rock in Mardin, SE Anatolia, Turkey by sequential extraction. Microchem J 91:63–69. https://doi.org/10.1016/j.microc.2008.08.001
He X, Huang R (2022) Effect of different carbon sources on vacuum carbothermal reduction of low-grade phosphorus ore. Chem Ind Chem Eng Q 28:29–37. https://doi.org/10.2298/CICEQ210219013H
Ma H, Feng X, Deng C (2018) Water–phosphorus nexus for wet-process phosphoric acid production. Ind Eng Chem Res 57:6968–6979. https://doi.org/10.1021/acs.iecr.7b05399
Ma H, Feng X, Zeng B (2018) Self-anticorrosion for the combustion tower of heat recovered thermal process phosphoric acid production. Process Saf Environ 118:330–347. https://doi.org/10.1016/j.psep.2018.07.008
Liu Q, Liu WZ, Lü L, Li RH, Liang B, Yue HR, Tang SW, Li C (2018) Study on reactions of gaseous P2O5 with Ca3(PO4)2 and SiO2 during a rotary kiln process for phosphoric acid production. Chin J Chem Eng 26:795–805. https://doi.org/10.1016/j.cjche.2017.11.016
Wu FH, Jin CY, Xie RS, Qu GF, Chen BJ, Qin J, Liu XX, Li HL, Kuang LR (2023) Extraction and transformation of elements in phosphogypsum by electrokinetics. J Clean Prod 385:1–12. https://doi.org/10.1016/j.jclepro.2022.135688
Qin XT, Cao YH, Guan HW, Hu QS, Liu ZH, Xu J, Hu B, Zhang ZY, Luo R (2023) J Clean Prod 387:1–14. https://doi.org/10.1016/j.jclepro.2023.135858
Zheng GY, Cao RF, Li Y, Xia JP, Chen ZJ (2020) The additive effect of K2CO3–NiSO4 on the carbothermal reduction process of phosphate rock and SiO2. SILICON 12:1985–1994. https://doi.org/10.1007/s12633-019-00311-5
Jacob KD, Reynolds DS, Hill WL (1929) Reduction of tricalcium phosphate by carbon. J Ind Eng Chem 21:1126–1132. https://doi.org/10.1021/ie50239a041
Li XF, Huang R, Wu QH, Xie SF, Li JQ (2021) Experimental approach for the characterization of low-grade phosphate ore performance in isothermal conditions. J Sustain Metall 7:1736–1747. https://doi.org/10.1007/s40831-021-00451-2
Yang MR, Lv XW, Bai CG, Wang HY (2022) Isothermal kinetics model for solid–solid reaction of powders through surface area and size distribution of particles. Metall Mater Trans B 53:968–980. https://doi.org/10.1007/s11663-021-02393-2
Phutke M, Raichur AR, Suresh AK (2022) Contact-point models for solid–solid reaction kinetics: a parameter estimation and derived insights. Ind Eng Chem Res 61:11636–11644. https://doi.org/10.1021/acs.iecr.2c00513
Shimizu A, Hao YJ, Tanaka T (1998) Representation of the concentric spherical powder reaction model in the form of linear regression analysis. Can J Chem Eng 76:69–75. https://doi.org/10.1002/cjce.5450760109
Phutke M, Dedhia J, Suresh AK (2019) Modelling solid–solid reactions: contact-point approach. Chem Eng J 377:1–10. https://doi.org/10.1016/j.cej.2018.12.030
Dalvi VV, Suresh AK (2011) A contact-point based approach for the analysis of reactions among solid particles. AIChE J 57:1329–1338. https://doi.org/10.1002/aic.12347
Lv YC, Li HB, Liu Y, Wang HB (2022) Effects of dephosphorization from solid 2CaO·SiO2–3CaO·P2O5 of steelmaking slag. In: International conference on advanced manufacturing technology and manufacturing systems (ICAMTMS), Shijiazhuang, China, 2022, p 123091X-4. https://doi.org/10.1117/12.2645818
Pahlevani F, Kitamura SY, Shibata H, Maruoka N (2010) Distribution of P2O5 between solid solution of 2CaO·SiO2–3CaO·P2O5 and liquid phase. ISIJ Int 50:822–829. https://doi.org/10.2355/isijinternational.50.822
Zhu B, Zhu MM, Luo J, Dou XF, Wang Y, Jiang HJ, Xie B (2020) Distribution behavior of phosphorus in 2CaO·SiO2–3CaO·P2O5 solid solution phase and liquid slag phase. Metals 10:1–13. https://doi.org/10.3390/met10081103
Wang ML, Yang WY (2020) Dephosphorization in the early stage of converter steelmaking. Ironmak Steelmak 47:1127–1134. https://doi.org/10.1080/03019233.2019.1673546
Sun H, Yang J, Lu XW, Liu WS, Ye GF, Zhang RH, Yang WK (2021) Dephosphorization in double slag converter steelmaking process at different temperatures by industrial experiments. Metals 11:1–23. https://doi.org/10.3390/met11071030
Acknowledgements
The authors are especially grateful for financial support from the National Natural Science Foundation of China (Grant No. 52064010), Outstanding Young Scientists and Technologists Program of Guizhou Province, China (Grant No. [2021]5644), the Key Nurturing Projects of Guizhou University (Grant No. [2019]07), and the Natural Science Research Project of Guizhou Provincial Department of Education ([2022]041).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
The contributing editor for this article was M. Akbar Rhamdhani.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, M., Zhu, R., Huang, R. et al. Effect of MgO on Vacuum Carbothermal Reduction Mechanism of Ca3(PO4)2 in SiO2–C–Ca3(PO4)2–MgO-Based System. J. Sustain. Metall. 9, 1429–1443 (2023). https://doi.org/10.1007/s40831-023-00735-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-023-00735-9