
Abstract
Purpose of Review
Hematopoietic stem cells (HSCs) propagate the hematopoietic system throughout the lifetime of an individual. Beyond homeostatic regulation, HSCs respond to many stressors, including infection, aging, irradiation, and chemotherapy, through distinct cellular and molecular pathways to restore homeostasis. Here, we review how HSCs and bone marrow niche cells respond to various stressors and their role in HSC regeneration.
Recent Findings
In this review, we summarize the manner in which HSCs respond to different stressors via intrinsic and extrinsic, and niche-driven mechanisms to support hematopoietic regeneration. We discuss recent work defining the cellular and molecular mechanisms by which HSCs respond to various forms of stress through specific alterations in cell cycling, DNA damage repair, and cell death. We also summarize the roles of recently defined bone marrow niche cell subtypes and niche-derived factors in mediating HSC regeneration.
Summary
Stress through aging, inflammation, and myelosuppressive treatments significantly alters hematopoietic homeostasis, requiring HSCs to quickly respond to restore order. In the event that an inadequate HSC response occurs, patients are at risk for life-threatening complications such as hemorrhage, infection and bone marrow failure. In this review, we summarize recent work defining how HSCs respond to stress and the role of the bone marrow niche in HSC regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
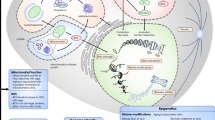
Hematopoietic stem cells (HSCs) are responsible for maintaining and regenerating the adult hematopoietic system throughout the lifetime of an individual. External and environmental stress incurred by HSCs leads to significant alterations in HSC behavior, often leading to permanent cellular and molecular consequences (Fig. 1). In this review, we will summarize how intrinsic and extrinsic mechanisms regulate the HSC response to stress and hematopoietic regeneration.

Hematopoietic stem cell response to stress. At the left, HSC properties during homeostasis. At the baseline, HSCs display self-renewal (curved arrow), increased quiescence, and repopulate the hematopoietic system with normal proportions of myeloid cells, B cells, T cells, and other hematopoietic cell subtypes. At right, HSC response to stress can lead to HSC exhaustion defined by a loss of self-renewal (dotted arrow). Stress induces DNA damage, which is inefficiently repaired. HSC apoptosis occurs, driven by p53, PUMA, and other effectors, along with HSC senescence, characterized by increased expression of p21 and p16. Stress induces unbalanced hematopoietic repopulation, characterized by an increased production of myeloid cells, or myeloid skewing
Mechanisms of HSC Stress
HSCs are at the top of the hematopoietic hierarchy, through which they support the maintenance and regeneration of the hematopoietic system via self-renewal and hematopoietic lineage specification [1]. HSCs primarily exist in the quiescent state [2] but are capable of entering cell cycle to generate mature hematopoietic cells to supplement hematopoietic cell turnover during homeostasis. Meanwhile, HSC activation and downstream hematopoietic cell production can occur in response to chemical and biological stressors, which will be detailed below.
Aging
Comprehensive studies in mice have enabled a deep analysis of the cellular and molecular consequences of aging on HSCs. First, in older mice, there are increased phenotypic long-term–HSCs (LT-HSCs) compared to young mice [3]. However, the long-term repopulating ability of aged HSCs is significantly reduced compared to young mice, revealing that HSC function is lost during aging. Another characteristic of aging is myeloid skewing, or the accumulation of myeloid cells in the peripheral blood compared to other hematopoietic lineage cells [3,4,5]. Following transplantation, aged HSCs also display increased myeloid engraftment and decreased B cell engraftment compared to young HSCs [3].
Other unique characteristics of aged HSCs include changes in polarity, cell cycle status, and DNA damage accumulation. For example, there is a decreased frequency of early hematopoietic progenitor cells exhibiting a polar distribution of microtubules in aged mice compared to young mice [6]. Aged LT-HSCs exhibit an apolar distribution of Cdc42 and tubulin compared to young LT-HSCs [7]. Aged HSCs exhibit increased accumulation of DNA damage compared to young HSCs. This can be explained by the broad reduction in DNA damage repair mechanisms in quiescent HSCs in aging mice and because aged HSCs enter cell cycle at a lower frequency compared to young HSCs [8, 9]. Induction of cell cycling promotes DNA repair in aged HSCs [9]. Consistent with these findings, HSCs from aged mice demonstrate increased γH2AX staining compared to HSCs from younger mice [10••]. HSCs from older humans also display increased DNA damage at baseline compared to HSCs from young donors [11]. While aged HSCs possess distinct cell morphology, proliferation, and DNA damage characteristics, the molecular mechanisms underlying these fundamental differences in HSC properties remain incompletely understood.
Inflammation
Inflammatory signals resulting from infection, injury, or aging can alter the maintenance of the HSC pool. Inflammatory signals promote both the expansion of the HSC compartment and differentiation of HSCs to generate immune cells. For example, following the administration of lipopolysaccharide to mice, c-Kit+Sca-1+Lineage− (KSL) hematopoietic stem/progenitor cells (HSPCs) undergo increased cell division [12, 13]. Mycobacterium avium infection was also shown to induce the proliferation of side population KSL HSCs [14]. Consistent with these findings, exposure of KSL CD34− CD48− CD150+ FLT3− LT-HSCs to IL-6, TNF and CC-chemokine ligand 2 (CCL2) promotes HSC proliferation [15]. While there is a clear role for inflammation in HSC expansion, this comes at the expense of HSC self-renewal, which is significantly diminished following inflammatory signaling of IL-6, TNF, TLRs, IFNs, and IL-1, through various molecular mechanisms [14,15,16,17, 18•, 19].
Although inflammation-induced HSC expansion diminishes HSC self-renewal capacity, HSC proliferation is necessary to generate immune cells, which respond to infection. Granulocytes serve a critical role in controlling infection but are short-lived due to their function in pathogen phagocytosis and degranulation [20]. As such, during infection, there is an intense demand for granulocyte production, termed emergency granulopoiesis. While hematopoietic progenitor cells are primarily responsible for producing granulocytes [21], granulocyte production can also be stimulated by HSC activation directly. HSCs that express the interferon gamma receptor have been shown to drive myeloid cell production [22, 23]. While inflammatory cytokines promote HSC proliferation, HSCs eventually return to their dormant state despite persistent inflammatory signaling [18•, 24, 25]. In the setting of sustained inflammatory stress, HSCs exhibit myeloid skewing and diminished lymphoid cell production [26].
Ionizing Radiation and Chemotherapy
Ionizing radiation (IR) and chemotherapy are routinely utilized in the treatment of patients with cancer and cause direct damage to HSCs. Analysis of the regenerative response of bone marrow (BM) hematopoietic stem and progenitor cells to irradiation has demonstrated significant variability in their sensitivities to stress [27,28,29,30]. The variation in stress response is likely due to the heterogeneity within the populations of hematopoietic stem and progenitor cells that were analyzed. With the recent identification of well-defined HSC surface markers [31,32,33] coupled with flow cytometry, additional studies have demonstrated a more clearly defined stress response in the HSC population [34, 35].
The damage incurred to hematopoietic stem and progenitor cells can result in acute or chronic bone marrow injury. For example, in the short-term, IR causes a rapid loss of proliferating hematopoietic progenitor cells due to apoptotic death, leading to a massive reduction in the ability to replenish mature hematopoietic elements over 1 to 4 weeks [36]. Meanwhile, IR also induces HSC apoptosis, which results in longer-term bone marrow injury and loss of HSC repopulating capacity [36]. Several studies have suggested an important role for apoptotic mechanisms in the response of HSCs to IR. For example, overexpression of Bcl2 protects HSCs and HSC function after lethal irradiation [37, 38], while Bcl2−/− mice exhibit decreased hematopoietic colony survival following irradiation compared to Bcl2+/+ mice [39]. While HSCs are more resistant to apoptosis than more proliferative hematopoietic progenitor cells, they are still susceptible to IR-induced cell death. For example, HSC-mediated apoptosis occurs through p53-mediated mechanisms [40,41,42,43]. Deletion of the pro-apoptotic protein, p53 upregulated modulator of apoptosis (PUMA), selectively protects HSPCs following IR injury [44]. To counterbalance the effects of PUMA, hematopoietic progenitor cells also upregulate expression of the transcriptional repressor, Slug, under the control of p53 in response to irradiation, thereby suppressing p53-mediated induction of PUMA [45]. While apoptosis has a clear role in the HSC response to irradiation, it is thought that a combination of programmed HSC death, indirect damage to the bone marrow microenvironment, and permanent HSC cell cycle arrest, or senescence, collectively impair HSC function, leading to long-term BM failure [36]. An important limitation of many studies performed to date has been the analysis of hematopoietic stem and progenitor populations, such as KSL cells, which are mostly progenitor cells, not HSCs, and may not therefore provide precise insight into the response of more purified HSC populations to IR. Importantly, Morhrin et al. evaluated the response of more purified ckit+sca-1+lin−flk2− HSPCs and found that this population of HSPCs displayed increased expression of pro-survival genes compared to committed myeloid progenitor cells and predominantly underwent growth arrest, rather than apoptosis, following 2 Gy IR [46••]. Further studies are required to analyze the interplay between apoptosis, necrosis, and senescence mechanisms in purified HSC populations, as well as assessment of niche-derived signals that contribute to the HSC response to IR.
Chemotherapy, like ionizing radiation, can induce HSC apoptosis and cell-cycle defects. The chemotherapeutic agent 5-flurouracil (5-FU) selectively kills cycling HSCs and diminishes HSC repopulating capacity as measured by competitive transplantation assays [47]. 5-FU treatment also leads to increased donor myeloid and decreased donor lymphoid cell production upon repopulation, similar to the hematopoietic lineage skewing seen in physiological aging. Additional studies have shown that induction of HSC cell cycle arrest prior to chemotherapy administration protects HSCs from exhaustion [48].
Cellular and Molecular Consequences of HSC Stress or Injury
Proliferation
As previously mentioned, LT-HSCs primarily exist in a quiescent state during homeostasis [49]. To enable hematopoietic regeneration following stress-induced depletion of mature hematopoietic cells, HSCs must exit quiescence to enter the cell cycle. Chronic stress can activate HSCs, leading to increased HSC proliferation and elevated numbers of bone marrow LT-HSCs and hematopoietic progenitor cells [50]. Several molecules regulate HSC proliferation in response to stress. For example, Inhibitor of DNA binding 1 (Id1) mediates LT-HSC proliferation; BrdU analyses revealed that Id1−/− HSCs are less proliferative than Id1+/+ HSCs following 5-FU chemotherapy administration. Further analyses demonstrated that Id1 expression protects HSC from exhaustion in response to chronic stressors such as aging and toll-like receptor (TLR) signaling [51]. Another key regulator of HSC response to stress is the kinase p38a, which is crucial for mouse recovery from 5-FU chemotherapy administration. Additionally, p38a is critical for induction of HSC cell cycling following BM transplantation [52].
Senescence
While acute and chronic stresses promote HSC proliferation to enable differentiation and ultimately, hematopoietic recovery, HSCs have a limited replicative capacity. Senescence is the state in which HSCs have lost their ability to proliferate and differentiate [53]. Two thoroughly characterized pathways that initiate cellular senescence are the p53-p21Cip1/Waf1 or p19Arf-Mdm2-p53-p21Cip1/Waf1 pathway and the p16Ink4a-Rb pathway [27, 54,55,56]. Specialized studies examining the expression of major components of these pathways in HSCs under stress demonstrated that irradiation induced the expression of p21Cip1/Waf1, p16Ink4a, and p19Arf in long-term BM cultures [27]. Additional analyses revealed that irradiation also induced the expression of SA-β-galactosidase, a well-characterized senescence marker [57]. Follow-up studies examining the functional role of p16Ink4a and p19Arf in regulating HSC self-renewal demonstrated that upon serial BM transplantation, there was a modest increase in survival in mice transplanted with HSCs that lacked Ink4a-Arf expression [58].
DNA Damage
Stressors such as irradiation, chemotherapy, or aging can lead to aberrations in chromosomal structure, which require repair to restore the integrity of the genome. Through molecular sensing of DNA damage, the DNA damage response (DDR) pathway is initiated to repair chromosomal insults and reestablish genome integrity. In response to gamma-irradiation, HSCs exhibit delayed double strand break repair and prolonged p53-mediated DNA damage response activation compared to hematopoietic progenitor cells [59]. Another study using mouse HSPCs demonstrated that long-lived HSCs exhibit distinct responses to irradiation, including expression of pro-survival genes, and induction of p53-mediated DNA damage response compared to short-lived myeloid progenitor cells, which were more prone to undergo apoptosis following genotoxic stress [46••]. Importantly, the authors further demonstrated that quiescent HSCs more frequently undergo non-homologous end joining (NHEJ) rather than homologous recombination (HR), which renders HSCs more susceptible to mutagenesis. Additional studies demonstrated that thrombopoietin (TPO) stimulates the DDR by promoting NHEJ at the time of γ-radiation or chemotherapy administration [60]. Recent work from our lab has also shown that epidermal growth factor (EGF) promotes HSC DNA repair and hematopoietic regeneration through augmentation of DNA-protein kinase-catalytic subunit (DNA-PKcs) activity and nonhomologous end-joining (NHEJ) repair [61].
In addition to irradiation, aging leads to the accumulation of DNA damage in HSCs [10••]. Nucleotide excision repair (NER) and NHEJ mechanisms decline in LT-HSCs during aging, leading to alterations in LT-HSC self-renewal, proliferation, and apoptosis. Human CD34+CD38− HSCs from elderly donors exhibit increased DNA damage compared to middle-aged patients [62]. As HSC quiescence is a critical contributor to the DDR, researchers analyzed HSC cell cycle status during aging and determined that older HSCs do not cycle more frequently than young counterparts [11]. This suggests that enhanced cycling is not the driving factor promoting the accumulation of DNA damage during aging [11]. Building upon these studies, it was shown that induction of HSC cycling by culturing cells in cytokine-rich media or in vivo induction via 5-FU treatment promoted DNA damage repair regardless of age [8]. Interestingly, Flach et al. showed that the accumulation of γH2AX foci in aged HSCs occurs through replication stress, rather than deficient DNA repair [63••]. Further studies will be required to uncover the intricacies of the molecular mechanisms through which DNA damage regulates HSC fate in response to different stressors.
HSC Intrinsic Responses to Stress
In addition to the pathways outlined above, HSPCs respond to stress by initiating signaling that promotes HSC recovery and hematopoietic regeneration. For example, human BM CD34+ cells express vascular endothelial growth factor (VEGF) and VEGF signaling promotes human cord blood HSPC survival following irradiation [64]. Other studies demonstrated that VEGF-deficient BM cells are incapable of restoring the hematopoietic system following lethal irradiation compared to wild-type cells [65]. Importantly, blockade of external VEGF activity had no effect on HSPC regeneration, suggesting that a VEGF–VEGFR2 internal autocrine loop regulates this process [65]. More recent studies by Chen et al. using Vav1-Cre;Vegfr2fl/fl mice demonstrated no significant change in transplantation efficiency compared to control mice [66]. However, upon diphtheria toxin–mediated depletion of hematopoietic cells that express Vegfr2 (Vegfr2ΔHC), the authors observed a significant increase in CD11b+ myeloid cells, indicating a role for hematopoietic-mediated VEGFR2 signaling in regulating hematopoietic cell differentiation. In a fascinating study, Zhou et al. demonstrated that HSCs secrete Angiopoietin-1 and that deletion of Angiopoietin-1 from hematopoietic cells decreased by vascular and hematopoietic recovery following irradiation in mice, at the expense of increased vascular leakiness [67]. Of note, Chen et al. also showed that VEGF produced by transplanted HSPCs likely promotes the regeneration of VEGFR2+ BM ECs, which also contributes to hematopoietic reconstitution in vivo. Another tyrosine kinase receptor involved in hematopoietic response to stress is the fibroblast growth factor receptor (FGFR). Using a Scl-Cre;Fgfr1fl/fl model, the authors demonstrated a significant reduction in KSL HSPCs in the BM compared to control littermates following 5-FU treatment, demonstrating a role for FGFR1 signaling in regulating hematopoietic recovery [68]. Further mechanistic analysis demonstrated that FGFR1 signaling critically regulates HSPC mobilization in response to BM damage [68].
Extrinsic Regulation of HSC Regeneration
Growth Factors
The HSC microenvironment, or niche, consists of cellular and soluble components which support HSC function. Following injury, the BM niche is significantly remodeled, leading to changes in the production of paracrine factors that impact HSC regeneration (Fig. 2). Previous studies have shown that systemic administration of hematopoietic cytokines, stem cell factor (SCF), TPO, or Flt3 ligand within 2 hours following high dose TBI caused radioprotection and increased survival in mice [69]. However, it remains less clear how myelosuppressive injury affects the endogenous concentrations of such factors in the BM and how specific BM niche cells regulate the production of such cytokines during hematopoietic regeneration in vivo. One such extrinsic mediator is SCF, which is the ligand for the c-Kit receptor tyrosine kinase. SCF expressed by BM adipocytes and leptin receptor (LepR)–expressing stromal cells was shown to be essential for HSC regeneration following myelosuppression, demonstrating a positive role SCF on regeneration [70, 71, 72•]. Although SCF has been previously shown to suppress HSC apoptosis and promote HSC self-renewal in combination with other cytokines [73,74,75], the mechanism of action of SCF in promoting HSC regeneration following total body irradiation remains unclear.

Bone marrow niche remodeling in response to stress. Schematic representation of extrinsic mechanisms of regulation of the HSC response to stress or injury. Major cellular components of the bone marrow niche, including endothelial cells, stromal cells, osteolineage cells, megakaryocytes, granulocytes, and adipocytes produce soluble proteins such as SCF, PTN, TPO, EGF, DKK1, Noradrenaline, TNFα, FGF1, TGFβ, Angiopoietin, and VEGF in response to injury
Another potential regulator of HSC regeneration is stromal cell-derived factor 1 (SDF-1, CXCL12), which is expressed by LepR+ stromal cells and Prx1+ mesenchymal progenitor cells and is essential for HSC maintenance in homeostasis [76, 77]. At homeostasis, SDF-1 binds to the C-X-C motif chemokine receptor 4 (CXCR4), which is expressed by HSCs and promotes HSC quiescence [78]. However, conditional loss of Cxcl12 expression improved hematopoietic regeneration and extended survival in mice following 5-FU treatment, demonstrating an inhibitory role in regeneration [79]. It remains unclear whether the cellular source of CXCL12 impacts HSC regeneration following myelotoxicity. Interestingly, chronic neurobehavioral stress increased HSPC proliferation in mice in association with an augmented inflammatory response [50]. HSPC proliferation in this model was caused by increased sympathetic nerve production of noradrenaline and activation of beta-3 adrenergic receptors on BM stromal cells, thereby decreasing stromal cell production of CXCL12 [50].
Inflammation plays a central role in the hematopoietic response to stresses such as infection and genotoxic stresses, including irradiation and chemotherapy. A key mediator of the inflammatory response is tumor necrosis factor-alpha (TNF-alpha), which can mediate either cellular apoptosis or necroptosis in target cells [80, 81]. In a fascinating study, Yamashita et al. demonstrated that TNF-alpha promoted HSC survival during inflammation via an NFkB-dependent mechanism, while myeloid progenitor cells underwent apoptosis [81]. Furthermore, the authors showed that TNF-alpha drives myeloid regeneration from the HSC pool in the early stages of inflammation. Taken together, this study revealed a powerful and complex role for TNF-alpha in regulating the HSC response to inflammation.
Endothelial Cells
BM endothelial cells (ECs) have an essential role in regulating HSC regeneration in vivo. Hooper et al. showed that deletion of VEGFR2 in Tie2+ ECs caused a delay in BM vascular recovery and concordant hematopoietic failure in irradiated mice [82••]. Salter et al. also showed that treatment of irradiated mice with anti-VE-cadherin antibody delayed BM vascular regeneration and hematologic recovery in mice [83]. More recently, Chen et al. described an important role for Apelin+ BM ECs in regulating hematopoietic reconstitution in the setting of BM transplantation [66]. We and other laboratories have shown that BM ECs secrete paracrine factors, including pleiotrophin (PTN), which promotes HSC regeneration via binding and inhibition of receptor protein tyrosine phosphatase–zeta (PTPZ) expressed on HSCs [84, 85]. Doan et al. showed that BM ECs also secrete epidermal growth factor (EGF) in response to TBI and EGF signaling is necessary for HSC regeneration to occur over time [61, 86]. Interestingly, BM osterix+ osteoprogenitor cells secrete the Wnt antagonist, Dickkopf-1 (Dkk1), in response to TBI, and Dkk1 acts on BM ECs to induce secretion of EGF, thereby also promoting HSC regeneration in vivo [87]. Guo et al. reported that EC-specific deletion of the Notch ligand, Jagged-2, had no effect on steady state hematopoiesis but suppressed HSPC regeneration following irradiation [88]. EC-specific deletion of Jagged 1 also suppressed hematopoietic progenitor cell recovery and decreased survival in irradiated mice [89]. Xu et al. recently utilized the combination of podoplanin and Sca-1 expression to discriminate arterial and sinusoidal BM ECs and reported that arterial BM ECs (CD45−Ter119−Sca-1brightPDPN−) were the primary EC source of SCF. Deletion of SCF from arterial ECs caused depletion of HSCs in steady state and suppressed hematopoietic regeneration following myeloablation [70]. Taken together, these studies demonstrate that BM ECs provide an orchestration of paracrine signals that regulate HSC regeneration following myelosuppression.
Mesenchymal Stromal Cells
BM mesenchymal stromal cells contribute to the BM perivascular niche and have been characterized by the expression of leptin receptor (LepR), nestin-GFP and platelet-derived growth factor–alpha (PDGFR-alpha) [90, 91]. In steady state, LepR+ stromal cells are essential sources of SCF and CXCL12, which maintain the HSC pool [76, 77, 92, 93]. LepR+ stromal cells also contribute indispensably to HSC maintenance by secretion of PTN [85]. In irradiated mice or mice treated with 5FU, LepR+ stromal cells also have an essential role via secretion of SCF, whereas SCF production by BM ECs or osteoblasts was shown to be non-essential following myelosuppression [72•].
CXCL12 production by BM mesenchymal stromal cells is essential for HSC maintenance in homeostasis [76, 77], but the specific requirement for CXCL12 from BM mesenchymal stromal cells in regulating HSC regeneration has not been resolved. Conditional deletion of CXCL12 in adult mice was associated with accelerated hematologic and progenitor cell recovery following 5FU chemotherapy, although the specific contributions of BM stromal cells or other niche cell sources of CXCL12 to this phenotype were not addressed [79]. Separately, Himburg et al. showed that BM LepR+ stromal cells secrete PTN in maintaining the HSC pool in homeostasis, but PTN expression by LepR+ stromal cells was not essential for HSC regeneration following TBI [85].
Adipocytes
BM adipocytes comprise a relatively small percentage of the BM niche cell population in young mice, but increase in relative frequency following myeloablation [72•]. Naveiras et al. showed that A-ZIP/F1 “fatless” mice displayed increased hematopoietic engraftment following BM transplantation, suggesting that adipocytes negatively regulate HSC function [94]. Subsequently, Zhou et al. demonstrated that BM adipocytes are a necessary source of SCF to promote hematopoietic regeneration following irradiation or 5FU treatment [72•]. It remains unclear whether adipocytes also secrete other HSC regenerative factors that influence hematopoietic reconstitution, but adipocytes clearly have an important role in the regulation of hematopoietic regeneration.
Osteolineage Cells
The general importance of BM osteolineage cells in promoting hematopoietic reconstitution following BM transplantation was shown by Caselli et al. [95]. Subsequent studies suggested that BM megakaryocytes promote BM osteolineage cell recovery following TBI, which in turn, promoted hematopoietic reconstitution following BM transplantation [96]. More recently, single-cell RNA sequencing analysis of BM osteolineage cells in proximity to engrafted HSCs revealed upregulation of an RNase, Angiogenin, which enhances hematopoietic regeneration of transplanted hematopoietic cells by preserving HSC stemness and promoting myeloid progenitor cell proliferation [97, 98]. In separate studies, Himburg et al. reported that BM osterix+ osteoprogenitor cells expressed the Wnt inhibitor, Dickkopf-1 (Dkk1), after TBI and Dkk1 promoted HSC regeneration in irradiated mice via direct effects on HSCs and indirectly via induction of EGF secretion by BM ECs [85].
Megakaryocytes
Megakaryocytes are multinucleated cells responsible for platelet production, and also regulate HSC quiescence in steady state via a variety of mechanisms [99]. Megakaryocytes secrete the chemokine, CXCL4, and also TGF-beta, which each directly promote HSC quiescence in the niche [71, 100]. Separately, megakaryocytes were demonstrated to promote HSC quiescence through production of thrombopoietin, which is under the control of CLEC-2 signaling in megakaryocytes [101, 102]. Megakaryocytes also indirectly regulate hematopoietic reconstitution following irradiation and BM transplantation via remodeling of the osteoblastic niche [96]. Importantly, Zhao et al. demonstrated that megakaryocytes also directly promote HSC regeneration following chemotherapy through secretion of FGF1 [71].
Myeloid Cells
Myeloid cells, particularly macrophages, have been shown to have an important role in regulating the retention and maintenance of HSCs in the niche and this topic has been reviewed elsewhere [99]. In the context of myelotoxicity or myeloablation, the role of differentiated myeloid cells is less well understood. However, granulocytes from transplanted donor BM were shown to mediate BM vascular and hematopoietic regeneration in myeloablated recipient mice [103]. The authors further showed that granulocytes produced TNF-alpha which acted on TNF receptor—expressing BM vascular ECs to promote vascular regeneration in the BM and that the vascular regeneration induced by transplanted granulocytes was dependent on TNF-alpha/TNF receptor signaling in vivo [103].
Sympathetic Nervous System
Seminal studies have demonstrated the critical role of the sympathetic nervous system (SNS) in regulating HSC retention and egress from the BM niche [99, 104, 105]. Sympathetic nerves regulate HSC retention via the secretion of noradrenaline and noradrenaline action on beta-3 adrenergic receptors expressed on Nestin-GFP+ BM stromal cells, which in turn regulate CXCL12 expression and gradient levels in the niche [106]. Importantly, the SNS has been shown to have an essential role in mediating both circadian oscillations of HSC egress from the BM and GCSF-mediated HSC mobilization [104, 105]. It has also been shown that multiple cycles of platinum-based chemotherapy causes sympathetic nerve injury in the BM [107]. Additional analyses demonstrated that depletion of sympathetic nerves or suppression of β-adrenergic signaling in the BM suppressed hematopoietic recovery following chemotherapy and this was mediated via the delayed recovery of BM Nestin+ stromal cells and ECs [107]. Genetic or pharmacologic neuroprotection was shown to improve hematopoietic recovery after myeloablation and improve recovery of BM stromal cells and ECs. Taken together, these studies suggest that targeting of the SNS could have therapeutic effects on hematopoietic regeneration following myelotoxicity.
Summary
In the past decade, extraordinary progress has been made toward elucidating the intrinsic and extrinsic mechanisms that control the HSC response to stress and the process of HSC regeneration. These discoveries have transformed our fundamental understanding of the hematopoietic response to genotoxic stresses such as chemotherapy and irradiation, infection, and aging, while also providing insights into longitudinal propensities for clonal hematopoiesis and malignant transformation over time [108, 109]. The next frontier will feature the development of targeted therapies which modulate the autonomous and non-cell autonomous responses of HSCs to stress for the purpose of augmenting hematologic and immune reconstitution in patients, while at the same time lessening risk for clonal hematopoiesis and premature aging.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2(6):640–53.
Venezia TA, et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2(10):1640–51.
Rossi DJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194–9.
Kim M, Moon HB, Spangrude GJ. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann N Y Acad Sci. 2003;996:195–208.
Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106(4):1479–87.
Kohler A, et al. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones. Blood. 2009;114(2):290–8.
Florian MC, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10(5):520–30.
Beerman I, et al. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15(1):37–50.
de Haan G, Nijhof W, Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89(5):1543–50.
•• Rossi DJ, et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9 Describes the role of DNA damage repair mechanisms in regulating HSC fitness during aging.
Rossi DJ, et al. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle. 2007;6(19):2371–6.
Nagai Y, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24(6):801–12.
Takizawa H, et al. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med. 2011;208(2):273–84.
Baldridge MT, et al. Quiescent hematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465(7299):793–7.
Chen C, et al. Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J Clin Invest. 2010;120(11):4091–101.
Essers MA, et al. IFNalpha activates dormant hematopoietic stem cells in vivo. Nature. 2009;458(7240):904–8.
Herman AC, et al. Systemic TLR2 agonist exposure regulates hematopoietic stem cells via cell-autonomous and cell-non-autonomous mechanisms. Blood Cancer J. 2016;6:e437.
• Pietras EM, et al. Chronic interleukin-1 exposure drives hematopoietic stem cells toward precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18(6):607–18 Demonstrated how chronic inflammation promotes myeloid cell production and reduces HSC self-renewal.
Takizawa H, et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. 2017;21(2):225–240 e5.
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–75.
Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14(5):302–14.
Matatall KA, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. 2016;17(10):2584–95.
Matatall KA, et al. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells. 2014;32(11):3023–30.
Pietras EM, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med. 2014;211(2):245–62.
Schuettpelz LG, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia. 2014;28(9):1851–60.
Hirche C, et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Rep. 2017;19(11):2345–56.
Meng A, et al. Ionizing radiation and busulfan inhibit murine bone marrow cell hematopoietic function via apoptosis-dependent and -independent mechanisms. Exp Hematol. 2003;31(12):1348–56.
van Bekkum DW. Radiation sensitivity of the hemopoietic stem cell. Radiat Res. 1991;128(1 Suppl):S4–8.
Wagemaker G. Heterogeneity of radiation sensitivity of hemopoietic stem cell subsets. Stem Cells. 1995;13(Suppl 1):257–60.
Wang Y, et al. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood. 2006;107(1):358–66.
Christensen JL, Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc Natl Acad Sci U S A. 2001;98(25):14541–6.
Kiel MJ, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–21.
Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13(1):102–16.
Singh S, Jakubison B, Keller JR. Protection of hematopoietic stem cells from stress-induced exhaustion and aging. Curr Opin Hematol. 2020;27(4):225–31.
Zhao JL, Baltimore D. Regulation of stress-induced hematopoiesis. Curr Opin Hematol. 2015;22(4):286–92.
Shao L, Luo Y, Zhou D. Hematopoietic stem cell injury induced by ionizing radiation. Antioxid Redox Signal. 2014;20(9):1447–62.
Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med. 2000;191(2):253–64.
Domen J, Gandy KL, Weissman IL. Systemic overexpression of BCL-2 in the hematopoietic system protects transgenic mice from the consequences of lethal irradiation. Blood. 1998;91(7):2272–82.
Hoyes KP, et al. Effect of bcl-2 deficiency on the radiation response of clonogenic cells in small and large intestine, bone marrow and testis. Int J Radiat Biol. 2000;76(11):1435–42.
Cui YF, et al. Apoptosis in bone marrow cells of mice with different p53 genotypes after gamma-rays irradiation in vitro. J Environ Pathol Toxicol Oncol. 1995;14(3–4):159–63.
Hirabayashi Y, et al. The p53-deficient hemopoietic stem cells: their resistance to radiation-apoptosis. but lasted transiently Leukemia. 1997;11(Suppl 3):489–92.
Komarov PG, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–7.
Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A. 1993;90(12):5742–6.
Shao L, et al. Deletion of proapoptotic Puma selectively protects hematopoietic stem and progenitor cells against high-dose radiation. Blood. 2010;115(23):4707–14.
Wu WS, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123(4):641–53.
•• Mohrin M, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–85 Characterized the distinct DNA repair responses in HSPCs compared to committed progenitor cells.
Beerman I, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12(4):413–25.
He S, et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med. 2017;9(387):eaaI3986.
Arai F, Suda T. Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci. 2007;1106:41–53.
Heidt T, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754–8.
Singh SK, et al. Id1 Ablation protects hematopoietic stem cells from stress-induced exhaustion and aging. Cell Stem Cell. 2018;23(2):252–265 e8.
Karigane D, et al. p38alpha activates purine metabolism to initiate hematopoietic stem/progenitor cell cycling in response to stress. Cell Stem Cell. 2016;19(2):192–204.
Chen J. Senescence of hematopoietic stem cells and bone marrow failure. Int J Hematol. 2005;82(3):190–5.
Marcotte R, Wang E. Replicative senescence revisited. J Gerontol A Biol Sci Med Sci. 2002;57(7):B257–69.
Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol. 2001;13(6):748–53.
Toussaint O, et al. Stress-induced premature senescence as alternative toxicological method for testing the long-term effects of molecules under development in the industry. Biogerontology. 2000;1(2):179–83.
Debacq-Chainiaux F, et al. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798–806.
Stepanova L, Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood. 2005;106(3):827–32.
Milyavsky M, et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell. 2010;7(2):186–97.
de Laval B, et al. Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell. 2013;12(1):37–48.
Fang T, et al. Epidermal growth factor receptor-dependent DNA repair promotes murine and human hematopoietic regeneration. Blood. 2020;136(4):441–54.
Yahata T, et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood. 2011;118(11):2941–50.
•• Flach J, et al. Replication stress is a potent driver of functional decline in aging hematopoietic stem cells. Nature. 2014;512(7513):198–202 Demonstrated the importance of replication stress in promoting the functional decline of aging HSCs.
Katoh O, et al. Expression of the vascular endothelial growth factor (VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res. 1995;55(23):5687–92.
Gerber HP, et al. VEGF regulates hematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002;417(6892):954–8.
Chen Q, et al. Apelin(+) endothelial niche cells control hematopoiesis and mediate vascular regeneration after myeloablative injury. Cell Stem Cell. 2019;25(6):768–783 e6.
Zhou B, et al. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting angiopoietin-1. eLIFE 2015;4:e05521.
Zhao M, et al. FGF signaling facilitates postinjury recovery of mouse hematopoietic system. Blood. 2012;120(9):1831–42.
Zhao Y, et al. Soluble factor(s) from bone marrow cells can rescue lethally irradiated mice by protecting endogenous hematopoietic stem cells. Exp Hematol. 2005;33(4):428–34.
Xu C, et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun. 2018;9(1):2449.
Zhao M, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20(11):1321–6.
• Zhou BO, et al. Bone marrow adipocytes promote the regeneration of stem cells and hematopoiesis by secreting SCF. Nat Cell Biol. 2017;19(8):891–903 Demonstrated the role of adipocytes in promoting hematopoietic stem cell regeneration in vivo following myelosuppression.
Bowie MB, et al. Steel factor responsiveness regulates the high self-renewal phenotype of fetal hematopoietic stem cells. Blood. 2007;109(11):5043–8.
Hassan HT, Zander Z. Stem cell factor as a survival and growth factor in human normal and malignant hematopoiesis. Acta Haematol. 1996;95(3–4):257–62.
Zhang CC, Lodish HF. Cytokines regulating hematopoietic stem cell function. Curr Opin Hematol. 2008;15(4):307–11.
Ding L, Morrison SJ. Hematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–5.
Greenbaum A, et al. CXCL12 in early mesenchymal progenitors is. required for hematopoietic stem-cell maintenance. Nature. 2013;495(7440):227–30.
Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205(4):777–83.
Tzeng YS, et al. Loss of Cxcl12/Sdf-1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood. 2011;117(2):429–39.
Brenner D, Blaser H, Mak TW. Regulation of tumor necrosis factor signaling: live or let die. Nat Rev Immunol. 2015;15(6):362–74.
Yamashita M, Passegue E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25(3):357 − +.
•• Hooper AT, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;4(3):263–74 Demonstrated the essential role of VEGFR+ endothelial cells in regulating hematopoietic regeneration following myelosuppression.
Salter AB, et al. Endothelial progenitor cell infusion induces hematopoietic stem cell reconstitution in vivo. Blood. 2009;113(9):2104–7.
Himburg HA, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16(4):475–82.
Himburg HA, et al. Distinct bone marrow sources of pleiotrophin control hematopoietic stem cell maintenance and regeneration. Cell Stem Cell. 2018;23(3):370–381 e5.
Doan PL, et al. Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat Med. 2013;19(3):295–304.
Himburg HA, et al. Dickkopf-1 promotes hematopoietic regeneration via direct and niche-mediated mechanisms. Nat Med. 2017;23(1):91–9.
Guo P, et al. Endothelial jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J Clin Invest. 2017;127(12):4242–56.
Poulos MG, et al. Endothelial jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 2013;4(5):1022–34.
Morrison SJ, Scadden DT. The bone marrow niche for hematopoietic stem cells. Nature. 2014;505(7483):327–34.
Wei QZ, Frenette PS. Niches for hematopoietic stem cells and their progeny. Immunity. 2018;48(4):632–48.
Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014;35(1):32–7.
Ding L, et al. Endothelial and perivascular cells maintain hematopoietic stem cells. Nature. 2012;481(7382):457–62.
Naveiras O, et al. Bone-marrow adipocytes as negative regulators of the hematopoietic microenvironment. Nature. 2009;460(7252):259–U124.
Caselli A, et al. IGF-1-mediated osteoblastic niche expansion enhances long-term hematopoietic stem cell engraftment after murine bone marrow transplantation. Stem Cells. 2013;31(10):2193–204.
Olson TS, et al. Megakaryocytes promote murine osteoblastic HSC niche expansion and stem cell engraftment after radioablative conditioning. Blood. 2013;121(26):5238–49.
Goncalves KA, et al. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell. 2016;166(4):894–906.
Silberstein L, et al. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell. 2016;19(4):530–43.
Pinho S, Frenette PS. Hematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. 2019;20(5):303–20.
Bruns I, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315–20.
Nakamura-Ishizu A, et al. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem Biophys Res Commun. 2014;454(2):353–7.
Nakamura-Ishizu A, et al. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow (vol 212, pg 2133, 2015). J Exp Med. 2015;212(13):2323.
Bowers E, et al. Granulocyte-derived TNF alpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat Med. 2018;24(1):95 − +.
Katayama Y, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–21.
Mendez-Ferrer S, et al. Hematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452(7186):442–U4.
Mendez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Skelet Biol Med. 2010;1192:139–44.
Lucas D, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. 2013;19(6):695–703.
Boettcher S, Ebert BL. Clonal hematopoiesis of indeterminate potential. J Clin Oncol. 2019;37(5):419–22.
Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98.
Funding
This work was supported by funding from the NIAID AI107333 (JPC), NIAID AI067769 (JPC), the CIRM Leadership Award LA1-08014 (JPC), the Damon Runyon Cancer Foundation DRG-2327-18 (CMT), the Burroughs Wellcome Fund PDEP #1018686 (CMT), and the UC President’s Postdoctoral Fellowship (CMT).
Author information
Authors and Affiliations
Contributions
CMT and JPC wrote the paper; CMT developed the figures.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Cancer and Stem Cells
Rights and permissions
About this article

Cite this article
Termini, C.M., Chute, J.P. Hematopoietic Stem Cell Stress and Regeneration. Curr Stem Cell Rep 6, 134–143 (2020). https://doi.org/10.1007/s40778-020-00181-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-020-00181-3