
Abstract
Sulfidic copper–lead–zinc tailings can pose a significant environmental threat, ranging from generation of acid mine drainage (AMD) to dam failures. On the other hand, they can also be considered as low-grade ore resources for zinc and copper provided that novel economically feasible metal extraction and metal recovery techniques are developed. Due to the low metal concentrations in these resources, the leaching will generate dilute leachates from which metal recovery is a challenge. Ion flotation is a foam separation technique capable of recovering metal ions from dilute aqueous leachates. In this paper, ion flotation was applied to separate copper from ammoniacal leachates of microwave-roasted sulfidic tailings samples. The sulfidic tailings were first roasted at 550 °C for 60 min, for the oxidation of sulfide minerals to more easily soluble sulfates using microwave-assisted irradiation as heating source. The microwave-roasted material was then leached with a mixture of ammonia and ammonium carbonate solutions. The optimum leaching efficiencies of zinc (86%) and copper (75%) were obtained under the following conditions: liquid-to-solid ratio = 10 mL g−1, T = 90 °C, [NH3 + NH4+] = 4 mol L−1, NH3:NH4+ = 2:1, t = 5 h. From the generated pregnant leach solution, it was possible to selectively separate 85% of copper to the foam phase by ion flotation, with sodium dodecyl sulfate (SDS) surfactant, as colloidal copper(II) tetraamine dodecyl sulfate under the optimized conditions: [SDS]total = 5.85 mmol L−1, [EtOH] = 0.5 % (v/v), ttotal = 5 h, flotation stages = 3. The zinc that remained in the solution after ion flotation was recovered by precipitation (95%) as basic zinc carbonate.
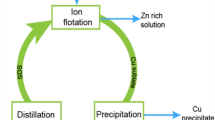
Graphical Abstract
Copper and zinc extraction by ammoniacal leaching from microwave-roasted Cu-Zn-Pb sulfidic tailings followed by recovery of copper and zinc by ion flotation and precipitation.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tailings are mixtures of fine-grained unwanted fractions after mineral beneficiation of ores at a mining site that are considered as a waste [1, 2]. Sulfidic tailings arise during the mining and mineral processing of copper–zinc–lead (Cu–Zn–Pb) ores. The resulting tailings are usually stored in tailing ponds for physical dewatering [3, 4]. Without proper management, sulfidic tailings can cause serious environmental threats, ranging from generation of acid mine drainage (AMD) to dam failures [5,6,7,8].
On the other hand, sulfidic tailings often contain valuable metals, such as copper, zinc, and lead, which can potentially be recovered. In order to simultaneously mitigate the environmental risks of the sulfidic tailings and valorize the material, the following strategy could be followed: (1) recovery of valuable metals, (2) removal of toxic elements, and (3) re-use of the matrix [9,10,11]. To tackle this challenge, new economically viable and environmentally friendly metallurgical techniques capable of treating wastes with low-grade valuable metal content must be developed.
Copper and zinc are usually present in the sulfidic tailings as stable metal sulfide phases (e.g., Cu2S, ZnS) [12]. By roasting sulfidic minerals, the sulfides are converted into more soluble sulfates or oxides and the unwanted ferrous minerals (e.g., FeS2) are oxidized to hematite [13,14,15]. Roasting is usually performed in conventional furnaces, where heating is achieved via conduction and/or convection. Metal sulfide ores are known to be very good microwave absorbers, so microwaves can efficiently heat the material [16]. Microwave roasting offers several advantages over the conventional process such as fast, volumetric, and selective heating, reduced energy consumption, elevated reproducibility, and ease of control [17]. Several studies about microwave roasting of sulfidic tailings showed that it is a very promising alternative roasting technique [18, 19].
Leaching with ammonia and ammonium salts, also known as ammoniacal leaching, is a hydrometallurgical process to extract copper and zinc very selectively from Cu–Zn–Pb sulfidic tailings, with respect to iron and lead [20]. Ammonia and ammonium salt mixtures are cheaper, less toxic, and easily regenerated compared to acids [21]. In addition, ammonia has been proven to be an efficient lixiviant for the leaching of zinc and copper from low-metal-grade secondary resources [22, 23]. However, leaching of low-grade secondary resources does generate dilute leachates and the metal recovery step might be challenging [10, 24, 25].
The state-of-the art method to selectively recover copper from leachates is solvent extraction [26,27,28]. However, in the case of dilute aqueous leachates, solvent extraction is not efficient due to the losses of the organic phases, which is caused by a certain solubility of extractants in the aqueous phase. Adsorption or ion exchange might be more suitable techniques to recover copper from dilute aqueous leachates [29, 30]. However, the main drawbacks of these techniques are the high cost of selective adsorbents and the formation of large amounts of secondary waste due to the elution step. Ion flotation is an adsorptive bubble separation technique capable of recovering or removing metal ions from dilute aqueous solutions [31,32,33,34,35]. For a dilute solution, ion flotation can potentially be an interesting alternative hydrometallurgical technique to recover and separate valuable metals [36]. In this technique, surfactants (collectors) are added and compressed air or nitrogen gas is sparged in the solution, to generate a mobile gas/liquid interface (bubbles). The targeted metal ions (colligends) are attached to the gas/liquid interface due to interactions with the oppositely charged collectors [37]. In most cases, the interaction between colligend and collector is electrostatic and forms a colloid, also known as sublate, which is concentrated to a foam phase as the bubbles ascend [38]. In addition, high metal recovery yields can be achieved from dilute aqueous solutions [39, 40]. However, the technique is not efficient for treating concentrated solutions and some of the collectors might not be suitable to use from an economic and/or environmental point of view [28]. Recent studies showed that the stripping of the collected metals and the re-use of the collector are possible [41, 42].
A considerable amount of literature has been published on the ion flotation of copper over the last years. Several studies investigated its recovery from synthetic solutions by using commercially available ionic collectors [43,44,45,46,47]. The results obtained in these studies were promising, since the recovery efficiency of copper was almost quantitative, both in acidic and in alkaline media. More recently, attention has been paid to the recovery of copper from synthetic solutions by employing novel chelating collectors or combing chelating agents with conventional collectors. Furthermore, it was shown that chelating systems in ion flotation can recover the targeted metal ions with better selectivity [48,49,50,51]. On the other side of the spectrum, there are very few studies in which the ion flotation of copper was tested in real material. To the best of our knowledge, only two studies have investigated so far the removal of copper from dilute mine wastewaters by ion flotation with quite promising results [52, 53].
The studies described above provide evidence that the recovery of copper by ion flotation is possible by a plethora of collectors. To date, research on real effluents or leachates is limited or missing. Unlike literature studies, solutions generated from hydrometallurgical processes have complex chemical compositions and contain several metal ions. In most cases, one is interested to selectively separate one metal ion over the others. Therefore, the feasibility of applying ion flotation for the recovery of valuable metals from dilute aqueous leachates needs to be investigated for realistic conditions. This work makes a step forward, towards the application of such separation technique from real leachates. Hence, the objective of this paper is to evaluate the efficiency of ion flotation for the recovery of copper from real leachates of sulfidic tailings. The leachates were generated after ammoniacal leaching of microwave-roasted Cu-Zn-Pb sulfidic tailings of the Neves Corvo mine area (Portugal), in the so-called Iberian Pyrite Belt.
Experimental
Chemicals
Sodium dodecyl sulfate (≥ 99%), nitric acid (65% a.r.), and zinc(II) sulfate monohydrate (99%) were purchased from Acros Organics (Geel, Belgium). Multi-element standard (100 mg L−1 in 2–5% HNO3), arsenic standard (1000 mg L−1 in 2–5% HNO3), sulfur standard (1000 mg L−1 in 2–5% HNO3), dysprosium standard (1000 mg L−1 in 2–5% HNO3), and ammonium carbonate (≥ 30% NH3 basis) were obtained from Sigma-Aldrich (Overijse, Belgium). Anhydrous copper(II) sulfate (a.r.), sulfuric acid solution (95 wt%), and ammonia solution (25 wt%) were purchased from ChemLab (Zedelgem, Belgium). Tetrafluoroboric acid solution (50% w/w) and ethanol (EtOH, 99.8+%, absolute) were obtained from Fisher Scientific (Thermo Fisher Scientific, Loughborough, United Kingdom). Hydrochloric acid solution (37 wt%) was purchased from VWR (Fontenay-sous-Bois, France). Water was always of ultrapure quality, deionized to a resistivity of 18.2 MΩ cm with a Millipore ultrapure water system. All chemicals were used as received without any further purification. The sulfidic tailings samples were kindly provided by SOMNICOR Lunding Mining S.A. from the tailings storage facilities of the company located in Neves Corvo mine area of the Iberian Pyrite Belt (Portugal).
Equipment
An IKA EUROSTAR 20 stirrer was used for the homogenization of the sulfidic tailings samples. The homogenized sulfidic tailings sample was dried in a Heraeus RVT vacuum oven, fed with continuous nitrogen flow in order to prevent oxidation. The thermal behavior of the dried sulfidic tailings sample was determined using a Netzsch STA449 C Jupiter thermogravimetric analyzer (TGA). Microwave roasting of sulfidic tailings was carried out in a Milestone PYRO advanced microwave (MW) oven (maximum MW power of 1800 W, frequency of 2.45 GHz) in air atmosphere. The microwave furnace was equipped with a silicon carbide (SiC) plate that absorbs microwave irradiation and releases heat at the top of the heating chamber in order to ensure a homogenous temperature, within the chamber. Leaching of the microwave-roasted material was carried out in 4 mL glass vials on a heating plate, RCT classic. Larger-scale leaching experiments at optimized conditions were carried out in 250 mL and 500 mL round-bottom flasks, which were fixed on a customized aluminum heating block and then stirred on a heating plate. After leaching, the solid phase was separated from the liquid phases by syringe filtration through a PET filter (0.45 μm) for the small-scale leaching experiments or by vacuum filtration for the larger-scale leaching experiments. Ion flotation experiments were carried out in an inhouse-built lab-scale set-up (Fig. 1), comprising a glass column of 45 cm in height and an internal diameter of 4.5 cm. The column was equipped with an adjustable bubble generation mechanism of a sintered glass disk (D4 pore size, ~ 10–15 μm), a sampling port, and a port for collecting the foam. The whole set-up was connected through a tube to a flowmeter for controlling the nitrogen gas stream that was introduced to the column. Precipitation of zinc via aeriation was performed in a 250 mL gas wash bottle, placed in a sand bath. The gas wash bottle was equipped with a sintered glass disk of D0 porosity grade and two glass tubing connectors of 11 mm external diameter. Teflon™ tubes were used to connect the bottle to the compressed air source and to another sintered glass disk of 1 porosity grade, which was mounted on a 300 mL Erlenmeyer flask. Concentrations of elements in the solutions were measured by inductively coupled plasma—optical emission spectroscopy (ICP-OES) by using a Perkin- Elmer Optima 8300 spectrometer equipped with an axial/ radial dual plasma view, a GemTip Cross-Flow II nebulizer, a Scott double pass with inert Ryton spray chamber, and a demountable one-piece Hybrid XLT ceramic torch with a sapphire injector (2.0 mm internal diameter). Dilutions were done with 5 vol% HNO3 solutions and all ICP-OES samples were measured in triplicate. The dilution factor was chosen so that the final concentration was lower than 25 mg L−1 and dysprosium was used as internal standard. The mineralogical composition of the sulfidic tailings, microwave-roasted material, and zinc precipitate was characterized by X-ray diffraction (XRD) analysis using a Bruker D2 Phaser Diffractometer. Diffractograms were recorded in the measurement range of 5° to 70° 2θ using CuKα radiation and applying an acceleration voltage of 45 kV, an electrical current of 30 mA, a step size of 0.020°, and a counting of 1 s per step. The raw data were processed with Bruker DiffractEVA software. The dried sulfidic tailings sample, microwave- roasted material, and leaching solid residue were digested in a CEM Mars 6 EasyPrep iWave microwave digester. For the digestion, 100 mg of material was weighted and placed in the EasyPrep vessels and 3 mL of HCl, 1 mL of HNO3, and 2 mL of HBF4 were added. The vials were properly closed and digested for 2 h at 105 °C. The digested sample was then transferred to 50 mL volumetric flask and diluted with 5 vol% HNO3. Each digestion was performed in triplicate and a blank sample was also digested to be used as reagent blank in the ICP-OES measurements. The sublate was analyzed by a Bruker Vertex 70 Fourier transform infrared-attenuated total reflectance (ATR-FTIR), operated at room temperature. The content of carbon, hydrogen and nitrogen of the sublate was measured in triplicate using a Thermo Scientific FLASH2000 CHN analyzer. The pH of the solutions was measured with a Mettler-Toledo pH meter SevenCompact pH/ ion S220 after calibration with standard buffer solutions of pH 1, 4, 7, and 12. A Heraeus D-6450 oven was employed to dry the solid samples. All experiments were done in duplicates and all data in figures represent averaged values of each duplicate.

Lab set-up used for the ion flotation experiments: (1) flowmeter, (2) bubble generation mechanism, (3) sampling port, (4) foam port
Calculations with HSC Chemistry 8
The simulation of the mineral phase transition as a function of roasting temperature of the examined sulfidic tailings was modeled using the software HSC Chemistry 8 (Metso Outotec Oyi., Finland).
Characterization of the Tailing Materials
The sulfidic tailings were received as a slurry. They were first homogenized at 100–200 rpm and subsample was taken and dried at 40 °C to constant mass. The dried sulfidic tailing materials were crushed and milled using a mortar and a pestle and characterized by TGA, XRD, and microwave digestion followed by ICP-OES.
Microwave Roasting Experiments
Microwave roasting was performed by weighting 20 g of dried sulfidic tailings placed in an alumina crucible in the heating chamber of the microwave furnace. The sample was roasted at 550 °C under the following conditions: 30 min ramp time, 60 min dwell time, and 1200 W maximum power. After reaching room temperature, the crucible was weighted to determine the mass loss ML (%) based on the following equation:
where mi is the mass of the dried sulfidic tailing sample (g) and mf is the mass of the microwave-roasted material (g). The obtained roasted material was characterized by XRD and microwave digestion followed by ICP-OES.
Leaching Experiments
The small-scale leaching experiments of the microwave-roasted material were carried out by adding 0.1 g of roasted material to a 4 mL glass vial with a magnetic stirring bar and 1 mL of the lixiviant (solution of {ammonia − ammonium}/[NH3 + NH4+]). The vial was sealed and then placed in a sand bath on a heating plate, equipped with a thermocouple for temperature control. Under these conditions, the parameters for leaching were optimized by varying the lixiviant concentration (2–10 mol L−1 of total [NH3 + NH4+]), the leaching temperature (60, 70 and 90 °C), and the leaching time (1–24 h). The scalability of the leaching step at the optimized conditions was also tested by adding 10 g or 20 g of roasted material with 100 mL or 200 mL of lixiviant in a 250 mL or 500 mL round-bottom flask, respectively, placed on an aluminum heating block with a thermocouple on a heating plate. Immediately after the filtration, aliquots from the pregnant leach solution (PLS) were withdrawn and diluted with HNO3 (5 vol%) for ICP-OES analysis. The leaching efficiency EL (%) was calculated according to Eq. (2):
where CPLS is the metal ion concentration in the PLS (mg L−1), VLIX is the volume of the lixiviant used for leaching (L), ms is the mass of the solid material used for leaching (kg), and Cs is the concentration of the metal in the roasted material before leaching (mg kg−1). The pH of the PLS was also measured after filtration. The solid residue after leaching was dried in an oven at 40 °C for 24 h and digested and characterized by ICP-OES.
Ion Flotation Experiments
For the ion flotation experiments, synthetic solutions were prepared by mixing the appropriate amounts of metal ions, surfactant, ethanol, and [NH3 + NH4+] solution from stock solutions. The solutions were stirred for 10 min with a magnetic stirring bar on a magnetic stirrer at low speed (200 rpm), in order to avoid the generation of foam. The solutions were then poured slowly in the flotation column by using a funnel; nitrogen gas was bubbled through the solution from the bottom. Aliquots of approximately 3 mL were withdrawn for ICP-OES analyses from the bulk solution before and after the flotation experiments. The parameters for ion flotation were optimized using synthetic solutions, by varying the concentration of the collector (1–6 mmol L−1), the flotation time (1–24 h), and multiple flotation stages (1–4 stages). For the optimization of the ion flotation stages, the required amount of sodium dodecyl sulfate (SDS) was added as a solid in the solution after the first flotation stage. The solutions were then stirred for 15 min at low speed to ensure that the added surfactant was dissolved and the ion flotation experiments were stopped when no further foam was produced. At optimized conditions, ion flotation was applied to the generated PLS in order to evaluate the efficiency of the technique with realistic conditions. The efficiency of flotation results was expressed as the recovery percent (Re%) according to the following equation:
where Ci and Cr are the initial and residual metal ion concentration of the bulk solution (mg L−1), respectively. In all the ion flotation experiments, 0.5 % (v/v) of ethanol (EtOH) was added as frother and the initial volume was 300 mL. The generated foam was collected in a beaker and allowed to physically collapse. The concentrated solution was then centrifuged in order to separate and collect the colloidal product (sublate), which was characterized by FTIR, CHN, and ICP-OES analysis (after digestion with HCl).
Precipitation of Zinc
For the precipitation of zinc from the treated leachate after the ion flotation by aeriation, compressed air was bubbled into a gas wash bottle. The excess of ammonia was trapped into another flask, containing 2 mol L−1 of H2SO4. At specified time intervals, a small aliquot of the sample was withdrawn for ICP-OES analysis. At the end of the experiment, the mixture was filtered by vacuum filtration. The precipitate was washed with MilliQ water, dried at 40 °C for 24 h, and analyzed by XRD and ICP-OES analysis. The precipitation efficiency EP (%) of zinc was calculated based on the following equation:
where Ci(Zn) and Cf(Zn) are the initial and residual zinc concentration in the solution.
Results and Discussion
Characterization of Sulfidic Tailings
The mineralogical analysis of the Neves Corvo sulfidic tailings (Fig. 2, Table 1), characterized by quantitative X-ray powder diffraction, showed high contents of pyrite (40.8 wt%), quartz (25.5 wt%), and clay minerals (e.g., clinochlore—17.2 wt%). There was also the presence of muscovite (6.9 wt%) and carbonate minerals (e.g., siderite—5.3 wt%), while galena (PbS), and sphalerite (ZnS) were detected, but the peak intensities of the diffraction peaks were too low for accurate quantification. Copper was most likely present as chalcocite (Cu2S), given the fact that the tailing material is a byproduct of sulfidic ores of the Iberian Pyrite Belt, with chalcocite as the main copper-bearing minerals [54]. The main elemental content (Table S1) of the examined sample was in agreement with the determined mineralogy, with iron (Fe), sulfur (S), and silica (Si) being the predominant elements (26 wt%, 24 wt%, and 11 wt% accordingly). The targeted metals, i.e., copper (Cu) and zinc (Zn), were present in concentrations of less than 1 wt%.

X-ray diffractogram of the dried Neves Corvo sulfidic tailings sample
The thermal behavior of the sample was examined by TGA (Fig. S1). Two main decomposition temperature ranges were observed. The first mass loss (± 11%; ~ 380 °C to 500 °C) was linked to the oxidation of pyrite, in which sulfur dioxide is released according to the following chemical reactions (Eqs. 5, 6, 7 and 8) [13, 55]:
The second mass loss (± 5.50%; ~ 500 °C to 730 °C) was related to the transformation of iron(II) sulfate phases to hematite (Eq. 9),
Microwave Roasting of Sulfidic Tailings
Based on the thermal behavior of the examined sample (Fig. S1), the conversion of pyrite to hematite occurred above 500 °C. Additionally, simulating the roasting conditions as a function of temperature by HSC Chemistry software (Fig. S2), it was observed that above 500 °C, the predominant iron phase was hematite, while copper and zinc were still present as sulfates, which are known to be more soluble at mild conditions than the corresponding oxides. Finally, previous studies conducted on the roasting of sulfidic tailings from the Iberian Pyrite Belt showed that the optimal leaching efficiencies for Cu and Zn were obtained after roasting of the solid material between 500 and 600 °C [56]. Therefore, 550 °C was selected as the roasting temperature for this study.
The experimental results supported the above hypothesis. From the XRD analysis of the microwave-roasted material (Fig. 3 and Fig. S3), the main iron phases were assigned to hematite, which indicated that pyrite (FeS2) was decomposed to hematite (Fe2O3) by microwave roasting at 550 °C for 1 h.

Comparative X-ray diffractograms of the examined sulfidic tailings sample (grey sample) and the microwave-roasted material (red sample) (Color figure online)
The mineral phase transition from iron(II) sulfide to iron(II) oxide can be verified by the mineralogy of the roasted material (Table 1, Fig. S3), which mainly consists of hematite (40.4 wt%), quartz (27.2 wt%), and mica minerals (10.8%). Pyrite was still present but at very low amounts (0.3 wt%) compared to the original sample (40.8%). Other iron-containing mineral phases (e.g., goethite, pyrrhotite) could not be detected by the used XRD diffractometer, equipped with a Cu-anode. It is known that applying CuKα radiation reduces the sensitivity towards iron phases as high background scattering occurs due to fluorescence of iron [57]. Based on similar studies, we could assume a minor presence of pyrrhotite (Fe(1−x)S) apart from hematite [58]. Zinc was present as zinc(II) sulfate (1.4 wt%). Given the origin of the non-roasted sample and based on the calculated simulation (Fig. S2), copper was probably also present as sulfate. Regarding the chemical composition of the microwave-roasted material (Table S1), the most notable difference with the non-roasted sample is the reduction of sulfur content from 23.7 to 2.4 wt%. This reduction can be attributed mainly to the formation and release of SOx gases during the roasting and resulted into a mass loss of 14.9% [59]. No major differences were observed in the zinc and copper content before and after microwave roasting (0.9 wt% and 0.4 wt%, respectively).
Ammoniacal Leaching of Microwave-Roasted Material
Based on the characterization of the microwave-roasted material, it can be assumed that zinc and copper were present in the divalent state. As such, they can be selectively solubilized with aqueous ammonia (NH3) as copper(II) and zinc(II) tetraammine complexes ([Cu(NH3)4]2+, ([Zn(NH3)4]2+) , according to the following reactions (Eqs. 10 and 11) [60]:
In addition, the presence of an ammonium salt gives a buffering effect and the anion acts as complexing agent, which stabilizes better the formed tetraammine complexes of the metals [61,62,63,64].
For this reason, ammonium carbonate ((NH4)2CO3) was selected as ammonium salt for the leaching of the microwave-roasted material in combination with aqueous ammonia. The effect of total {ammonia + ammonium} concentration ([NH3 + NH4+]), leaching temperature, and leaching time on the leaching efficiencies of copper and zinc is shown in Fig. 4. A total [NH3 + NH4+] concentration above 4 mol L−1 had a negligible effect on the leaching efficiencies of copper and zinc (Fig. 4a, Table S2). In particular, at a total [NH3 + NH4+] concentration of 4 mol L−1, copper and zinc leachabilities were 58.3% and 55.2%, respectively. Therefore, this concentration was considered as optimal. The effect of temperature was investigated at [NH3 + NH4+] = 4 mol L−1 and by keeping the other conditions constant (Fig. 4b, Table S3). Upon leaching at 90 °C, the leaching efficiency of zinc significantly increased from 58.3 to 66.2% and of copper from 55.2 to 63.7%. After 5 h of leaching (Fig. 4c, Table S4), zinc and copper were extracted for about 88.6% and 78.7%, respectively. However, the leaching efficiencies were decreased by almost 10% over the course of 7 and 24 h of leaching. The filtered leachate after 5 h of leaching was not stable; after standing for 2 days at room temperature, a brown precipitate was observed and the targeted metal content was reduced by approximately 10% compared to the fresh filtered leachate. Therefore, this decrease in leaching efficiency over time might be attributed to a partial precipitation of the targeted metals from the PLS.

a Effect of total ammonia concentration [NH3 + NH4+] on the leaching efficiency of zinc and copper from the microwave-roasted sample. Leaching conditions: L/S = 10 mL g−1, T = 60 °C, t = 3 h, NH3:NH4+ = 2:1. b Effect of leaching temperature on the leaching efficiency of zinc and copper from the microwave-roasted sample. Leaching conditions: L/S = 10 mL g−1, [NH3 + NH4+] = 4 mol L−1, t = 3 h, NH3:NH4+ = 2:1. c Effect of leaching time on the leaching efficiency of zinc and copper from the microwave-roasted sample. Leaching conditions: L/S = 10 mL g−1, [NH3 + NH4+] = 4 mol L−1, T = 90 °C, NH3:NH4+ = 2:1
Based on the experimental results described above and by keeping constant the liquid-to-solid (L/S) ratio (10 mL g−1), the ratio of NH3:NH4+ (2:1), and the stirring speed (900 rpm), the optimal ammoniacal leaching conditions for the extraction of Cu and Zn from the microwave-roasted material were found to be as follows: [NH3 + NH4+] = 4 mol L−1, T = 90 °C, and t = 5 h. Under these conditions, the scalability of leaching was tested from 1 to 100 mL and 200 mL. The obtained results (Fig. 5, Table S5) were comparable to those of the small-scale leaching. In particular, the leaching efficiencies of zinc were 83.7% (100 mL) and 86.7% (200 mL) and that for copper 71.1% and 73.6%, instead of 88.6% and 78.7%, respectively. The PLS had a pH of 10.2 and relatively small content of metal/metalloid impurities (Table S5). The solubilization of iron (Fe) and arsenic (As) was below 1 mg L−1, while that of calcium (Ca), lead (Pb), and aluminum (Al) was below 5 mg L−1 and of magnesium (Mg) was at around 70 mg L−1. The zinc and copper content in the leaching residue (Table S2) were reduced from 0.9 to 0.09 wt% and from 0.4 to 0.08 wt%, respectively.

Comparative leaching efficiencies of copper and zinc from the microwave-roasted material with mixture of ammonia- ammonium solution at small scale (1 mL), 100 and 200 times upscale (100 mL and 200 mL). Leaching conditions: L/S = 10 mL g−1, T = 90 °C, [NH3 + NH4+] = 4 mol L−1, NH3:NH4+ = 2:1, t = 5 h
Separation of Copper Over Zinc from the Pregnant Leaching Solution by Ion Flotation
As discussed in the previous section, the measured pH of the filtered leachate obtained under the optimized ammoniacal leaching conditions was 10.20. Based on the species distribution diagrams of the Cu–NH3–H2O and Zn–NH3–H2O systems at pH around 10, copper and zinc are mainly present as copper and zinc tetraammine complexes [65, 66]. Since [Cu(NH3)4]2+ and [Zn(NH3)4]2+ are positively charged complexes, their floatability is favored by the presence of an anionic surfactant such as sodium dodecyl sulfate (SDS). In principle, [Cu(NH3)4]2+ and [Zn(NH3)4]2+ complexes should be absorbed to the gas/liquid interface due to electrostatic interactions with the hydrophilic group of the collector (DS¯). This interaction results in the formation of a colloidal phase (sublate) according to the suggested reactions (Eqs. 12 and 13):
Nevertheless, this adsorption interface will occur if the collector is fully solubilized and does not form micelles. Since the collector has to chemically react with the metal ions, to achieve the highest recovery efficiencies, it has to be present at least at stoichiometric amount with respect to the metal ions [28, 29]. Therefore, a concentrated solution should be diluted before employing the technique. This will ensure not only that the collector does not exceed its critical micelle concentration (CMC), but also that it is present in a sufficient amount for the recovery of the targeted metal ions.
The CMC value of SDS is around 8 mmol L−1, depending on the purity of the dodecyl sulfate salt [67]. The PLS obtained under the optimized conditions had a concentration of zinc and copper of 800 mg L−1 (12 mmol L−1) and 250 mg L−1 (4 mmol L−1), respectively. Therefore, in order to ensure that the SDS concentration does not exceed its CMC value, a simulated ammoniacal leachate was prepared based on the concentrations obtained from the optimized leaching conditions, but assuming that it was diluted by a factor of two (i.e., [Cu2+] = 2 mmol L−1 and [Zn2+] = 6 mmol L−1). The effect of SDS concentration, time, and multiple flotation steps on the recovery efficiency of copper and zinc by ion flotation from synthetic leachates is illustrated in Fig. 6.

a Effect of SDS concentration on the recovery of zinc and copper from simulated ammoniacal leachate. Flotation conditions: [NH3 + NH4+] = 2 mol L−1, NH3:NH4+ = 2:1, [Zn2+] = 6 mmol L−1, [Cu2+] = 2 mmol L−1, [EtOH] = 0.5 % (v/v), t = 4 h, pH = 10.15. b Effect of flotation time on the recovery of zinc and copper from simulated ammoniacal leachate. Flotation conditions: [NH3 + NH4+] = 2 mol L−1, NH3:NH4+ = 2:1, [Zn2+] = 6 mmol L−1, [Cu2+] = 2 mmol L−1, [EtOH] = 0.5 % (v/v), [SDS] = 5 mmol L−1, pH = 10.18. c Effect of multiple ion flotation stages on the recovery of zinc and copper from simulated ammoniacal leachate. Flotation conditions: [NH3 + NH4+] = 2 mol L−1 , NH3:NH4+ = 2:1, [Zn2+] = 6 mmol L−1, [Cu2+] = 2 mmol L−1, [EtOH] = 0.5 % (v/v), [SDS]total = 5.85 mmol L−1 (5 mmol L−1, 0.7 mmol L−1, and 0.15 mmol L−1 for the first, second, and third flotation stage, respectively), ttotal = 5 h (3.5 h, 1 h, and 0.5 h for the first, second, and third flotation stage, respectively)
With the increase in SDS concentration (Fig. 6a, Table S6), copper was selectively concentrated to the foam phase over zinc. An optimal copper recovery efficiency of 78.7% was obtained at [SDS] = 5 mmol L−1, which is 25% in excess of the stoichiometric amount (based on Eq. 12) and selected for the subsequent experiments. The selectivity for copper over zinc cannot be easily explained. Thermodynamic studies proved that ion flotation depends on the Gibbs free energy for adsorption (\(\Delta G_{{{\text{ads}}}}^{0}\)) between a colligend and a collector at the gas/liquid interface. Depending on the examined system \(\Delta G_{{{\text{ads}}}}^{0}\) is governed by various factors such as electrical, hydrophobic, and dehydration interactions as well as chelating contributions. The more negative the \(\Delta G_{{{\text{ads}}}}^{0}\), the more favorably a colligend is concentrated to the foam phase over another [50, 51, 68,69,70]. So it would be plausible that the overall \(\Delta G_{{{\text{ads}}}}^{0}\) of the SDS complex of [Cu(NH3)4]2+ was more negative than that of the SDS complex of [Zn(NH3)4]2+ and therefore copper was selectively separated over zinc from the solution.
From Fig. 6b and Table S7, it can been observed that there was a steep increase in the recovery rate of copper within the first 3 h (78.7%) and a plateau was reached after 3.5 h of flotation time (71.2%). At the same time, the recovery efficiency of zinc remained almost negligible.
In an effort to further increase the recovery of Cu, additional ion flotation stages were carried out in the same solution (Fig. 6c, Table S8). This was performed by assuming that 75% of copper was recovered in each step and 125% of the stoichiometric amount of SDS was added as a solid. The experimental results showed that after three flotation steps, the overall recovery efficiency of copper was increased from 81.2 to 91.3%. The addition of extra steps increased the overall SDS concentration ([SDS]total) to 5.85 mmol L−1 and total time (ttotal) to 5 h (see Fig. 6c).
From the optimization of the ion flotation parameters, it can be concluded that copper was best separated over zinc under the following conditions: [SDS]total = 5.85 mmol L−1, ttotal = 5 h and three ion flotation steps. To evaluate the efficiency of the optimized technique on realistic conditions, the recovery of copper from the ammoniacal leachates (as described in the previous section) was tested. The overall recovery efficiency of copper from the ammoniacal PLS was 84.2%, slightly lower compared to that obtained from the synthetic ammoniacal leachates (91.3%). This can be attributed to the increase in ionic strength due to the presence of other metal ions in the PLS (Table S5). Prior studies demonstrated that the efficiency of ion flotation was negatively affected by the increase of ionic strength, because the foreign ions acted competitively with the colligend at the interface [71]. In the present study, calcium, magnesium, and aluminum were also recovered, but in small percentages of 1.2%, 0.8%, and 2.1%, respectively. Although these recovery efficiencies were negligible, this behavior indicated a competition between these metal ions and copper with SDS at the gas/liquid interface.
Equation (12) suggests that copper is concentrated to the foam phase as [Cu(NH3)4](DS)2 sublate (Fig. S4). The analysis results (Table 2) of the collected sublate confirmed this hypothesis. The measured elemental wt% showed very small deviations from the ones calculated using the proposed chemical formula. Furthermore, the presence of N–H bonds (Fig. 7) was confirmed by four strong infrared absorption bands at 3348 cm−1, 1630 cm−1, 1280 cm−1, and 1053 cm−1 [71]. The CH3 (2955 cm−1), CH2 (2918 cm−1, 2850 cm−1), SO3 (1220 cm−1), and C–S–O (988 cm−1) bands present in the FTIR spectrum of SDS were also found in the spectrum of the sublate, indicating interactions of the surfactant with the copper(II) tetraamine complex [72].

Comparative FTIR spectra of solid sodium dodecyl sulfate (SDS) and the sublate of ion flotation
From the optimization of the process in simulated leachates and its implementation to real leachates, it can be concluded that copper was selectively separated to the foam phase as [Cu(NH3)4](DS)2 sublate. Even though these results were promising for the implementation of ion flotation as metal recovery technique in hydrometallurgy, it must be noted that a considerable amount of SDS (5.85 mmol L−1) was required to obtain 85% recovery efficiency for copper. Based on the proposed chemical formula, at least two molecules of SDS are required for the adsorption of one copper(II) tetraamine complex to the gas/liquid interface. Hence, from an engineering point of view, ion flotation can be introduced to an overall metallurgical flowchart as metal separation technique only if the regeneration and re-use of the collector are possible. The decomposition of copper–dodecyl sulfate sublates has been already investigated [42]. The techniques that were tested after the breakage of the foam fraction were as follows: precipitation of metals as hydroxides or sulfides, chemical stripping, and direct electrolysis. The latter technique appeared to be the most promising method for recovering the colligend in metallic form and regenerating the collector, which remained in the solution after the electrolysis. Although the chemical structure of the formed sublate in this process was different ([Cu(NH3)4](DS)2 instead of Cu(DS)2), the same collector was used. Therefore, one of the above-mentioned techniques could be potentially applied for the regeneration and re-use of the surfactant.
Zinc Recovery by Precipitation
The zinc left in the leachate after the flotation was recovered by precipitation. The solution was heated up to 60 °C and compressed air was bubbled through the solution. The excess of ammonia was removed by the flow of compressed air and the pH of the solution was lowered. This treatment was expected to precipitate zinc as basic carbonate. Another solution, containing 2 mol L−1 of H2SO4, was used to trap the removed ammonia. The experimental results (Fig. 8, Table S9) showed that 95% zinc was recovered as a white precipitate over the course of 270 min. XRD analysis of the powder showed that it consisted of basic zinc carbonate, Zn5(CO3)2(OH)6, and the XRD diffractogram of which was resembling that of hydrozincite (Fig. S5) and it is in agreement with previous findings [73]. No other metal impurities were detected by ICP-OES analysis. The remaining solution had very low metal/ metalloid content (Table S10) and its pH was 8.67. Other techniques to recover zinc from ammoniacal solutions, such as sulfide precipitation, could be also applied [74].

Effect of bubbling time on the precipitation efficiency of zinc from the solution after ion flotation
Conclusions
In this work, the separation of copper by ion flotation from real ammoniacal leachates of microwave-roasted sulfidic tailings was investigated. The sulfidic tailings were first roasted at 550 °C for 60 min in the microwave oven. At this temperature, pyrite was oxidized to hematite and the metal sulfide phases were converted to more soluble metal sulfates. The microwave-roasted material was then leached with a mixed solution of ammonia and ammonium carbonate. The optimum leaching efficiencies of zinc (86%) and copper (75%) were obtained under the following conditions: L/S = 10 mL g−1, T = 90 °C, [NH3 + NH4+] = 4 mol L−1, NH3:NH4+ = 2:1, t = 5 h. From the generated PLS, it was possible to separate 85% of copper to the foam phase as a colloidal copper(II) tetraamine dodecyl sulfate sublate, with formula [Cu(NH3)4](DS)2. The optimal ion flotation conditions were found to be as follows: [SDS]total = 5.85 mmol L−1, [EtOH] = 0.5 % (v/v), ttotal = 5 h, flotation stages = 3. Zinc was recovered from the remaining solution as basic zinc carbonate at a yield of 95%, after heating up the solution at 60 °C and bubbling compressed air through the solution for 270 min. These results were found to be very promising for recovering copper from dilute aqueous ammoniacal leachates of low-grade sulfidic copper–lead–zinc tailings by ion flotation.
References
Lottermoser BG (2010) Mine wastes: characterization, treatment and environmental impacts. Springer, Berlin. https://doi.org/10.1007/978-3-642-12419-8
Gupta A, Yan D (2006) Mineral processing design and operation. Elsevier B.V., Amsterdam
Fuerstenau MC, Chander S, Woods R (2007) Sulfide mineral flotation. In: Fuerstenau MC, Jameson G, Yoon RH (eds) Froth flotation: a century of innovation. Society for Mining, Metallurgy and Exploration Inc., Littleton, pp 425–465
Kossoff D, Dubbin WE, Alfredsson M, Edwards SJ, Macklin MG, Hudson-Edwards KA (2014) Mine tailings dams: characteristics, failure, environmental impacts, and remediation. Appl Geochem 51:229–245. https://doi.org/10.1016/j.apgeochem.2014.09.010
Naidu G, Ryu S, Thiruvenkatachari R, Choi Y, Jeong S, Vigneswaran SA (2019) Critical review on remediation, reuse, and resource recovery from acid mine drainage. Environ Pollut. https://doi.org/10.1016/j.envpol.2019.01.085
Skousen JG, Ziemkiewicz PF, McDonald LM (2019) Acid mine drainage formation, control and treatment: approaches and strategies. Extr Ind Soc 6(1):241–249. https://doi.org/10.1016/J.EXIS.2018.09.008
Kefeni KK, Msagati TAM, Mamba BB (2017) Acid mine drainage: prevention, treatment options, and resource recovery: a review. J Clean Prod 151:475–493. https://doi.org/10.1016/j.jclepro.2017.03.082
Nieto JM, Sarmiento AM, Olías M, Canovas CR, Riba I, Kalman J, Delvalls TA (2006) Acid mine drainage pollution in the Tinto and Odiel Rivers (Iberian Pyrite Belt, SW Spain) and bioavailability of the transported metals to the Huelva Estuary. Environ Int 33(4):445–455. https://doi.org/10.1016/j.envint.2006.11.010
Park I, Tabelin CB, Jeon S, Li X, Seno K, Ito M, Hiroyoshi NA (2019) Review of recent strategies for acid mine drainage prevention and mine tailings recycling. Chemosphere 219:588–606. https://doi.org/10.1016/j.chemosphere.2018.11.053
Spooren J, Binnemans K, Björkmalm J, Breemersch K, Dams Y, Folens K, González-Moya M, Horckmans L, Komnitsas K, Kurylak W, Lopez M, Mäkinen J, Onisei S, Oorts K, Peys A, Pietek G, Pontikes Y, Snellings R, Tripiana M, Varia J, Willquist K, Yurramendi L, Kinnunen P (2020) Near-zero-waste processing of low-grade, complex primary ores and secondary raw materials in Europe: technology development trends. Resour Conserv Recycl 160(May):104919. https://doi.org/10.1016/j.resconrec.2020.104919
Binnemans K, Jones PT, Blanpain B, van Gerven T, Pontikes Y (2015) Towards zero-waste valorisation of rare-earth-containing industrial process residues: a critical review. J Clean Prod 99:17–38. https://doi.org/10.1016/j.jclepro.2015.02.089
Martín-Crespo T, Gómez-Ortiz D, Martín-Velázquez S (2019) Geoenvironmental characterization of sulfide mine tailings. In: Mazadiego LF, De Miguel Garcia E, Barrio-Parra F, Izquierdo-Diaz M (eds) Applied geochemistry with case studies on geological formations, exploration techniques and environmental issues. IntechOpen, London, pp 1–25
Ozer M, Acma E, Atesok G (2017) Sulfation roasting characteristics of copper-bearing materials. Asia-Pacific J Chem Eng 12(3):365–373. https://doi.org/10.1002/apj.2078
Khairuzzaman MQ (2016) Roasting of sulfide minerals. Physical chemistry of metallurgical processes. John Wiley & Sons Inc, New Jersey, pp 39–69
Khalafalla SE, Evans JW, Koo C-H, Sohn HY, Turkdogan ET, Cutler IB, Pitt CH (1979) Pyrometallurgical processes. Rate processes of extractive metallurgy. Plenum Press, New York, pp 245–428
Kingman SW, Rowson NA (1998) Microwave treatment of minerals—a review. Miner Eng 11(11):1081–1087. https://doi.org/10.1016/s0892-6875(98)00094-6
Yang K, Li S, Zhang L, Peng J, Chen W, Xie F, Ma A (2016) Microwave roasting and leaching of an oxide-sulphide zinc ore. Hydrometallurgy 166:243–251. https://doi.org/10.1016/j.hydromet.2016.07.012
Moravvej Z, Mohebbi A, Daneshpajouh S (2018) The microwave irradiation effect on copper leaching from sulfide/oxide ores. Mater Manuf Process 33:1–6. https://doi.org/10.1080/10426914.2016.1244850
Liu Z, Lei HY, Bai T, Zheng W, Chen K, Chen J, Jian H, Wen Q (2015) Microwave-assisted arsenic removal and the magnetic effects of typical arsenopyrite-bearing mine tailings. Chem Eng J 272:1–11. https://doi.org/10.1016/j.cej.2015.02.084
Meng X, Han KN (1996) Principles and applications of ammonia leaching of metals—a review. Miner Process Extr Metall Rev 16(1):23–61. https://doi.org/10.1080/08827509608914128
Williamson AJ, Verbruggen F, Chavez Rico VS, Bergmans J, Spooren J, Yurramendi L, Laing G, Boon N, Hennebel T (2021) Selective leaching of copper and zinc from primary ores and secondary mineral residues using biogenic ammonia. J Hazard Mater 403:123842. https://doi.org/10.1016/j.jhazmat.2020.123842
Jha MK, Kumar V, Singh RJ (2001) Review of hydrometallurgical recovery of zinc from industrial wastes. Resour Conserv Recycl 33(1):1–22. https://doi.org/10.1016/S0921-3449(00)00095-1
Prasad S, Pandey BD (1998) Alternative processes for treatment of chalcopyrite—a review. Miner Eng 11(8):763–781. https://doi.org/10.1016/S0892-6875(98)00061-2
Tang J, Liu S, Zheng C, Hu H, Ji X (2020) Zinc recovery from dilute ammoniacal media using an integrated solvent extraction and electrolysis process. Hydrometallurgy 198:105510. https://doi.org/10.1016/j.hydromet.2020.105510
Deep A, Kumar P, Carvalho JM (2010) Recovery of copper from zinc leaching liquor using ACORGA M5640. Sep Sci Technol 76(1):21–25. https://doi.org/10.1016/j.seppur.2010.09.015
Kordosky GA (1992) Copper solvent extraction: the state of the art. JOM 44(5):40–45. https://doi.org/10.1007/BF03223049
Sole KC, Hiskey JB (1995) Solvent extraction of copper by Cyanex 272, Cyanex 302 and Cyanex 301. Hydrometallurgy 37(2):129–147. https://doi.org/10.1016/0304-386X(94)00023-V
Doyle FM (2003) Ion flotation—its potential for hydrometallurgical operations. Int J Miner Process 72(1–4):387–399. https://doi.org/10.1016/S0301-7516(03)00113-3
Fu F, Wang Q (2011) Removal of heavy metal ions from wastewaters: a review. J Environ Manage 92:407–418. https://doi.org/10.1016/j.jenvman.2010.11.011
van Nguyen N, Lee J, Chun, J, Yoo MK, Jeong K (2009) Copper recovery from low concentration waste solution using Dowex G-26 resin. J Hydrometallurgy 97(3–4):237–242. https://doi.org/10.1016/j.hydromet.2009.03.003
Lemlich R (1972) Ion flotation. Adsorptive bubble separation techniques. Academic Press, New York, pp 53–68. https://doi.org/10.1016/b978-0-12-443350-2.x5001-1
Grieves RB (1975) Foam separations: a review. Chem Eng J 9:93–106. https://doi.org/10.1016/0300-9467(75)80001-3
Peleka EN, Matis KA (2011) Water separation processes and sustainability. Ind Eng Chem Res 50(2):421–430. https://doi.org/10.1021/ie100079s
Eivazihollagh A, Tejera J, Svanedal I, Edlund H, Blanco A, Norgren M (2017) Removal of Cd2+, Zn2+, and Sr2+ by Ion flotation, using a surface-active derivative of DTPA (C12-DTPA). Ind Eng Chem Res 56(38):10605–10614. https://doi.org/10.1021/acs.iecr.7b03100
Xanthopoulos P, Binnemans K (2021) Removal of cadmium, zinc, and manganese from dilute aqueous solutions by foam separation. J Sustain Metall. https://doi.org/10.1007/s40831-020-00322-2
Nicol SK, Galvin KP, Engel MD (1992) Ion flotation—potential applications to mineral processing. Miner Eng 5(10–12):1259–1275. https://doi.org/10.1016/0892-6875(92)90163-4
Sebba F (1959) Concentration by ion flotation. Nature 184:1062–1063. https://doi.org/10.1038/1841062a0
Shouci L, Pugh RJ (2005) Gas/liquid interfacial separation. Studies in interface science. Elsevier B.V., Amsterdam, pp 559–645
Chang L, Cao Y, Fan G, Li C, Peng WA (2019) Review of the applications of ion floatation: wastewater treatment, mineral beneficiation and hydrometallurgy. RSC Adv 9(35):20226–20239. https://doi.org/10.1039/c9ra02905b
Peng W, Chang L, Li P, Han G, Huang Y, Cao Y (2019) An overview on the surfactants used in ion flotation. J Mol Liq. https://doi.org/10.1039/c9ra02905b
Eivazihollagh A, Bäckström J, Norgren MEH (2018) Electrochemical treatment of copper complexed by chelating agent and chelating surfactant in alkaline solution using a membrane cell. J Chem Technol Biotechnol 93(5):1421–1431. https://doi.org/10.1002/jctb.5510
Doyle FM, Duyvesteyn KS (1995) The use of ion flotation for detoxification of metal-contaminated waters and process effluents. In: Proceedings of the XIX International Mineral Processing Congress, San Francisco, CA (United States), pp 22–27.
Masuyama A, Okano T, Okahara M, Section E (1990) Application of surface-active amide oximes to the collectors for gallium ion in an ion-flotation system. Ind Eng Chem Res 29:290–294. https://doi.org/10.1021/ie00098a02
Yenial Ü, Bulut G (2017) Examination of flotation behavior of metal ions for process water remediation. J Mol Liq 241:130–135. https://doi.org/10.1016/j.molliq.2017.06.011
Polat H, Erdogan D (2007) Heavy metal removal from waste waters by ion flotation. J Hazard Mater 148(1–2):267–273. https://doi.org/10.1016/j.jhazmat.2007.02.013
Mcdonald C, Suleiman A (1979) Ion flotation of copper using ethylhexadecyldimethyl-ammonium bromide. Sep Sci Technol 14(3):219–225. https://doi.org/10.1080/01496397908066960
Strel’tsov, K. A., Abryutin, D. v. (2010) Investigation of regularities of ion flotation of copper with the use of sodium diethyldithiocarbamate. Russ J Non-Ferrous Met 51(2):85–88. https://doi.org/10.3103/S106782121002001X
Stoica L, Lacatusu I (2012) Cu(II) Separation from diluted aqueous solutions by flotation with atypical collectors anti and syn 2-hydroxy-3,5-di-tert-butyl-benzaldoxime. Int J Environ Waste Manage 9(3–4):293–312. https://doi.org/10.1504/IJEWM.2012.046394
Eivazihollagh A, Svanedal I, Edlund H, Norgren M (2019) On chelating surfactants: molecular perspectives and application prospects. J Mol Liq 278:688–705. https://doi.org/10.1016/j.molliq.2019.01.076
Doyle FM, Liu Z (2003) The effect of triethylenetetraamine (Trien) on the ion flotation of Cu2+ and Ni2+. J Colloid Interface Sci 258(2):396–403. https://doi.org/10.1016/S0021-9797(02)00092-9
Liu Z, Doyle FM (2009) Ion flotation of Co2+, Ni2+, and Cu2+ using dodecyldiethylenetriamine (Ddien). Langmuir 25(16):8927–8934. https://doi.org/10.1021/la900098g
Lazaridis NK, Peleka EN, Karapantsios TD, Matis KA (2004) Copper Removal from effluents by various separation techniques. Hydrometallurgy 74(1–2):149–156. https://doi.org/10.1016/j.hydromet.2004.03.003
Jafari M, Abdollahzadeh AA, Aghababaei F (2017) Copper ion recovery from mine water by ion flotation. Mine Water Environ 36:323–327. https://doi.org/10.1007/s10230-016-0408-2
Carvalho JRS, Relvas JMRS, Pinto AMM, Frenzel M, Krause J, Gutzmer J, Pacheco N, Fonseca R, Santos S, Caetano P, Reis T, Gonçalves M (2018) Indium and selenium distribution in the Neves-Corvo deposit, Iberian Pyrite Belt, Portugal. Miner. Mag. 82(S1):S5–S41. https://doi.org/10.1180/minmag.2017.081.079
Rezvanipour H, Mostafavi A, Ahmadi A, Karimimobarakabadi M, Khezri M (2018) Desulfurization of iron ores: processes and challenges. Steel Res Int 89(7):1–14. https://doi.org/10.1002/srin.201700568
Everaert M, Lemmens V, Atia TA, Spooren J (2020) Sulfidic Mine Tailings and marl waste rock as compatible resources in a microwave-assisted roasting process. J Clean Prod 274:122628. https://doi.org/10.1016/j.jclepro.2020.122628
Mos YM, Vermeulen AC, Buisman CJ, Weijma J (2018) X-ray diffraction of iron containing samples: the importance of a suitable configuration. Geomic J 35(6):511–517. https://doi.org/10.1080/01490451.2017.1401183
Wang L, Fan BW, He YT, Li P, Yin DQ, Hu YH (2014) Characteristics of minerals and their associations of transformation processes in pyrite at elevated temperatures: an X-ray diffraction study. Ironmak Steelmak 41(2):147–152. https://doi.org/10.1179/1743281213y.0000000113
Tullin C, Ljungström E (1989) Reaction between calcium carbonate and sulfur dioxide. Energy Fuels 3(3):284–287. https://doi.org/10.1021/ef00015a003
Dean JA (1999) Lange’s handbook of chemistry, 15th edn. McGraw-Hill, New York
Mena M, Olson FA (1985) Leaching of chrysocolla with ammonia-ammonium carbonate solutions. Metall Trans B 16(3):441–448. https://doi.org/10.1007/BF02654842
Rodriguez Rodriguez N, Gijsemans L, Bussé J, Roosen J, Önal MAR, Masaguer Torres V, Manjón Fernández Á, Jones PT, Binnemans K (2020) Selective removal of zinc from BOF sludge by leaching with mixtures of ammonia and ammonium carbonate. J Sustain Metall 6(4):680–690. https://doi.org/10.1007/s40831-020-00305-3
Han J, Liu W, Xue K, Qin W, Jiao F, Zhu L (2017) Influence of NH4HF2 activation on leaching of low-grade complex copper ore in NH3-NH4Cl solution. Sep Purif Technol 181:29–36. https://doi.org/10.1016/j.seppur.2017.03.012
Liu X, Jiang T, Xu B, Zhang Y, Li Q, Yang Y, He Y (2020) Thiosulphate leaching of gold in the Cu–NH3–S2O32- -H2O system: an updated thermodynamic analysis using predominance area and species distribution diagrams. Miner Eng 151:106336. https://doi.org/10.1016/j.mineng.2020.106336
Ding ZY, Chen QY, Yin ZL, Liu K (2013) Predominance diagrams for Zn(II)-NH3-Cl-H2O System. Trans Nonferrous Met Soc China (English Ed) 23(3):832–840. https://doi.org/10.1016/s1003-6326(13)62536-4
Motin MA, Hafiz Mia MA, Nasimul Islam AKM (2015) Thermodynamic properties of sodium dodecyl sulfate aqueous solutions with methanol, ethanol, n-propanol and iso-propanol at different temperatures. J Saudi Chem Soc 19(2):172–180. https://doi.org/10.1016/j.jscs.2012.01.009
Liu Z, Doyle FM (2001) Modeling metal ion removal in alkylsulfate ion flotation systems. Mining Metall Explor 18(3):167–171. https://doi.org/10.1007/bf03402891
Liu Z, Doyle FM (2001) A thermodynamic approach to ion flotation. II. metal ion selectivity in the SDS-Cu-Ca and SDS-Cu-Pb systems. Colloids Surf A Physicochem Eng Asp 178(1–3):93–103. https://doi.org/10.1016/S0927-7757(00)00554-9
Liu Z, Doyle FMA (2001) Thermodynamic approach to ion flotation I Kinetics of cupric ion flotation with alkylsulfates. Colloids Surf A Physicochem Eng Asp 178(1–3):79–92. https://doi.org/10.1016/S0927-7757(00)00555-0
Caballero M, Cela R, Perez-Bustamante JA (1990) Analytical applications of some flotation techniques—a review. Talanta 37(3):275–300. https://doi.org/10.1016/0039-9140(90)80056-L
Zubrick WJ (1984) The organic chem lab survival manual. John Wiley & Sons Inc, New York
Hoseinian FS, Rezai B, Kowsari E, Safari M (2018) Kinetic study of Ni(ii) removal using ion flotation: effect of chemical interactions. Miner Eng 119:212–221. https://doi.org/10.1016/j.mineng.2018.01.028
Sinhamahapatra A, Giri AK, Pal P, Pahari SK, Bajaj HC, Panda AB (2012) A rapid and green synthetic approach for hierarchically assembled porous ZnO nanoflakes with enhanced catalytic activity. J Mater Chem 22(33):17227–17235. https://doi.org/10.1039/C2JM32998K
Harvey TG (2006) The hydrometallurgical extraction of zinc by ammonium carbonate: a review of the Schnabel process. Miner Process Extr Metall Rev 27(4):231–279. https://doi.org/10.1080/08827500600815271
Acknowledgements
The research leading to these results has received funding from the European Community’s Horizon 2020 Programme under Grant Agreement No. 812580 (MSCA- ETN SULTAN). This publication reflects only the authors’ view, exempting the Community from any liability. The authors thank Bart van Huffel (KU Leuven) for helping with the CHN analysis and Myriam Mertens (VITO) for the Rietveld refinement of the sulfidic tailings and the roasted material. The authors would like also to express their gratitude to Dženita Avdibegović (KU Leuven) and Srećko Bevandić (KU Leuven) for the fruitful scientific discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
The contributing editor for this article was D. Panias.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
40831_2021_363_MOESM1_ESM.pdf
Supplementary file1 The supporting information is available on the Springer Nature website and includes the elemental composition of sulfidic tailings, microwave-roasted material, and solid leaching residue (Table S1), the TGA analysis of sulfidic tailings sample (Fig. S1), the HSC simulation (Fig. S2), the XRD of the microwave-roasted material (Fig. S3), the metal concentration (mg L‒1) in the PLS (Tables S2–S5), the residual metal concentration (mmol L‒1) of ion flotation experiments (Tables S6–S8), a picture of the collected sublate (Fig. S4), the residual zinc concentration (mg L‒1) of the precipitation experiments (Table S9), the XRD of the precipitate (Fig. S5), and the metal concentration (mg L‒1) of the final solution (Table S10). (PDF 435 kb)
Rights and permissions
About this article

Cite this article
Xanthopoulos, P., Kalebić, D., Kamariah, N. et al. Recovery of Copper from Ammoniacal Leachates by Ion Flotation. J. Sustain. Metall. 7, 1552–1564 (2021). https://doi.org/10.1007/s40831-021-00363-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-021-00363-1