
Abstract
Purpose of Review
Human induced pluripotent stem cells (iPSCs) represent an attractive source to generate in vitro-derived red blood cells, megakaryocytes, and platelets for transfusion support. We review the progress made and challenges remaining for generating terminally differentiated red cells and platelets suitable for clinical application.
Recent Findings
Human iPSC hematopoietic differentiation protocols primarily recapitulate the primitive stage of hematopoiesis, but a different hematopoietic progenitor that mimics the second wave of hematopoiesis has been identified that generates definitive blood cells. Coupled with strategies to improve maturation and expansion, this provides new opportunities to generate red cells and platelets that can mature, enucleate, and proliferate to clinical scale.
Summary
The major challenges of human iPSC-derived transfusion products are terminal differentiation and scalability. Despite these challenges, iPSCs offer a new source for unlimited generation of red cells and platelets with rare phenotypes for transfusion, blood bank reagents, and novel drug delivery systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
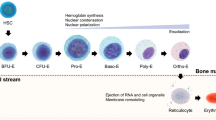
Human induced pluripotent stem cells (iPSCs) are renewable, potentially unlimited cell sources from which red cells and platelets may be derived. Donor red blood cells (RBCs) and platelets are anucleate cells and can undergo irradiation, which mitigate concerns about tumorigenicity of iPSC-derived transfusion products. iPSC-derived RBCs (iRBCs), megakaryocytes (iMegs), and platelets (iPlts) can complement existing donor-derived cells and offer new transfusion strategies (Fig. 1), particularly for patients who are alloimmunized to RBCs or human leukocyte antigens (HLA). These in vitro-derived cells can also be developed for blood bank reagent cells and novel drug delivery systems. The major challenges for iRBC and iPlt production for transfusion purposes include their ability to terminally differentiate and the need to generate precursors with high proliferative capacity, as 2 × 1012 cells are needed for one RBC unit and 3 × 1011 cells for one platelet unit.

iPSC-derived red blood cells and platelets for clinical applications. With a potentially unlimited capacity for renewal, iPSCs are an attractive source for in vitro generation of red blood cells, megakaryocytes, and platelets. Differentiation and expansion of iPSC-derived blood cells to clinically significant levels may complement donor-derived transfusions as well as provide rare blood bank reagents, novel strategies for drug delivery, disease modeling, and drug screening.
Human iPSC or embryonic stem cell (ESC) hematopoietic differentiation protocols primarily recapitulate the primitive or embryonic stage of hematopoiesis [1]. The red cells primarily express the embryonic globins, are larger in size than adult donor red cells, and achieve minimal enucleation [2, 3]. Ideally, iRBCs would produce adult- or fetal-type hemoglobin that appropriately binds and delivers oxygen. Whether in vitro-derived iPlts, or primitive iMegs that subsequently shed platelets in vivo once transfused, have similar properties and functionality of donor-derived platelets is not well established. Despite these challenges, human pluripotent stem cells (PSCs) offer an attractive source for unlimited generation of red cells and platelets to be used in the laboratory and the clinic.
Induced Pluripotent Stem Cell-Derived Red Blood Cells
Clinical Need for New Sources of RBCs
For most developed countries, an adequate and safe volunteer donor blood supply is available, but there remains a need for additional sources of RBCs to supplement blood donations, particularly for RBCs with rare or uncommon combinations of antigens. Patients who require frequent transfusions, including those with sickle cell disease and thalassemia, are often alloimmunized to multiple red cell antigens, which makes identification of compatible units challenging. Finding compatible blood can also be hindered by a lack of reagent red cells to rapidly and efficiently identify antibody specificities. Human iPSCs are an attractive source for the generation of RBCs for transfusion or as reagents since they are renewable, can be genetically characterized for their extended blood group antigen profiles, and are amenable to genome-editing techniques to produce rare phenotypes. One French study demonstrated that only three human iPSC lines used to generate RBCs with specific antigen profiles would be sufficient to match > 99% of 16,486 alloimmunized patients in their study cohort, and 15 such lines would provide matches for 100% of patients of European ancestry registered with the French National Registry of Rare Blood Phenotype or Genotype [4].
Hematopoietic Differentiation of iPSCs: Primitive vs. Definitive
In vitro protocols for the generation of red cells and other blood lineages recapitulate the hematopoietic programs that occur in vivo during normal mammalian development. The first hematopoietic cells to develop both in mouse and human develop from an extra-embryonic mesodermal population that is specified to the hematopoietic fate in the yolk sac (reviewed in [5,6,7]). These yolk sac progenitors generate the primitive wave of hematopoiesis that is comprised of nucleated red cells that express embryonic forms of globin (ε and ζ), macrophages, and primitive megakaryocytes. The second wave of hematopoiesis, termed definitive, is initiated at specific sites in the arterial vasculature of the developing embryo with the best characterized location being the aorta, gonad, and mesonephros (AGM) region. This program generates all of the hematopoietic lineages including long-term reconstituting hematopoietic stem cells (HSCs), enucleated red cells that express fetal hemoglobin (α and γ), myeloid cells, lymphocytes, and megakaryocytes [8]. Subsequently, definitive hematopoiesis occurs in the fetal liver and ultimately transitions to the bone marrow where most blood cells are generated during adult life, at which time mature RBCs express adult-type globins (α and β).
Methods for the Generation of iPSC-Derived Red Blood Cells
In vitro differentiation of iPSCs into mature blood cell types is typically based on sequential addition of cytokines at defined concentrations. Established protocols to generate iRBCs vary in technical details, but fall into two major categories to induce hematopoietic differentiation: suspension cultures with embryoid body (EB) formation and adherent differentiation with and without stromal cell co-culture. EB formation is typically performed by plating small aggregates of iPSCs in ultralow attachment plates, but forced aggregation of a defined number of disaggregated single cells can be also be achieved by plating a defined number of cells in untreated U- or V-bottom 96- or 384-well plates followed by centrifugation [9, 10]. Different protocols harvest cells from EBs at different time points for further erythroid differentiation. EBs may be disaggregated to form hemangioblasts [11], or the cells may be subsequently cultured in liquid media with [11] or without co-culture with stromal cells [12,13,14]. Alternatively, PSCs are differentiated via hematopoietic progenitors into erythroid cells by direct co-culture with the murine stromal cell line OP9 or human stromal cells [15,16,17].
Globin Expression in iRBCs
Most methods to culture iRBCs mimic the first developmental wave of hematopoiesis via generation of discrete yolk sac-like primitive hematopoietic progenitor cells (HPCs). These HPCs generate primitive red cells that arise prior to the onset of blood circulation in the embryo and express embryonic globins with little to no enucleation [2, 3]. The ability to detect definitive PSC-derived red cells with higher expression of fetal globins has been demonstrated [18]. However, this required 1 month of co-culturing PSCs with an immortalized cell line and an additional 3-week expansion of mixed populations of primitive and definitive hematopoietic progenitors. Similarly, others have demonstrated a more adult-like α/ζ globin ratio with increasing culture duration [19]. Co-culture with murine fetal liver-derived stromal cells led to a gradual increase in adult β-globin-containing erythroid cells with prolonged culture duration, but α and ε globin-containing cells were detected simultaneously consistent with a mixed population of primitive and definitive red cells [20]. Recently, a different PSC-derived HPC that mimics the second wave of hematopoiesis has been identified that generates definitive or fetal liver-like red cells [21••, 22]. By using this new HPC, a relatively pure population of definitive red cells can be generated that expresses predominantly fetal hemoglobin but their ability to mature, enucleate, and proliferate has not yet been fully characterized.
Generating iRBCs that are capable of switching to adult globin chains is likely advantageous for transfusion products, but fetal-type hemoglobin may be adequate. The switch from embryonic to fetal to adult hemoglobin synthesis is a major mechanism by which the developing fetus adapts from the relatively hypoxic intrauterine environment to the relatively oxygen-rich extrauterine environment. Adult globin chains have a lower affinity for oxygen and thus deliver oxygen more readily to the tissues. While high fetal hemoglobin is not typical for adult donor-derived RBCs, individuals with hereditary persistence of fetal hemogloblin have high levels of fetal hemoglobin (20–35% in heterozygotes and 100% for homozygotes) and are typically asymptomatic.
Red cells generated from varied PSC differentiation protocols predominantly demonstrate similar CO binding and O2 dissociation kinetics to that of cord blood [12, 20], but have also been shown to be comparable to adult red cells when co-cultured with mesenchymal stem cells (MSCs) [11]. iRBCs generated by prolonged culture conditions that produced primarily fetal globins exhibited similar oxygen affinity to cord blood red cells and, as expected, showed significantly higher oxygen affinity compared to adult red cells [23]. Notably, iPSC-derived reticulocytes that expressed predominantly fetal hemoglobin were shown to be capable of further globin switching in vivo [23]. Three days post-injection into immunodeficient mice, carboxyflouroscein succinimidyl ester (CSFE)-labeled cells contained over 50% adult β-globin chains. While the use of an in vivo murine system for maturation remains unrealistic for clinical applications, these studies suggest that iRBCs are capable of globin switching and terminal maturation, but may require an adult hematopoietic microenvironment.
Maturation and Enucleation
Terminal maturation and enucleation of iRBCs remains limited since most differentiation protocols recapitulate the primitive wave of hematopoiesis [18, 24]. Although RBC units can be irradiated prior to transfusion, iRBC enucleation is desirable to decrease the fear that the cells may be harboring cancer-inducing mutations. The degree of iRBC enucleation varies considerably depending on culture conditions, duration, and type of stromal co-culture. iRBCs may achieve up to 30% enucleation when derived from hemangioblasts without stromal cell co-culture, 30% enucleation when co-cultured with MSCs, and 65% enucleation when co-cultured with OP9 cells, suggesting the role of the stromal cells in facilitating enucleation [11]. However, enucleation has been demonstrated in up to 66% of human ESC-derived red cells and 26% of iRBCs without stromal cells by supplementing the culture media with human plasma, although this requires prolonged culture (up to 52 days) [12, 23]. Manipulating transcription factors is another potential strategy to enhance maturation of iRBCs as activation of KLF1 in iPSC-derived hematopoietic progenitors enhanced erythroid commitment and differentiation, including improved enucleation [25].
The Challenge of iRBC Yield
Large-scale culture of iRBCs would be necessary to produce a transfusion product, but significantly fewer cells are needed to generate reagent red cells. Final iRBC yield can be increased at various stages of hematopoietic differentiation, from HPC generation to expansion of erythroblasts. Co-culturing undifferentiated human ESCs with mouse fetal liver stromal cells has been shown to generate 100 erythroid progenitor cells from a single ESC [20]. Similar yield is achieved with stromal co-culture of iPSC-derived CD34+ progenitor cells with murine fetal hepatocytes, which generates ~ 80 orthochromatic erythroblasts from each progenitor cell after 24 days of liquid culture [18]. In the absence of stromal co-culture, EB differentiation and subsequent liquid culture has demonstrated the potential of each single iPSC to generate 440 mature erythroid cells at the end of 5 weeks of culture [12]. Extended cultures (7+ weeks) generated 1500–2830 erythroblasts with ~ 26% enucleation from each iPSC [23]. Similarly, one iPSC can yield 4000 erythroblasts by culturing CD34+ progenitor cells in suspension and up to 200,000 erythroblasts by co-culturing with MS5 bone marrow stromal cells [24]. Despite the increased yield, the long culture time (70–120 days) and dependence on murine stromal cells for co-culture are obstacles for clinical application. Recently, a feeder-free and serum-free multistep protocol that uses a combination of cytokines and small molecules showed that a single human PSC could generate 150 HPCs to subsequently produce 50,000–200,000 erythroblasts after 31 days of culture [26].
Several laboratories have manipulated particular transcription factors to increase cell yield. CRISPR/Cas9-mediated SH2B3 inactivation, a negative regulator of cytokine signaling for which naturally occurring loss-of-function variants increase RBC counts in humans, resulted in a threefold increase in erythroid cell production while maintaining similar morphology, surface marker, and globin expression as SH2B3WT ESCs [14]. Ectopic RUNX1a expression in human PSCs also expands the CD31+34+ hematopoietic progenitor cell population while retaining the ability to differentiate into multi-lineage cells [27].
Novel bioengineering solutions, such as three-dimensional scaffolds to re-create the bone marrow niche, will be necessary to improve the quality and quantity of iRBCs required for transfusion. The use of bioreactors to culture undifferentiated human PSCs provides a more efficient system to expand iPSCs than typical adhesion culture for large-scale use [28,29,30]. The use of bioreactors for erythroid differentiation has been tested with cord blood, suggesting a similar potential for iRBC generation. Cord blood HPCs can yield 107-fold expansion by day 21 of culture in 1-L stirred bioreactors with near pure populations of reticulocytes and > 90% enucleation frequency [31]. Maturation of cord blood-derived erythroblasts in stirred bioreactors also demonstrated higher enucleation (74%) than in static culture (54%) and similar size (8.3 μm diameter) and morphology to adult donor RBC (8.5 μm) [32]. Ex vivo large-scale generation of RBCs from cord blood CD34+ cells using a bottle-turning device system produced 2 × 108 RBCs from one CD34+ cell, indicating that one cord blood unit could, in theory, be equivalent to 500 RBC units [33].
Induced Pluripotent Stem Cell-Derived Megakaryocytes and Platelets
The Clinical Need for New Sources of Platelets in Clinical Practice
Platelets are circulating cytoplasmic fragments released from megakaryocytes that are highly differentiated structures whose main function is to support normal hemostasis. Isolation of donor-derived functional platelets for transfusion became sufficiently advanced by the early 1980s to encourage widespread clinical usage. Platelet transfusions not only support patients with qualitative or quantitative disorders of platelets, but those with other hemostatic challenges. Without such platelet transfusions, modern-day, aggressive care of patients with cancer and cardiovascular dysfunction would not be possible. As the population in the USA increasingly ages, the number of platelet transfusions administered has been rising despite more stringent platelet count thresholds for transfusion [34]. Finding a source of platelets other than donor-based will become increasingly important over the coming years.
An alternative source of platelets is in vitro-generated platelets from cultured megakaryocytes, the polyploid, terminally differentiated large cells that release platelets. When thrombopoietin, the primary cytokine for megakaryopoiesis was identified [35], it became possible to culture megakaryocytes in large numbers. Early studies identified proplatelet extensions from cultured megakaryocytes, as well as ex vivo-generated platelet-like particles (EV-PLPs) [36]. By light and electron microscopy, some of EV-PLPs were similar to donor-derived platelets and functionally, they responded to platelet agonists. In vitro-derived platelets offer a number of potential advantages over donor-derived platelets including uniform hemostatic quality, decreased risk of known or unknown transmissible infection, and specific HLA phenotypes. In vitro-derived platelets can also be developed as a drug delivery system to release stored proteins during platelet degranulation [37, 38].
Platelet Bioreactors
The development of platelet bioreactors advanced the ability to generate large numbers of EV-PLPs from cultured megakaryocytes. The simplest model used gradient fractions to harvest EV-PLPs that were released from megakaryocytes in a stationary culture dish, but the overall yield and purity were low [36]. An improved platelet yield can be achieved by exposing cultured megakaryocytes to turbulent shear forces [39••, 40]. A different strategy is to recapitulate the marrow environment by culturing megakaryocytes on silk strands with flow over a scaffold that allows platelet release by mechanical disruption via the shear of the silk network [41]. A platelet “bioreactor-on-a-chip” was designed for iMegs to traverse pores between endothelial cells similar to the intramedullary/vascular interface of the bone marrow, which triggers proplatelet initiation and platelet production [42, 43]. Although endothelial lined systems likely promote platelet generation from human iMegs [43], most systems currently use various polymeric surfaces.
The Quantitative and Qualitative Challenge in In Vitro Platelet Production
One major challenge of in vitro platelet production has been the quantitative and qualitative analysis of the final EV-PLP product. Platelet yield per in vitro-grown megakaryocytes has been in the 100–2 range, which is substantially less than the estimated 103 platelets produced per megakaryocyte in vivo [44]. It is important to note that the yield is sometimes reported per total large megakaryocytes rather than all megakaryocytes, which can artificially increase the number.
The functionality of EV-PLPs or in vitro-derived platelets should be compared to donor-derived platelets. In some studies, stored platelets that had reached their allowed shelf life were used as the control, but fresh donor samples are the gold standard. EV-PLPs also vary widely in size and lack the Gaussian-size distribution of donor platelets. Therefore, many studies focus only on EV-PLPs that are of similar size and granularity as donor-derived platelets, as determined by flow cytometry [45]. In our experience, using stationary grown CD34+-derived human cultures, less than 10% of EV-PLPs are actually generated from double-positive CD41+42a+ mature megakaryocytes. Moreover, only 10% of megakaryocyte-derived EV-PLPs are not apoptotic as measured by annexin V binding [46]. We demonstrated that these EV-PLPs infused into immunodeficient mice have a half-life of approximately 2 h compared to donor-derived human platelets that have a 12- to 24-h half-life [46], consistent with most EV-PLPs that are non-megakaryocyte-derived particles or are injured platelets.
To improve in vitro platelet production, identifying the site where platelets are released in vivo by megakaryocytes is critical. It has been suggested that platelets are released in the intramedullary bone marrow cavity during “stress thrombopoiesis” [47] or in certain abnormal conditions such as Wiskott-Aldrich syndrome [48]. While this is unlikely to be a major site of platelet formation, this would suggest that a stationary megakaryocyte growth model may suffice. The prevailing concept is that megakaryocytes migrate from the intramedullary osteoblastic region to the perivascular niche during differentiation and subsequently extend proplatelet processes through inter- or intra-endothelial pores to release proplatelet and platelet cytoplasmic fragments into the sinuses. Multi-photon microscopy of the calvarium medullar space supports this view [49, 50]. A third mechanism by which megakaryocytes can release platelets is by migrating out of the bone marrow and medullary sinuses to the pulmonary beds where they shed their cytoplasm. This concept, first developed in the 1930s by William Howell [50], has received recent support from in situ pulmonary vascular studies that suggest that ~ 50% of all platelets come from shedding by entrapped megakaryocytes in the lungs [51••].
Infusion of both mouse and human in vitro-grown megakaryocytes into recipient mice shows that these cultured megakaryocytes can shed platelets in vivo over a period of 2–4 h [46, 52]. The released platelets have a near-identical Gaussian-size distribution as donor-derived platelets with approximately the same half-life. The shed platelets also demonstrate similar responses to agonists in vitro and in vivo. The major drawback to these megakaryocyte infusion studies is that platelet yield per megakaryocyte remains low (101–2). We have shown that in vitro HPCs differentiate into mature CD41+42a+ megakaryocytes that can take up coagulation factor V into their alpha granules. Subsequently, these megakaryocytes undergo apoptosis [53]. However, if the megakaryocytes are infused into a recipient mouse instead, platelets are released within 5 min, suggesting that harvesting these peak megakaryocytes may optimize platelet yield.
Sources of In Vitro-Generated Megakaryocytes
One unit of platelets contains approximately 3 × 1011 platelets which would require 108–9 starting megakaryocytes or approximately 106 self-renewing intermediate cells as described [39]. For the 3 × 107 platelet units transfused per year in the USA, that would equate to ~ 1013 of such renewing cells. Thus, isolation of CD34+ HPCs from primary marrow would require an inordinate number of donors, likely exceeding the number of present-day platelet donors. Cord blood samples have been suggested as a primary source of starting material with the potential for expansion using valproic acid [54]. The functionality of EV-PLPs or megakaryocytes for infusion from cord blood-derived material needs exploration as neonatal platelets have decreased functionality compared to adult platelets [55].
Megakaryocyte-like cells have also been derived from cell lines and from adipocytes in the presence of thrombopoietin [56]. This is a very renewable and inexpensive source of cells. The biggest challenge with these approaches is whether they can generate primary-like, adult megakaryocytes and whether released EV-PLPs have similar properties of donor-derived platelets. Studies to generate megakaryocytes and platelets beginning with self-replicating endothelial-derived cells have also been recently reported [57], but the final megakaryocyte and platelet-like products have yet to be well studied. The most promising approach has been the use of human iPSCs. Multiple groups have shown that iPSCs can be driven into HPCs, yielding 100–1 HPCs per iPSC and perhaps 100–1 megakaryocytes from each iPSC. The final yield of EV-PLPs or platelets released in vivo in recipient mice is also low [46]. An additional concern with iMegs, EV-PLPs, and platelets is that they may be primitive or embryonic in nature and be even less functional than neonatal platelets, which have decreased agonist responsiveness compared to adult platelets [58].
Since the yield is low at each step and costs for cytokines to carry out differentiation are high, the focus by many groups to generate iPlts for standard transfusions seems misplaced. Instead, iPSCs can be manipulated to study and/or alter their genetics and, ultimately, to modify the final product. The use of iMegs to study aspects of megakaryocyte biology has complemented animal studies and CD34+-derived human megakaryocytes [59]. Additionally, iMegs have been useful to develop gene correction approaches for inherited platelet disorders [60]. These efforts both support that the final released EV-PLPs and/or released iPlts from infused iMegs may be clinically relevant and offer novel strategies for addressing the challenges of iMegs. There have been efforts to find alternative strategies to produce definitive, more adult megakaryocytes from iPSCs [61]. Others have focused on creating a late, terminally differentiated, but replicative cell line that upon appropriate signaling goes on to produce mature megakaryocytes [62, 63]. For example, using exogenous overexpression of three transcription factors, GATA1, FLI1, and TAL1, up to 2 × 105 iMegs can be generated from one single undifferentiated iPSC [39, 62, 63]. Such a strategy can reduce the cost of generating sufficient terminally differentiated megakaryocytes and perhaps meet clinical platelet number needs as well.
Conclusions
The hope to achieve clinically relevant numbers of red cells and platelets from hematopoietic differentiation of iPSCs is a reasonable expectation. Studies over the past 20 years have greatly advanced our understanding of erythropoiesis, megakaryopoiesis, and thrombopoiesis. They have also improved our understanding of how to judge success at each step. The importance of careful comparison to the best donor-derived cell products as the gold standard is intuitive but has not been consistently pursued. Studies of select platelet-like particles within the overall EV-PLP population had led to over-optimistic expectations. The ability of iRBCs to bind and release oxygen and iPlts to respond to agonists requires direct comparison to donor-derived cells. Establishment of clinically relevant in vivo assays to test functionality is still needed. Going forward, significant challenges remain at producing large numbers of highly functional red cells and platelets at acceptable costs, but likely can be overcome.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Kennedy M, Awong G, Sturgeon CM, Ditadi A, LaMotte-Mohs R, Zuniga-Pflucker JC, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2(6):1722–35. https://doi.org/10.1016/j.celrep.2012.11.003.
Olivier EN, Qiu C, Velho M, Hirsch RE, Bouhassira EE. Large-scale production of embryonic red blood cells from human embryonic stem cells. Exp Hematol. 2006;34(12):1635–42. https://doi.org/10.1016/j.exphem.2006.07.003.
Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood. 2005;106(3):860–70. https://doi.org/10.1182/blood-2004-11-4522.
Peyrard T, Bardiaux L, Krause C, Kobari L, Lapillonne H, Andreu G, et al. Banking of pluripotent adult stem cells as an unlimited source for red blood cell production: potential applications for alloimmunized patients and rare blood challenges. Transfus Med Rev. 2011;25(3):206–16. https://doi.org/10.1016/j.tmrv.2011.01.002.
Baron MH, Fraser ST. The specification of early hematopoiesis in the mammal. Curr Opin Hematol. 2005;12(3):217–21.
Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–36. https://doi.org/10.1016/j.stem.2012.01.006.
Tavian M, Peault B. Embryonic development of the human hematopoietic system. Int J Dev Biol. 2005;49(2–3):243–50. https://doi.org/10.1387/ijdb.041957mt.
Medvinsky AL, Dzierzak EA. Development of the definitive hematopoietic hierarchy in the mouse. Dev Comp Immunol. 1998;22(3):289–301.
Hong SH, Werbowetski-Ogilvie T, Ramos-Mejia V, Lee JB, Bhatia M. Multiparameter comparisons of embryoid body differentiation toward human stem cell applications. Stem Cell Res. 2010;5(2):120–30. https://doi.org/10.1016/j.scr.2010.04.007.
Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106(5):1601–3. https://doi.org/10.1182/blood-2005-03-0987.
Lu SJ, Feng Q, Park JS, Vida L, Lee BS, Strausbauch M, et al. Biologic properties and enucleation of red blood cells from human embryonic stem cells. Blood. 2008;112(12):4475–84. https://doi.org/10.1182/blood-2008-05-157198.
Lapillonne H, Kobari L, Mazurier C, Tropel P, Giarratana MC, Zanella-Cleon I, et al. Red blood cell generation from human induced pluripotent stem cells: perspectives for transfusion medicine. Haematologica. 2010;95(10):1651–9. https://doi.org/10.3324/haematol.2010.023556.
Byrska-Bishop M, VanDorn D, Campbell AE, Betensky M, Arca PR, Yao Y, et al. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J Clin Invest. 2015;125(3):993–1005. https://doi.org/10.1172/JCI75714.
Giani FC, Fiorini C, Wakabayashi A, Ludwig LS, Salem RM, Jobaliya CD, et al. Targeted application of human genetic variation can improve red blood cell production from stem cells. Cell Stem Cell. 2016;18(1):73–8. https://doi.org/10.1016/j.stem.2015.09.015.
Ledran MH, Krassowska A, Armstrong L, Dimmick I, Renstrom J, Lang R, et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell. 2008;3(1):85–98. https://doi.org/10.1016/j.stem.2008.06.001.
Klimchenko O, Mori M, Distefano A, Langlois T, Larbret F, Lecluse Y, et al. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood. 2009;114(8):1506–17. https://doi.org/10.1182/blood-2008-09-178863.
Qiu C, Hanson E, Olivier E, Inada M, Kaufman DS, Gupta S, et al. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Exp Hematol. 2005;33(12):1450–8. https://doi.org/10.1016/j.exphem.2005.09.003.
Chang CJ, Mitra K, Koya M, Velho M, Desprat R, Lenz J, et al. Production of embryonic and fetal-like red blood cells from human induced pluripotent stem cells. PLoS One. 2011;6(10):e25761. https://doi.org/10.1371/journal.pone.0025761.
Qiu C, Olivier EN, Velho M, Bouhassira EE. Globin switches in yolk sac-like primitive and fetal-like definitive red blood cells produced from human embryonic stem cells. Blood. 2008;111(4):2400–8. https://doi.org/10.1182/blood-2007-07-102087.
Ma F, Ebihara Y, Umeda K, Sakai H, Hanada S, Zhang H, et al. Generation of functional erythrocytes from human embryonic stem cell-derived definitive hematopoiesis. Proc Natl Acad Sci U S A. 2008;105(35):13087–92. https://doi.org/10.1073/pnas.0802220105.
•• Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014;32(6):554–61. https://doi.org/10.1038/nbt.2915 This paper established simple selective differentiation strategies for the generation of primitive or definitive hematopoietic progenitors by Wnt-ß-catenin manipulation, providing access to enriched populations.
Ditadi A, Sturgeon CM, Tober J, Awong G, Kennedy M, Yzaguirre AD, et al. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat Cell Biol. 2015;17(5):580–91. https://doi.org/10.1038/ncb3161.
Kobari L, Yates F, Oudrhiri N, Francina A, Kiger L, Mazurier C, et al. Human induced pluripotent stem cells can reach complete terminal maturation: in vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica. 2012;97(12):1795–803. https://doi.org/10.3324/haematol.2011.055566.
Dias J, Gumenyuk M, Kang H, Vodyanik M, Yu J, Thomson JA, et al. Generation of red blood cells from human induced pluripotent stem cells. Stem Cells Dev. 2011;20(9):1639–47. https://doi.org/10.1089/scd.2011.0078.
Yang CT, Ma R, Axton RA, Jackson M, Taylor AH, Fidanza A, et al. Activation of KLF1 enhances the differentiation and maturation of red blood cells from human pluripotent stem cells. Stem Cells. 2017;35(4):886–97. https://doi.org/10.1002/stem.2562.
Olivier EN, Marenah L, McCahill A, Condie A, Cowan S, Mountford JC. High-efficiency serum-free feeder-free erythroid differentiation of human pluripotent stem cells using small molecules. Stem Cells Transl Med. 2016;5(10):1394–405. https://doi.org/10.5966/sctm.2015-0371.
Ran D, Shia WJ, Lo MC, Fan JB, Knorr DA, Ferrell PI, et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood. 2013;121(15):2882–90. https://doi.org/10.1182/blood-2012-08-451641.
Olmer R, Lange A, Selzer S, Kasper C, Haverich A, Martin U, et al. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng Part C Methods. 2012;18(10):772–84. https://doi.org/10.1089/ten.TEC.2011.0717.
Serra M, Brito C, Sousa MF, Jensen J, Tostoes R, Clemente J, et al. Improving expansion of pluripotent human embryonic stem cells in perfused bioreactors through oxygen control. J Biotechnol. 2010;148(4):208–15. https://doi.org/10.1016/j.jbiotec.2010.06.015.
Amit M, Chebath J, Margulets V, Laevsky I, Miropolsky Y, Shariki K, et al. Suspension culture of undifferentiated human embryonic and induced pluripotent stem cells. Stem Cell Rev. 2010;6(2):248–59. https://doi.org/10.1007/s12015-010-9149-y.
Timmins NE, Athanasas S, Gunther M, Buntine P, Nielsen LK. Ultra-high-yield manufacture of red blood cells from hematopoietic stem cells. Tissue Eng Part C Methods. 2011;17(11):1131–7. https://doi.org/10.1089/ten.TEC.2011.0207.
Bayley R, Ahmed F, Glen K, McCall M, Stacey A, Thomas R. The productivity limit of manufacturing blood cell therapy in scalable stirred bioreactors. J Tissue Eng Regen Med. 2018;12(1):e368–e78. https://doi.org/10.1002/term.2337.
Zhang Y, Wang C, Wang L, Shen B, Guan X, Tian J, et al. Large-scale ex vivo generation of human red blood cells from cord blood CD34(+) cells. Stem Cells Transl Med. 2017;6(8):1698–709. https://doi.org/10.1002/sctm.17-0057.
Thiagarajan P, Afshar-Kharghan V. Platelet transfusion therapy. Hematol Oncol Clin North Am. 2013;27(3):629–43. https://doi.org/10.1016/j.hoc.2013.03.004.
Lok S, Kaushansky K, Holly RD, Kuijper JL, Lofton-Day CE, Oort PJ, et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature. 1994;369(6481):565–8. https://doi.org/10.1038/369565a0.
Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood. 1995;85(2):402–13.
Yarovoi HV, Kufrin D, Eslin DE, Thornton MA, Haberichter SL, Shi Q, et al. Factor VIII ectopically expressed in platelets: efficacy in hemophilia A treatment. Blood. 2003;102(12):4006–13. https://doi.org/10.1182/blood-2003-05-1519.
Kufrin D, Eslin DE, Bdeir K, Murciano JC, Kuo A, Kowalska MA, et al. Antithrombotic thrombocytes: ectopic expression of urokinase-type plasminogen activator in platelets. Blood. 2003;102(3):926–33. https://doi.org/10.1182/blood-2003-01-0054.
•• Ito Y, Nakamura S, Sugimoto N, Shigemori T, Kato Y, Ohno M, et al. Turbulence activates platelet biogenesis to enable clinical scale ex vivo production. Cell. 2018;174(3):636–48 e18. https://doi.org/10.1016/j.cell.2018.06.011 This paper offers a first approach to generating sufficient cell progenitors to produce functional platelets sufficient to be able to supply a significant portion of the platelet transfused population.
Dunois-Larde C, Capron C, Fichelson S, Bauer T, Cramer-Borde E, Baruch D. Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood. 2009;114(9):1875–83. https://doi.org/10.1182/blood-2009-03-209205.
Pallotta I, Lovett M, Kaplan DL, Balduini A. Three-dimensional system for the in vitro study of megakaryocytes and functional platelet production using silk-based vascular tubes. Tissue Eng Part C Methods. 2011;17(12):1223–32. https://doi.org/10.1089/ten.tec.2011.0134.
Nakagawa Y, Nakamura S, Nakajima M, Endo H, Dohda T, Takayama N, et al. Two differential flows in a bioreactor promoted platelet generation from human pluripotent stem cell-derived megakaryocytes. Exp Hematol. 2013;41(8):742–8. https://doi.org/10.1016/j.exphem.2013.04.007.
Thon JN, Mazutis L, Wu S, Sylman JL, Ehrlicher A, Machlus KR, et al. Platelet bioreactor-on-a-chip. Blood. 2014;124(12):1857–67.
Paulus JM, Deschamps JF, Prenant M, Casals FJ. Kinetics of platelets, megakaryocytes and their precursors: what to measure? Blood Cells. 1980;6(2):215–28.
Takayama N, Eto K. In vitro generation of megakaryocytes and platelets from human embryonic stem cells and induced pluripotent stem cells. Methods Mol Biol. 2012;788:205–17. https://doi.org/10.1007/978-1-61779-307-3_15.
Wang Y, Hayes V, Jarocha D, Sim X, Harper DC, Fuentes R, et al. Comparative analysis of human ex vivo-generated platelets vs megakaryocyte-generated platelets in mice: a cautionary tale. Blood. 2015;125(23):3627–36. https://doi.org/10.1182/blood-2014-08-593053.
Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell. 2015;17(4):422–34. https://doi.org/10.1016/j.stem.2015.07.007.
Sabri S, Foudi A, Boukour S, Franc B, Charrier S, Jandrot-Perrus M, et al. Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood. 2006;108(1):134–40. https://doi.org/10.1182/blood-2005-03-1219.
Junt T, Schulze H, Chen Z, Massberg S, Goerge T, Krueger A, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317(5845):1767–70. https://doi.org/10.1126/science.1146304.
Zhang L, Orban M, Lorenz M, Barocke V, Braun D, Urtz N, et al. A novel role of sphingosine 1-phosphate receptor S1pr1 in mouse thrombopoiesis. J Exp Med. 2012;209(12):2165–81. https://doi.org/10.1084/jem.20121090.
•• Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105–9. https://doi.org/10.1038/nature21706 This paper provides visual proof that megakaryocytes can normally shed platelets outside the medullary system.
Fuentes R, Wang Y, Hirsch J, Wang C, Rauova L, Worthen GS, et al. Infusion of mature megakaryocytes into mice yields functional platelets. J Clin Invest. 2010;120(11):3917–22. https://doi.org/10.1172/JCI43326.
Sim X, Jarocha D, Hayes V, Hanby HA, Marks MS, Camire RM, et al. Identifying and enriching platelet-producing human stem cell-derived megakaryocytes using factor V uptake. Blood. 2017;130(2):192–204. https://doi.org/10.1182/blood-2017-01-761049.
Perdomo J, Yan F, Leung HHL, Chong BH. Megakaryocyte differentiation and platelet formation from human cord blood-derived CD34+ cells. J Vis Exp. 2017;(130). doi:https://doi.org/10.3791/56420.
Ferrer-Marin F, Stanworth S, Josephson C, Sola-Visner M. Distinct differences in platelet production and function between neonates and adults: implications for platelet transfusion practice. Transfusion. 2013;53(11):2814–21; quiz 3. https://doi.org/10.1111/trf.12343.
Matsubara Y, Saito E, Suzuki H, Watanabe N, Murata M, Ikeda Y. Generation of megakaryocytes and platelets from human subcutaneous adipose tissues. Biochem Biophys Res Commun. 2009;378(4):716–20. https://doi.org/10.1016/j.bbrc.2008.11.117.
Lis R, Karrasch CC, Poulos MG, Kunar B, Redmond D, Duran JGB, et al. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature. 2017;545(7655):439–45. https://doi.org/10.1038/nature22326.
Sola-Visner M. Platelets in the neonatal period: developmental differences in platelet production, function, and hemostasis and the potential impact of therapies. Hematology Am Soc Hematol Educ Program. 2012;2012:506–11. https://doi.org/10.1182/asheducation-2012.1.506.
Vo KK, Jarocha DJ, Lyde RB, Hayes V, Thom CS, Sullivan SK, et al. FLI1 level during megakaryopoiesis affects thrombopoiesis and platelet biology. Blood. 2017;129(26):3486–94. https://doi.org/10.1182/blood-2017-02-770958.
Sullivan SK, Mills JA, Koukouritaki SB, Vo KK, Lyde RB, Paluru P, et al. High-level transgene expression in induced pluripotent stem cell-derived megakaryocytes: correction of Glanzmann thrombasthenia. Blood. 2014;123(5):753–7. https://doi.org/10.1182/blood-2013-10-530725.
Leung A, Zulick E, Skvir N, Vanuytsel K, Morrison TA, Naing ZH et al. Notch and aryl hydrocarbon receptor signaling impact definitive hematopoiesis from human pluripotent stem cells. Stem Cells. 2018. doi:https://doi.org/10.1002/stem.2822.
Nakamura S, Takayama N, Hirata S, Seo H, Endo H, Ochi K, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014;14(4):535–48. https://doi.org/10.1016/j.stem.2014.01.011.
Moreau T, Evans AL, Vasquez L, Tijssen MR, Yan Y, Trotter MW, et al. Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nat Commun. 2016;7:11208. https://doi.org/10.1038/ncomms11208.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Stella T. Chou reports grants from National Institutes of Health, during the conduct of the study. Hyun Hyung An and Mortimer Poncz declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cellular Therapies: Preclinical and Clinical
Rights and permissions
About this article

Cite this article
An, H.H., Poncz, M. & Chou, S.T. Induced Pluripotent Stem Cell-Derived Red Blood Cells, Megakaryocytes, and Platelets: Progress and Challenges. Curr Stem Cell Rep 4, 310–317 (2018). https://doi.org/10.1007/s40778-018-0144-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-018-0144-6