
Abstract
The chemical structure of a polyacrylamide (PA), synthesized by inverse emulsion polymerization, was confirmed by FTIR and 1H NMR. The experimental conditions were conducted to produce high molecular weight of 1.4 × 106 g mol−1 determined by both gel permeation chromatography and viscosity average molecular weight methods. The efficiency of PA was evaluated, on corrosion inhibition of C-steel in 1.0 M HCl solution, by means of electrochemical impedance spectroscopy, potentiodynamic polarization (PP), and mass-loss (ML) measurements, and a very good concordance was obtained from the three techniques. The inhibiting efficiency increased up to 4 × 10−6 mol L−1 and dropped down afterward. The PP curves revealed that PA was of mixed-type, influencing predominantly the anodic process. The electrochemical interface was satisfactorily modeled with the electrical circuit (RS + Qdl/Rct) using CPE(α,Q) reflecting one-time constant. This fact, testifying the dominant character of charge transfer control of the C-steel corrosion process, was well described in both Nyquist along with Bode diagrams which take into account the frequency dependence, justifying the suitable choice of the electrical circuit. The adsorption of PA involved both physical and chemical interactions of non-protonated and protonated molecules with a deeper discussion on the suitable sites of protonation. The performance of PA was maintained if not slightly ameliorated in a more aggressive medium such as 0.5 M H2SO4 at 298 K. Two-time constants were needed to model the electrochemical interface (RS + Qdl/(Rct + RL/L)).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
A survey of literature shows the abundantly usefulness of polymers, from both natural and synthetic sources, as corrosion inhibitors for various metals and alloys in different aggressive service environments, especially in acidic media [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. In fact, polymers have the advantage of being stable in acidic media, non-toxic, biodegradable, abundant, renewable, and capable of reacting with other molecules. Their inhibiting efficiency depends on their larger size, the presence of a greater number of functional groups as well as cyclic and aromatic rings in their chemical structure. Accordingly, polymers present the ability to either donate or accept electrons from the metallic surface. Through these possible double exchanges, polymers can form complexes with metal ions [29] and easily get adsorbed onto the metallic surface and so cover considerably more surface when compared to the corresponding monomers [30]. Thus, a relatively high surface coverage could be achieved even at low concentrations of polymers due to their good adsorption properties. In addition, as polymer size increases, inhibition efficiency also increases, when solubility is obviously achieved [31].
Previous studies involving polyacrylamide (PA) were focused on the problem of the conformational behavior on its adsorption on iron in 1.0 M H2SO4 [8] and in 3.0 M HCl [12]. A synergistic effect between PA and iodide ions is also reported on the corrosion inhibition of pure iron in 0.5 M H2SO4 [9]. Furthermore, the same synergism was also studied on aluminum corrosion in 0.5 M HCl solution [10]. Besides, Manimaran et al. [13] have tried to understand the impact of Zn2+, when added to PA, in controlling C-steel corrosion in ground water, as well as Srivastava et al. [14] who have undertaken the inhibitive effect of PA grafted with fenugreek mucilage on the corrosion of mild steel in 0.5 M H2SO4 at 35 °C. However, practically no works, in our knowledge, have so far been reported with regard to C-steel corrosion inhibition in 1.0 M HCl by PA and the impact of interfacial properties on its adsorption behavior at the C-steel/acidic solution interface.
In this optic, a recently synthesized PA by free radical polymerization of acrylamide monomer, using inverse emulsion polymerization, is investigated in terms of corrosion inhibition properties on C-steel in 1.0 M HCl. PA is a synthetic chemical, non-toxic, completely soluble in water, which can be tailored to some many industrial processes. It should be noted that PA, under study, was conducted as a stabilizer of the sand dunes in Al-Ain town of the United Arab Emirates in the arid regions as reported elsewhere [32].
The present study (part 1) aims at the synthesis and evaluation of corrosion inhibition properties of PA in 1.0 M HCl by means of mass-loss (ML) measurements, potentiodynamic polarization (PP), and electrochemical impedance spectroscopy (EIS) techniques. Besides, a comparative study is also done, at a suitable content of PA less than the optimum concentration, in 0.5 M H2SO4, to evaluate the performance of PA in a medium with higher corrosiveness and in order to find pathways to understand the performance of PA corrosion inhibitor. Subsequent paper will cover, in part 2, the effects of temperature, immersion time period as well as a confrontation of six adsorption isotherm models. Hence, the kinetic–thermodynamic parameters are evaluated, analyzed, and discussed.
2 Experimental Details
2.1 Synthesis of PA Inhibitor
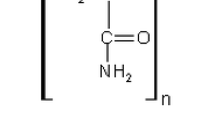
PA was synthesized according to the reaction in Fig. 1 by inverse emulsion polymerization of acrylamide, according to the procedure reported by Mohsin et al. [32]. Typical recipe to produce high molecular weight PA of 1.4 × 106 g mol−1 has been given in Table 1.

Scheme of PA synthesis
The chemical structure of a PA synthesized was confirmed by FTIR and 1H NMR. FTIR spectra for PA were recorded by NEXUS470 FTIR spectrometer at room temperature in the range of wave-number from 4000 to 500 cm−1 during 32 scans with 2 cm−1 resolution. 1H NMR spectra were obtained using a Varian 400 instrument operating at 400 MHz, with D2O–DMSO as solvent and TMS as internal standard. All chemicals were purchased from Sigma-Aldrich Chemicals Company.
2.2 Materials and Methods
2.2.1 Materials
Prior to all measurements, the C-steel samples containing in wt%: 0.210% C, 0.360% Si, 1.250% Mn, 0.046% S, 0.025% P, 0.160% Cr, 0.160% Ni, 0.081% V, 0.017% Sn, 0.410% Cu, 0.003 Al, 0.017% Mo, and the remainder iron Fe were mechanically polished on wet SiC paper (400, 600, 1000, and 1200), rinsed with distilled water, degreased in acetone in an ultrasonic bath immersion for 5 min, washed again with bi-distilled water, and then dried at room temperature before use. The acid solutions used were made from Riedel-de-Haën. The aggressive solutions of 1.0 M HCl and of 0.5 M H2SO4 were prepared by dilution of analytical grade 37% HCl and 98% H2SO4, respectively, with bi-distilled water. Appropriate concentrations of PA inhibitor were prepared with 1.0 M HCl addition.
2.2.2 Mass-Loss Tests
Gravimetric measurements were performed on C-steel samples with a rectangular form (1 cm × 4 cm × 0.25 cm) in 1.0 M HCl solution at 5 × 10−7 to 9 × 10−6 mol L−1 of PA. The duration of the immersion period was 6 h. The ML method was used to evaluate the corrosion rate (Wcorr) in an average value.
2.2.3 Electrochemical Tests
Electrochemical measurements were carried out in a conventional three-electrode glass cell and were conducted by a potentiostat/galvanostat (Radiometer-analytical PGZ 100) and controlled with analysis software (Voltamaster 4). The experimental set-up is given by Fig. 2. The working electrode (WE) was used as a rectangular specimen of C-steel with only one face of the electrode with a constant 1 cm2 surface area exposed. Ag/AgCl (3 M KCl) was used as reference electrode (RE) and a rectangular platinum foil was used as counter electrode. The WE was immersed in 50 mL of the test solution during 30 min until a steady state of the open-circuit potential (Eocp) was obtained.

Scheme of the experimental set-up for electrochemical measurements
Polarization curves were recorded by scanning the electrode potential at first in the cathodic direction from − 900 to Eocp + 20 mVAg/AgCl and then in the anodic direction from Eocp − 20 mVAg/AgCl to − 100 mV with the same scanning rate of 1 mV s−1. The corrosion parameters such as cathodic Tafel slope (βc), corrosion current density (icorr) and corrosion potential (Ecorr) were evaluated by using Ec-Lab software.
Impedance spectra were recorded, at the open-circuit potential, in the frequency range, from 100 kHz to 100 mHz with 10 points per decade, at alternating current amplitude of ± 10 mV ac voltage peak-to-peak. The impedance spectra, given in the Nyquist and Bode representations, were analyzed in terms of an appropriate equivalent electrical circuit implemented in the Ec-Lab software.
3 Results and Discussion
3.1 Characterization of PA Inhibitor
The FTIR spectrum for linear PA is shown in Fig. 3. It can be seen that a broad transmittance band at round 3494 cm−1 is due to the stretching frequency of the hydrogen bonding after polymerization which occur in the N–H groups of the polymer. A band at 2995 cm−1 is associated with the C–H asymmetric stretching vibration. The two bands around 1667 and 1519 cm−1 are due to amidic carbonyl groups stretching and bending vibrations. In the case of transmittance, bands at 1325 and 1120 cm−1 are for the C–N stretching vibration frequencies.

FTIR spectra of linear PA
The methylene group –CH2 backbone produces a characteristic peak at 1444 cm−1. It should be mentioned that the effectiveness of monomer conversion to polymer can be confirmed because of the disappearance of a single band at 1675 cm−1 due to vinylic (C=C) vibration which was present in the monomer before polymerization. PA was also characterized using proton NMR spectroscopy in deuterium oxide (D2O). Figure 4 is a typical example of spectrum of the PA. The two peaks with chemical shift of 1.29–2.05 ppm correspond to the hydrogen in the main chain namely methylene and methine groups, respectively. The signal at 4.6 ppm corresponds to the hydrogen of the NH and NH2 groups and the water in the solvent D2O (99.9%) and the moisture in the sample.

1H NMR spectrum of PA
Finally, the molecular weight of the PA prepared and tested in corrosion inhibition was of 1.4 × 106 g mol−1. It was determined by the gel permeation chromatography and the viscosity average molecular weight methods.
3.2 Effect of PA Inhibitor Concentration
3.2.1 Mass-Loss Measurements
The effect of the addition of different concentrations of PA, on the corrosion of C-steel, in 1.0 M HCl solution, is studied by ML measurements at 298 K after 6-h immersion period. For every concentration, the mean value of the corrosion rate Wcorr and the inhibition efficiency ηML % are calculated using Eqs. (1) and (2), respectively:
where \({\nu _{\text{b}}}\) and \({\nu _{\text{a}}}\) are the specimen weight before and after immersion in the aggressive solution, respectively. Wcorr,0 and Wcorr,inh are the values of corrosion mass-losses of C-steel in uninhibited and inhibited solutions, respectively. S is the area of the C-steel specimen exposed to the aggressive solution (cm2), and t is the exposure time (h). The evolution of corrosion rate and inhibition efficiency is given in Fig. 5.

Evolution of corrosion rate and inhibition efficiency versus of PA concentration
Figure 5 reveals that the corrosion rate Wcorr decreases up to 4 × 10−6 mol L−1 and then increases afterward at higher concentrations of PA. Simultaneously, it can be observed an appreciable degree of protection testified by the evolution of the inhibiting efficiency. Then, a relatively high surface coverage can be achieved, even at low concentrations of PA, indicating the good adsorption characteristics of PA on C-steel in hydrochloride acid. It is to be noted that the inhibiting efficiency reached at 3 × 10−6 mol L−1 is very comparable to that obtained at 4 × 10−6 mol L−1. Hence, 75% of the PA product can be economized for any deeper investigations. In this context, we remain in coherence with the suitable definition of an inhibitor.
The observed increase in inhibition efficiency with rise of PA concentration can be attributed to the increase in surface area covered by the adsorbed polymer molecules onto the C-steel surface which reduces the direct contact between C-steel and the corrosive environment. Indeed, the presence of adsorption centers (oxygen and nitrogen heteroatom) in the PA molecular structure is expected to cause an easier electron transfer from the functional groups to the C-steel surface which provides greater adsorption ability and thereby inhibitory efficiency. In addition to this mode of adsorption of the molecular form, the hypothesis that PA can be present in protonated form in an acidic solution is not to be neglected. Indeed, the amides may be partially protonated at the oxygen atom in acidic media [33]. In fact, two possibilities of protonation may be considered as shown in Fig. 6a. The resonance within these protonated forms appears to be the decisive factor in determining the site of protonation. So, the protonation at the oxygen allows resonance stabilization of the positive charge while N-protonation destroys conjugation (Fig. 6b).

Protonation reactions of PA molecules
Indeed, the formation of positively charged protonated species facilitates adsorption of PA compound on the metal surface through electrostatic interaction with the negatively charged metal surface. In fact, it was reported that the surface charge of C-steel at Ecorr in hydrochloride solution is expected to be positive [34]. In fact, this charge was determined from the potential difference Ecorr − EPZC, where EPZC is the potential of zero charge, which is the metal potential measured against the RE under conditions of zero charge on the metal. Relationship between charge transfer resistance Rct values and the applied potential for C-steel electrode in 1.0 M HCl solution at 298 K showed that the potential of zero charge was reported to be more negative than the corrosion potential so the metal surface acquires slight positive charge. Thus, the Cl− ions are first adsorbed on the C-steel surface creating an excess of negative charges, which, in turn, facilitates physical adsorption of the inhibitor cations [35].
Furthermore, the larger molecular size and polymeric chain of PA molecule are to be considered which can induce greater coverage of the metallic surface and thus also could contribute to its inhibitory efficiency. However, the observed decrease in inhibition efficiency with increase in PA concentrations is probably due to desorption of PA from the metal surface. Inhibitor molecules probably start to desorb due to interaction between the inhibiting molecules already adsorbed on the surface and those yet dissolved in solution.
3.2.2 Potentiodynamic Polarization Measurements
PP measurements are carried out in order to gain knowledge concerning the kinetics of the anodic and cathodic reactions. Figure 7 illustrates the steady-state current–voltage characteristics for C-steel in 1.0 M HCl solution, at 298 K after half an hour of immersion period at open-circuit potential, in the presence and absence of restricted and representative concentrations of PA ranging from 1 × 10−6 to 6 × 10−6 mol L−1. In fact, at 5 × 10−7 and 9 × 10−6 mol L−1, the values of ηML% are so low what justifies the uselessness of their study with further techniques.

Polarization curves for C-steel in 1.0 M HCl containing different concentrations of PA and at 298 K
Figure 7 clearly shows that the addition of PA into the aggressive solution decreases the anodic along with the cathodic current densities. This implies that both the anodic metal dissolution and the cathodic hydrogen evolution reactions are inhibited. It is apparently shown that the cathodic current densities considerably reduce even at low inhibitor concentrations. Thus, the cathodic active sites of the C-steel may be almost covered by PA molecules at the lower inhibitor concentration, whereas at higher concentrations, PA has little effect on the rate of the hydrogen evolution reaction. The cathodic curves present a Tafelian behavior. Thus, the reduction of H+ ions at the cathodic sites of the C-steel surface takes place through a charge transfer mechanism or the process is activation controlled. Besides, an obvious ennoblement of potential is observed with addition of PA inhibitor at all studied concentrations without any particular trend.
The electrochemical kinetic parameters Ecorr, icorr, and βc are reported in Table 2. The icorr values are used to calculate the inhibition efficiency ηpp% using Eq. (3):
where icorr,0 and icorr,inh are the corrosion current densities for C-steel electrode in the absence and presence of PA inhibitor, respectively.
The results in Table 2 clearly indicate that the increase of PA concentration shifts slightly the values of Ecorr towards more anodic potentials but the magnitude of the displacement is not sufficient enough to ascertain the type of inhibitor being anodic or cathodic [36, 37]. The largest displacement of Ecorr value observed at 6 × 10−6 mol L−1 of PA is 77 mV which is much < 85 mV; value required for inhibitor to be recognized as anodic-type inhibitor [38]. However, the visual examination of polarization curves reveals that anodic curves are significantly more altered in the presence of inhibitor than cathodic curves. This indicates that anodic reaction is predominantly retarded on the addition of the inhibitor, and therefore, this compound could be classified as mixed-type corrosion inhibitor with predominant control of the anodic reaction.
It is to be mentioned that the PA inhibitor influences the anodic reaction only at over potentials more negative than − 300 mVAg/AgCl. In this domain, the increase of PA concentration leads to a marked decrease in the current-density so the adsorption rate of PA remains higher than its desorption rate. However, the current-density increases at more positive potentials than − 300 mV which can be ascribed to desorption of PA on the anodic sites of the electrode. This potential is generally defined as the desorption potential [39]. This behavior was the object of some controversies, according to similar results reported in the literature [40, 41]. It is explained by the deformation of inhibitor film over the metal surface and could also be the potential of molecules rearrangement forming the inhibitor film [42,43,44,45]. It is noteworthy from the shape of the anodic curves that it will be erroneous to extract the anodic Tafel slopes βa in order to evaluate the corrosion current densities. Thus, the anodic dissolution of C-steel is not activation controlled. However, it remains possible to deduce the corresponding values (not reported here). Simultaneously, the changed Tafel slopes βc, in the presence of PA, indicate that the inhibitor acts probably by merely blocking the catholic reaction sites of the metal surface with modification of the mechanism of hydrogen discharge.
Figure 8 exemplifies that the increase in PA concentrations slows down significantly the corrosion current densities icorr with respect to the uninhibited medium and thus increases the inhibition efficiencies ηpp%. The lowest current density occurs at 4 × 10−6 mol L−1, which is a result of the C-steel surface being covered by more inhibitor molecules due to their adsorption. On the other hand, at concentrations higher than 4 × 10−6 mol L−1, the corrosion current density increases, and thereby ηpp% decreases, indicating the desorption of PA inhibitor on the active sites of the electrode as reported elsewhere [46, 47].

Evolution of current density and inhibition efficiency as function of PA concentration
Hence, this behavior is in accordance with that derived from the gravimetric method. Furthermore, the results, at 3 × 10−6 mol L−1, are narrower to those obtained at 4 × 10−6 mol L−1 without any real improvement neither on icorr (2 µA cm−2) nor on ηpp% (0.4%).
3.2.3 EIS Measurements
A better understanding of the mechanism taking place at the electrode surface can be attained through EIS measurements. The EIS impedance is performed under potentiostatic conditions at Ecorr and at 298 K in the uninhibited and inhibited acidic solutions containing various concentrations of PA. Before each measurement, the electrode is left at the open-circuit conditions during 30 min. The electrode system does not evolve significantly during the impedance measurements. The Nyquist and Bode diagrams of C-steel, obtained in 1.0 M HCl solution in the absence and presence of various concentrations of PA, are shown in Fig. 9a, b, respectively.

Nyquist (a) and Bode (b) diagrams for C-steel in 1.0 M HCl solution at different concentrations of PA after half an hour of immersion period at Ecorr and at 298 K
It is clear from Fig. 9a that all the Nyquist diagrams exhibit one single capacitive loop, which indicates that the corrosion of C-steel is mainly controlled by charge transfer process [48]. Moreover, the capacitive loops are slightly depressed as often obtained in acidic media [49, 50]; their centers are located below the real axis which implies the measured capacitive response is not ideal. The deviation of Nyquist plots from an ideal semicircle is generally ascribed to Cole–Cole [51, 52] and/or Cole–Davidson [53] representations inherent to frequency dispersion. This phenomenon is generally attributed to different physical processes such as the non-homogeneity of the electrode surface or its roughness during the corrosion process [54, 55], adsorption of inhibitors [56], and formation of porous layers [57]. On the other hand, the diameters of these loops increase with rise of PA concentration, up to 4 × 10−6 mol L−1, without any change in the shape of the whole diagrams. This can suggest a similar corrosion mechanism in the presence of PA as the one in uninhibited medium.
In the optic of confirmation of the proposed mechanism, it was very useful to draw the Bode diagrams which explicitly show frequency information at high frequencies. The slope of transition between low-frequency and high-frequency asymptotes can also provide useful information concerning the nature of the impedance response if characteristic time constants are well separated. In fact, the Bode diagrams, reported in Fig. 9b, indicate the existence of three main regions. The first one, at low frequencies (from 10 Hz to 100 MHz); i.e., the frequency f → 0, the second region, at high frequencies (from 6 to 100 kHz); i.e. f → ∞,the measured impedances Z are practically not influenced by the frequency accompanied with a decrease of the phase angle (φ → 0). Thus, these two regions show typical ohmic behavior where the measured impedances correspond to an ohmic resistance. However, the region ranged from 10 Hz to 1 kHz shows a linear dependence of log|Z|–log f with a slope, p = ∂(log|Z|)/(∂log f), and a phase angle φ far from − 1° to 90°, respectively, showing then a non-ideal condenser. Both of these two parameters depend on PA concentration in the solution. Hence, in the above region, the impedance can be represented as follows (Eq. 4) with a constant phase element CPE:
where Q is a proportional factor whose unit is F cm−2 sα−1 and α is related to the phase shift of \({Z_{{\text{CPE}}}}\) (− 1 ≤ α ≤ +1), which is usually used as a gauge of the heterogeneity or roughness of the surface [58]. j2 = − 1 is an imaginary number and ω is the angular frequency.
Accordingly, the experimental data are then fitted and the best data-fitting equivalent circuit obtained to model the C-steel interface is shown in Fig. 10, characterized by parallel combination term (Rct//CPE) in series with the resistor RS.

Equivalent circuit model used to fit the EIS experiment data for C-steel in 1.0 M HCl
The impedance which corresponds to such an equivalent circuit can be expressed by Eq. (5):
where \({\tau _1}={R_{{\text{ct}}}} \times Q\) and \({\tau _2}=\frac{{{R_{\text{S}}} \times {R_{{\text{ct}}}} \times Q}}{{{R_{\text{S}}}+{R_{{\text{ct}}}}}}\).
The corresponding reduced impedance is given by Eq. (6):
where \(u=\tau _{{_{1}}}^{{1/\alpha }}\omega\) and \(T=\frac{{{\tau _2}}}{{{\tau _1}}}=\frac{{{R_{\text{S}}}}}{{{R_{\text{S}}}+{R_{{\text{ct}}}}}}<1\).
For any impedance, the expression is given as follows:
Simultaneously, the real Z′ and the imaginary − Z″ component of the reduced impedance as well as the phase angle or the argument φ are defined by Eqs. (8)–(10), respectively.
where \({c_\alpha }=\cos ({{\pi \times \alpha } \mathord{\left/ {\vphantom {{\pi \times \alpha } {2)}}} \right. \kern-0pt} {2)}}\) and \({s_\alpha }=\sin ({{\pi \times \alpha } \mathord{\left/ {\vphantom {{\pi \times \alpha } {2)}}} \right. \kern-0pt} {2)}}\).
So, at high frequencies, the impedance ZCPE acts like a short-circuit and the equivalent circuit can be modeled by Scheme 1 of Fig. 11, thus the measured impedance, at high frequencies, corresponds to the ohmic resistance of the electrolytic solution (φ → 0) determined between the working and the REs, whereas the ones measured at low frequencies correspond to the sum of charge transfer and electrolytic resistance (φ → 0) since, in this case, the impedance ZCPE acts like an open-circuit and the equivalent circuit can be modeled by Scheme 2 of Fig. 11. Evidently, the measured impedances in these two regions allow the identification of both solution resistance and charge transfer resistance.

Equivalent circuits corresponding at high frequencies (1) and at low frequencies (2), respectively
On the other hand, the increase in the peak heights in the phase-angle plots, depicted in Fig. 9b, indicates a more capacitive response of the interface due to the presence of the inhibitor molecules at the interface when compared to the blank solution. On the other hand, the phase angle is more important probably due to the formation of protective layer by PA inhibitor on C-steel surface, which changed the electrode interfacial structure.
The charge transfer resistance Rct has been used to calculate inhibition efficiency ηEIS% of PA given by Eq. (11):
where Rct,0 and Rct,inh are the charge transfer resistances in the absence and presence of PA inhibitor, respectively.
However, the double layer pseudo-capacitance Cdl obtained by fitting experimental data is defined by Eq. (12) [59]:
Table 3 collects the overall electrochemical parameters issued from the suitable electrical circuit of Fig. 10 along with the goodness of fit χ2 is also reported in Table 3, defined as follows in Eq. (13) [60, 61]:
where Zmeas(i) is the measured impedance at the fi frequency; Zmodel(fi, param) is function of the chosen model and param is the model parameters (RS, Rct, Q). Di is the normal deviation.
It can be shown that the RS values are very small, which confirms that the IR-drop can be neglected in the present system. Table 3 reports also the calculated “relaxation time” values, τ, according to Eq. (14):
As it can be seen from Bode plots, only one-time constant appears, which confirms that the corrosion of C-steel in 1.0 M HCl solution is mainly controlled by a charge transfer process.
Figure 12 exemplifies the evolution of the charge transfer resistance Rct along with the double layer pseudo-capacitance Cdl. The Rct values in uninhibited 1.0 M HCl solution increase significantly after the addition of PA inhibitor. The higher Rct values are generally associated with slower corroding system characterized by a difficult electron transfer crossing the metal surface. The Rct values increase while Cdl values reduce with rise of PA concentration up to 4 × 10−6 mol L−1 in 1.0 M HCl (Fig. 12), which led to an increase in ηEIS% (Fig. 13). This is due to the gradual replacement of water molecules by the adsorption of the inhibitor molecules on the metal surface, and decreasing the extent of dissolution reaction. This situation suggests that more inhibitor molecules are adsorbed on the metal surface and provide better surface coverage which reduces the pseudo-capacitance by either a decrease in local dielectric constant or an increase in the thickness of the electrical double layer. This result can be confirmed by the increase in the value of α through the addition of PA in the corrosive solution which indicates reduction of surface inhomogeneity due to the adsorption of inhibitor molecules on the active adsorption sites at C-steel/solution interface [62, 63].

Variation of the charge transfer resistance and the double layer capacitance versus the PA concentration at 298 K

Evolution of charge transfer resistance and inhibition efficiency as function of PA concentration at 298 K
On the other hand, the addition of PA in 1.0 M HCl increases slightly the relaxation time constant value with an opposite trend of Cdl (cf. Table 3). Indeed, in the uninhibited and at 4 × 10−6 mol L−1 of PA, the interface τ parameter increases from 11.585 to 13.09 ms while the capacitance of double layer decreases from 306.585 to 49.49 µF cm−2, signifying that the charge and discharge rates on mild C-steel/solution interface are significantly decreased. This shows that there is conformity between the discharge velocity at the interface and the amount of charge that can be stored (i.e., capacitance). It is worth mentioning that the value of the relaxation time constant remains almost constant despite the slight increase of circa 2 ms in inhibited media. This finding justifies the choice of the equivalent circuit reported in Fig. 10.
Furthermore, it is worth noting that the inhibition efficiency values, calculated from EIS measurements, show the same trend as those obtained from ML and PP measurements.
3.3 Comparative Study of PA Behavior at 3 × 10−6 mol L−1 in Both Acidic Media 1.0 M HCl and 0.5 M H2SO4
The comparison of the inhibition behavior in both acids has been performed using EIS technique, in order to investigate the impact of the sulfate ions characterized by their smaller degree of hydration compared to chloride ones. The Nyquist and Bode diagrams of C-steel, obtained in 1.0 M hydrochloric and 0.5 M sulfuric acids in the absence and presence of 3 × 10−6 mol L−1of PA at 298 K, are shown in Fig. 14a–d, respectively.

Nyquist (a, c) and Bode (b, d) diagrams for C-steel in 1.0 M HCl and 0.5 M H2SO4 solutions, respectively, at 3 × 10−6 mol L−1 of PA after half an hour of immersion period at Ecorr and at 298 K
First of all, the Nyquist and Bode diagrams obtained in the absence and the presence of 3 × 10−6 mol L−1 of PA have similar shape in sulfuric acid, indicating that almost no change in the corrosion mechanism occurred as a result of PA addition. In contrast, when compared to 1.0 M HCl, these diagrams show, in contrast, two-time constants consisting of large capacitive loop at high to medium frequencies and an inductive loop at low frequencies. The capacitive loop, as obtained in chloride acid, is a depressed semicircle shown in Nyquist plots and the phase angle is lower than 90° in Bode phase plots. This capacitive loop is usually related to the time constant of the charge transfer of the corrosion process and double layer behavior. On the other hand, the cause of the inductive loop is still uncertain. It may be attributed to the relaxation process of adsorbed charged intermediates include Hads+, acid anions or inhibitor species on the electrode surface. It might also be attributed to the re-dissolution of the passivated surface at low frequencies [64]. The best data-fitting equivalent circuit obtained to model the C-steel interface is shown in Fig. 15. It is composed, in addition to RS, Rct, and CPE, the inductive elements, RL and L. Electrochemical parameters issued from this suitable electrical circuit are given in Table 4.

Equivalent circuit model used to fit the EIS experiment data for C-steel in 0.5 M H2SO4 solution at 3 × 10−6 mol L−1 of PA at 298 K
It is also apparent from the Nyquist plots that the impedance response of mild steel in uninhibited HCl and H2SO4 solutions has significantly changed after the addition of PA. This indicates that the impedance of inhibited substrate increases in both acids suggesting that corrosion inhibition of C-steel is afforded by the adsorption of the PA molecules and blocking the active sites. The greatest effect, compared to inhibitor-free solution, was observed in 0.5 M H2SO4 (Table 4). Indeed, in inhibited media, the charge transfer resistance is 8 and 6.8 times greater in 0.5 M H2SO4 and 1.0 M HCl, respectively. Thus, the inhibitive ability of PA in H2SO4 is comparatively more than in HCl. This may be due to the availability of more sites on the metal surface in H2SO4 solution because of the lesser adsorption of the sulfate ions on the C-steel surface [65]. In addition, the electrostatic influence on the inhibitor adsorption may be the reason for a decreased protective effect in chloride containing solution.
Indeed, it has been reported that PA molecules exist in a protonated form in acidic media [33]. Therefore, it has been proposed in the literature [66] that the protonated inhibitor loses its associated proton when entering the double layer and chemisorbs by donating electrons to the metal (competitive adsorption). In addition, the protonated inhibitor electrostatically adsorbs onto the anion covered surface through its cationic form (cooperative adsorption). Moreover, the chloride ions are more strongly adsorbed on the C-steel surface than sulfate ions and the cationic form of the PA molecule can so jointly adsorb on the metal surface without much difficulty in HCl when compared to H2SO4 [67]. However, the inhibition efficiency obtained by PA was found to be higher in H2SO4 solution. It can be concluded that the availability of more sites on the metal surface, in H2SO4 solution, because of the lesser adsorption of the sulfate ions on C-steel surface, may be the reason for an increased protective effect in sulfuric acid solution. Furthermore, the presence of carbonyl (–C=O) and amino (–NH2) groups in the structure of PA repeat unit is expected to cause, in the neutral form, formation of an iron–nitrogen coordination bond (Fig. 16).

Schematic representations of competitive and cooperative adsorption of PA in acid solution
It remains to be noted that in the continuation of the present study, part 2 will focus some insights into the PA adsorption on C-steel through the confrontation of six adsorption isotherms by evaluating and analyzing the corresponding kinetic–thermodynamic parameters and will also cover the effects of temperature as well as immersion time period.
4 Conclusions
From the above analyses and discussion of the experimental results, the following main conclusions are drawn:
-
1.
High molecular weight PA is successfully synthesized by inverse emulsion polymerization. It chemical structure is characterized by different spectroscopic techniques and molecular weight, of 1.4 × 106 g mol−1, determined by the gel permeation chromatography and the viscosity average molecular weight methods.
-
2.
The PA acts as a good inhibitor for the corrosion of C-steel in 1.0 M HCl solution and its inhibition efficiency depends on its concentration.
-
3.
PP measurements indicate that PA is a mixed-type inhibitor predominantly anodic type.
-
4.
The EIS data testified that the corrosion of C-steel is mainly controlled by charge transfer on heterogeneous and irregular surface.
-
5.
The corrosion inhibition takes place by adsorption of PA protonated molecules on the C-steel surface through electrostatic interactions as well as charge sharing or transfer between PA non-protonated molecules and the metal surface.
-
6.
EIS measurements related to C-steel in 0.5 M H2SO4 solution at 298 K indicate that PA, at 3 × 10−6 mol L−1, show at high frequencies capacitive loop related to the charge transfer process of the metal corrosion and the double layer behavior and at low frequencies an inductive part attributed to the relaxation of adsorbed species on the metal surface.
-
7.
The inhibition efficiency is better in 0.5 M H2SO4 than in 1.0 M HCl at 3 × 10−6 mol L−1 and 298 K.
References
Kowalski D, Ueda M, Ohtsukat T (2007) The effect of counter anions on corrosion resistance of steel covered by bi-layered polypyrrole film. Corros Sci 49:3442–3452. https://doi.org/10.1016/j.corsci.2007.03.007
Jeyaprabha C, Sathiyanarayanan S, Venkatachari G (2006) Polyaniline as corrosion inhibitor for iron in acid solutions. J Appl Polym Sci 101:2144–2153. https://doi.org/10.1002/app.22579
Elbakari M, Touir R, Ebn Touhami M, Srhiri A, Benmessaoud M (2008) Electrosynthesis of adherent poly(3-amino-1,2,4-triazole) films on brass prepared in nonaqueous solvents. Corros Sci 50:1538–1545. https://doi.org/10.1016/j.corsci.2008.02.014
Lehr IL, Saidman SB (2007) Corrosion protection of iron by polypyrrole coatings electrosynthesised from a surfactant solution. Corros Sci 49:2210–2225. https://doi.org/10.1016/j.corsci.2006.10.033
Migahed MA, Aly RO, Al-Sabagh AM (2004) Impact of gamma-ray-pre-irradiation on the efficiency of corrosion inhibition of some novel polymeric surfactants. Corros Sci 46:2503–2516. https://doi.org/10.1016/j.corsci.2004.01.013
Titz T, Horzenberger F, Bergh KVD, Grundmeier G (2010) Correlation of interfacial electrode potential and corrosion resistance of plasma polymer coated galvanized steel. Part 1: ultra-thin plasma polymer films of varying thickness. Corros Sci 52:369–377. https://doi.org/10.1016/j.corsci.2009.09.024
Umoren SA, Ogbobe O, Okafor PC, Ebenso EE (2007) Polyethylene glycol and polyvinyl alcohol as corrosion inhibitors for aluminium in acidic medium. J Appl Polym Sci 105:3363–3370. https://doi.org/10.1002/app.26530
Umoren SA, Obot IB (2008) Polyvinylpyrollidone and polyacrylamide as corrosion inhibitors for mild steel in acidic medium. Surf Rev Lett 15:277–286. https://doi.org/10.1142/S0218625X08011366
Umoren SA, Lia Y, Wang FH (2010) Electrochemical study of corrosion inhibition and adsorption behaviour for pure iron by polyacrylamide in H2SO4: synergistic effect of iodide ions. Corros Sci 52:1777–1786. https://doi.org/10.1016/j.corsci.2010.01.026
Umoren SA, Solomon MM (2010) Effect of halide ions additives on the corrosion inhibition of aluminum in HCl by polyacrylamide. Arab J Sci Eng 35(2):115. https://www.researchgate.net/publication/259284202
Umoren SA, Li Y, Wang FH (2011) Influence of iron microstructure on the performance of polyacrylic acid as corrosion inhibitor in sulfuric acid solution. Corros Sci 53:1778–1785. https://doi.org/10.1016/j.corsci.2011.01.052
Chamovska D, Cvetkovska M, Grchev T (2007) Corrosion inhibition of iron in hydrochloric acid by polyacrylamide. J Serb Chem Soc 72(7):687–698
Manimaran N, Rajendran S, Manivannan M, John Mary S (2012) Corrosion inhibition of carbon steel by polyacrylamide. Res J Chem Sci 2(3):52–57. http://www.isca.in (ISSN 2231-606X)
Srivastava V, Banerjee S, Singh MM (2010) Inhibitive effect of polyacrylamide grafted with fenugreek mucilage on corrosion of mild steel in 0.5 M H2SO4 at 35 °C. J Appl Polym Sci 116(2):810–816. https://doi.org/10.1002/app.31559
Amin MA, Abd EI-Rehim SS, El-Sherbini Essam EF, Hazzazi OA, Abbas Mohsen N (2009) Polyacrylic acid as a corrosion inhibitor for aluminium in weakly alkaline solutions. Part I: weight loss, polarization, impedance EFM and EDX studies. Corros Sci 51:658–667. https://doi.org/10.1016/j.corsci.2008.12.008
Finšgar M, Fassbender S, Nicolini F, Milošev I (2009) Polyethyleneimine as a corrosion inhibitor for ASTM 420 stainless steel in near-neutral saline media. Corros Sci 51:525–533. https://doi.org/10.1016/j.corsci.2008.12.006
Gupta G, Birbilis N, Khanna (2013) Polyaniline-lignosulfonate/epoxy coating for corrosion protection of AA2024-T3. Corros Sci 67:256–267. https://doi.org/10.1016/j.corsci.2012.10.022
Arthur DE, Jonathan A, Ameh PO, Anya C (2013) A review on the assessment of polymeric materials used as corrosion inhibitor of metals and alloys. Int J Ind Chem 4:2–9. https://doi.org/10.1186/2228-5547-4-2
Saliyan, VR, Adhikari AV (2008) Inhibition of corrosion of mild steel in acid media by N′-benzylidene-3-(quinolin-4-ylthio)propanohydrazide. Bull Mater Sci 31:699–711. https://doi.org/10.1007/s12034-008-0111-4
Benabdellah M, Ousslim A, Hammouti B, Elidrissi A, Aouniti A, Dafali A, Bekkouch K, Benkaddour M (2007) The effect of poly(vinyl caprolactone-co-vinyl pyridine) and poly(vinyl imidazol-co-vinyl pyridine) on the corrosion of steel in H3PO4 media. J Appl Electrochem 37:819–826. https://doi.org/10.1007/s10800-007-9317-1
Müller LB, Schmelich T (1995) High-molecular weight styrene maleic acid copolymers as corrosion inhibitors for aluminium pigments. Corros Sci 37:877–883. https://doi.org/10.1016/0010-938X(94)00171-2
Bereket G, Yurt A, Turk H (2003) Inhibition of corrosion of low carbon steel in acidic solution by selected polyelectrolytes and polymers. Anti-Corros Methods Mater 50:422–435. https://doi.org/10.1108/00035590310501585
Shukla SK, Quraishi MA, Prakash R (2008) A self-doped conducting polymer polyanthranilic acid an efficient corrosion inhibitor for mild steel in acidic solution. Corros Sci 50:2867–2872. https://doi.org/10.1016/j.corsci.2008.07.025
Ren Y, Luo Y, Zhang K, Zhu G, Tan X (2008) Lignin terpolymer for corrosion inhibition of mild steel in 10% hydrochloric acid medium. Corros Sci 50:3147–3153. https://doi.org/10.1016/j.corsci.2008.08.019
Solomon MM, Umoren SA, Udosoro II, Udoh AP (2010) Inhibitive and adsorption behaviour of carboxymethyl cellulose on mild steel corrosion in sulphuric acid solution. Corros Sci 52:1317–1325. https://doi.org/10.1016/j.corsci.2009.11.041
Bhandari H, Srivastav R, Choudhary V, Dhawan SK (2010) Enhancement of corrosion protection efficiency of iron by poly(aniline-co-amino-naphthol-sulphonic acid) nanowires coating in highly acidic medium. Thin Solid Films 519:1031–1039. https://doi.org/10.1016/j.tsf.2010.08.038
Yurt A, Buetuen V, Duran B (2007) Effect of the molecular weight and structure of some novel water-soluble triblock copolymers on the electrochemical behaviour of mild steel. Mater Chem Phys 105:114–121. https://doi.org/10.1016/j.matchemphys.2007.04.009
Abd El-Maksoud SA, Fouda AS (2005) Some pyridine derivatives as corrosion inhibitors for carbon steel in acidic medium. Mater Chem Phys 93:84–90. https://doi.org/10.1016/j.matchemphys.2005.02.020
Rajendran S, Sridevi SP, Anthony N, John Amalraji A, Sundaravadivedi M (2005) Corrosion behavior of carbon steel in polyvinyl alcohol. Anti-Corros Methods Mater 52:102–107. https://doi.org/10.1108/00035590510584816
Jeyaprabha C, Sathiyanarayanan S, Phani KLN, Venkatachari G (2005) Influence of poly(aminoquinone) on corrosion inhibition of iron in acid media. Appl Surf Sci 252:966–975. https://doi.org/10.1016/j.apsusc.2005.01.098
Small PA (1953) Some factors affecting the solubility of polymers. J Appl Chem 3:71–80. https://doi.org/10.1002/jctb.5010030205
Mohsin MA, Attia NF (2015) Inverse emulsion polymerization for the synthesis of high molecular weight polyacrylamide and its application as sand stabilizer. Int J Polym Sci. https://doi.org/10.1155/2015/436583
Fraenkel G, Niemann C (1958) The mode of protonation of amides. Proc Natl Acad Sci USA 44(7):688–691
Morad MS (2008) Inhibition of iron corrosion in acid solutions by Cefatrexy: behaviour near and at the corrosion potential. Corros Sci 50:436–448. https://doi.org/10.1016/j.corsci.2007.08.018
Bentiss F, Jama C, Mernari B, El Attari H, El Kadi L, Lebrini M, Traisnel M, Lagrenée M (2009) Inhibition of iron corrosion in acid solutions by Cefatrexyl: behaviour near and at the corrosion potential. Corros Sci 51:1628–1635. https://doi.org/10.1016/j.corsci.2009.04.009
Bentrah H, Rahali Y, Chala A (2014) Gum Arabic as an eco-friendly inhibitor for API 5L X42 pipeline steel in HCl medium. Corros Sci 82:426–431. https://doi.org/10.1016/j.corsci.2013.12.018
Soltani N, Tavakkoli N, Khayatkashani M, Jalali MR, Mosavizade A (2012) Green approach to corrosion inhibition of 304 stainless steel in hydrochloric acid solution by the extract of saliva officinalis leaves. Corros Sci 62:122–135. https://doi.org/10.1016/j.corsci.2012.05.003
Afia L, Salghi R, Bammou L, Hammouti B, Bazzi L, Bouyanzer A (2014) Anti-corrosive properties of Argan oil on C38 steel in molar HCl solution. J Saudi Chem Soc 18:19–25. https://doi.org/10.1016/j.jscs.2011.05.008
Zarrouk A, Zarrok H, Salghi R, Hammouti B, Bentiss F, Touir R, Bouachrine M (2013) Evaluation of N-containing organic compound as corrosion inhibitor for carbon steel in phosphoric acid. J Mater Environ Sci 4(2):177–192 (ISSN 2028-2508 CODEN: JMESCN)
Bentiss F, Traisnel M, Vezin H, Hildebrand HF, Lagrenée M (2004) 2,5-Bis(4-dimethylaminophenyl)-1,3,4-oxadiazole and 2,5-bis(4-dimethylaminophenyl)-1,3,4-thiadiazole as corrosion inhibitors for mild steel in acidic media. Corros Sci 46:2781–2792. https://doi.org/10.1016/j.corsci.2004.04.001
Qian B, Wang J, Zheng M, Hou B (2013) Synergistic effect of polyaspartic acid and iodide ion on corrosion inhibition of mild steel in H2SO4. Corros Sci 75:184–192. https://doi.org/10.1016/j.corsci.2013.06.001
Zerga B, Hammout Bi, Ebn Touhami M, Touir R, Taleb M, Sfaira M, Bennajeh M, Forssal I (2012) Comparative inhibition study of new synthesized pyridazine derivatives towards mild steel corrosion in hydrochloric acid. Part-II: thermodynamic proprieties. Int J Electrochem Sci 7:471–483. http://www.electrochemsci.org/papers/vol7/7010471.pdf
Solmaz R (2010) Investigation of the inhibition effect of 5-((E)-4-phenylbuta-1, 3-dienylideneamino)-1, 3, 4-thiadiazole-2-thiol Schiff base on mild steel corrosion in hydrochloric acid. Corros Sci 52:3321–3330. https://doi.org/10.1016/j.corsci.2010.06.001
Znini M, Cristofari G, Majidi L, Ansari A, Bouyanzer A, Paolini J, Costa J, Hammouti B (2012) Green approach to corrosion inhibition of mild steel by essential oil leaves of Asteriscus graveolens (Forssk.) in sulphuric acid medium. Int J Electrochem Sci 7:3959–3981. http://www.electrochemsci.org/papers/vol7/7053959.pdf
Bentiss F, Outirite M, Traisnel M, Vezin H, Lagrenée M, Hammouti B, Al-Deyab SS, Jama C (2012) Improvement of corrosion resistance of carbon steel in hydrochloric acid medium by 3,6-bis(3-pyridyl)pyridazine. Int J Electrochem Sci 7:1699–1723. http://www.electrochemsci.org/papers/vol7/7021699.pdf
Wang J, Cao C, Chen J, Zhang M, Ye G, Lin H (1995) Anodic desorption of inhibitors. J Chin Soc Corros Prot 15:241–248. http://www.jcscp.org/EN/Y1995/V15/I4/241
Feng Y, Siow KS, Teo WK, Hsieh AK (1999) The synergistic effects of propargyl alcohol and potassium iodide on the inhibition of mild steel in 0.5 M sulfuric acid solution. Corros Sci 41:829–852. https://doi.org/10.1016/S0010-938X(98)00144-9
Behpour M, Ghoreishi SM, Mohammadi N, Soltani N, Salavati-Niasari M (2010) Investigation of some Schiff base compounds containing disulfide bond as HCl corrosion inhibitors for mild steel. Corros Sci 52:4046–4057. https://doi.org/10.1016/j.corsci.2010.08.020
Ramananda MS (2013) A green Approach: a corrosion inhibition of mild steel by adhatoda vasica plant extract in 0.5 M H2SO4. J Mater Environ Sci 4:119–126 (ISSN 2028-2508 CODEN: JMESCN)
Gualdrón AF, Becerra EN, Peña DY, Gutiérrez JC, Becerra HQ (2013) Inhibitory effect of Eucalyptus and Lippia alba essential oils on the corrosion of mild steel in hydrochloric acid. J Mater Environ Sci 4:143–158 (ISSN 2028-2508 CODEN: JMESCN)
Cole KS, Cole RH (1941) Dispersion and absorption in dielectrics. I. Alternating current characteristics. J Chem Phys 9:341–351. https://doi.org/10.1063/1.1750906
Duval S, Keddam M, Sfaira M, Srhiri A, Hakenouti H (2002) Electrochemical impedance spectroscopy of epoxy-vinyl coating in aqueous medium analyzed by dipolar relaxation of polymer. J Electrochem Soc 149:B520–B529. https://doi.org/10.1149/1.1512667
Davidson DW, Cole RH (1951) Dielectric relaxation in glycerol, propylene glycol, and n-propanol. J Chem Phys 19:1484–1490. https://doi.org/10.1063/1.1748105
Juttner K (1990) Electrochemical impedance spectroscopy (EIS) of corrosion processes on inhomogeneous surfaces. Electrochim Acta 35(90):1501–1508. https://doi.org/10.1016/0013-4686(90)80004-8
Deflorain F, Miscovic-Stankovic VB, Bonora PL, Fedrizzi L (1994) Degradation of epoxy coatings on phosphatized zinc-electroplated steel. Corrosion 50:438–446. https://doi.org/10.5006/1.3293522
Bentiss F, Traisnel M, Gengembre L, Lagrenée M (1999) A new triazole derivative as inhibitor of the acid corrosion of mild steel: electrochemical studies, weight loss determination, SEM and XPS. Appl Surf Sci 152:237–249. https://doi.org/10.1016/S0169-4332(99)00322-0
Bentiss F, Traisnel M, Lagrenée M (2000) Inhibitor effects of triazole derivatives on corrosion of mild steel in acidic media. Br Corros J 35:315–320. https://doi.org/10.1179/000705900101501326
Schultze JW, Wippermann K (1987) Inhibition of electrode processes on copper by AHT in acid solutions. Electrochim Acta 32:823–831. https://doi.org/10.1016/0013-4686(87)85115-0
Jovic VD, Jovic BM BM (2003)EIS and differential capacitance measurements onto single crystal faces in different solutions. Part I: Ag(111) in 0.01 M NaCl. J Electroanal Chem 541:1–11. https://doi.org/10.1016/S0022-0728(02)01309-8
Press WH, Teukolsky SA, Vetterling WT, Flannery BP (1992) Numerical recipes in C: the art of the scientific computing, 2nd edn. Cambridge University Press, New York. ISBN 0-521-43108-5
Barsoukov E, Ross Macdonald J (2005) Impedance spectroscopy: theory, experiment and applications, 2nd edn. Wiley, New York. ISBN 978-0-471-64749-2
Zarrok H, Zarrouk A, Hammouti B, Salghi R, Jama C, Bentiss F (2012) Corrosion control of carbon steel in phosphoric acid by purpald-weight loss, electrochemical and XPS studies. Corros Sci 64:243–252. https://doi.org/10.1016/j.corsci.2012.07.018
Popova A, Christov M, Vasilev A (2007) Inhibitive properties of quaternary ammonium bromides of N-containing heterocycles on acid mild steel corrosion. Part II: EIS results. Corros Sci 49:3290–3302. https://doi.org/10.1016/j.corsci.2007.03.012
Yadav FK, Quraishi MA, Maiti B (2012) Inhibition effect of some benzylidenes on mild steel in 1 M HCl: an experimental and theoretical correlation. Corros Sci 55:254–266. https://doi.org/10.1016/j.corsci.2011.10.030
Lorenz WJ, Mansfeld F (1981) Determination of corrosion rates by electrochemical. DC and AC methods. Corros Sci 21:647–672. https://doi.org/10.1016/0010-938X(81)90015-9
Bentiss F, Traisnel M, Lagrenée M (2001) Influence of 2,5-bis(4-dimethylaminophenyl)-1,3,4-thiadiazole on corrosion inhibition of mild steel in acidic media. J Appl Electrochem 31:41–48. https://doi.org/10.1023/A:1004141309795
Muralidharan S, Phani KLN, Pitchumani S, Ravichandran S, Iyer SVK (1995) Polyamino–benzoquinone polymers: a new class of corrosion inhibitors for mild steel. J Electrochem Soc 142:1478–1483. https://doi.org/10.1149/1.2048599
Funding
This study was supported by BENIKEN.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Beniken, M., Driouch, M., Sfaira, M. et al. Anticorrosion Activity of a Polyacrylamide with High Molecular Weight on C-Steel in Acidic Media: Part 1. J Bio Tribo Corros 4, 38 (2018). https://doi.org/10.1007/s40735-018-0155-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40735-018-0155-y