
Abstract
Purpose of Review
Sleep-disordered breathing (SDB) is highly prevalent in heart failure (HF), and the coexistence of these two disease conditions increases mortality. The purpose of this review is to examine the mechanistic interactions and associations between SBD, specifically obstructive sleep apnea (OSA), obesity hypoventilation (OHS), left ventricular (LV) diastolic heart function, and pulmonary hypertension (PH), as factors that directly contribute to the development of HF.
Recent Findings
The mechanistic interactions may be thought of as direct and indirect effects. The direct effects are related to abnormalities caused by sleep-disordered breathing that induce LV remodeling. Recent studies support the notion that the severity of OSA and recurrent hypoxia enhance LV remodeling even after accounting for age, weight, and underlying cardiorespiratory comorbid conditions. Indirect effects are due to the mechanical alterations intrathoracic organs during sleep due to negative intrathoracic pressure, obesity, as well as multiple derangements causing activation of the sympathetic nervous system and endothelium. The review also delineates multiple mechanisms that eventually lead to pulmonary vascular remodeling and PH.
Summary
The severity of daytime arterial oxygen levels and/or duration of nocturnal hypoxia, rather than the severity of SDB, seems to be the major determinants of PH in SBD, after adjusting for other cardiorespiratory diseases. Mechanisms by which hypercapnia contributes to PH and diastolic dysfunction need to be explored. Moreover, interactions between central sleep apnea and diastolic heart function are not well delineated. Further investigations are needed to elucidate any direct effect of hypercapnia on pulmonary pressures and potential cardiovascular consequences. There is limited evidence on the impact of PAP or oxygen therapy on these intermediary mechanisms of HF in SDB populations. The collection of evidence indicates that PAP may enhance diastolic function and reduce pulmonary artery pressure in OSA whereas studies in OHS populations are sparse and inconclusive. Thus, future research should delineate whether pathophysiology-based therapies can alleviate HF and HF related mortality in these SDB populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) impacts 5.7 million adults in the USA with a projected prevalence increase of 46% from 2012 to 2030, resulting in > 8 million adults with HF [1]. The cost of HF to the US is an estimated $30.7 billion each year and projected to increase to $77.7 billion in 2030 [2]. HF is closely linked to obstructive sleep apnea (OSA) [3] and HF in patients with OSA is associated with increased mortality [4] and morbidity. OSA is a highly prevalent disease (2–9%) [5], characterized by repetitive closure of the upper airway with associated arousals and intermittent hypoxia, and whose prevalence has increased 14–55% over the last two decades [6] in concert with the recognized obesity epidemic [7]. The obesity epidemic likely also increased the risk for obesity hypoventilation syndrome (OHS), where obesity-related daytime hypoventilation and hypoxia coexist with sleep-disordered breathing [8], and confers a risk for developing pulmonary hypertension (PH) and HF [9]. This review focuses on the pathophysiology of diastolic dysfunction and PH as pathways for the development of HF in OSA and OHS. Studies that are observational and cross-sectional provide the bulk of the evidence, in addition to mechanistic studies in humans and animal models.
Diastolic Dysfunction and OSA
Cross-Sectional Associations
Multiple studies have evaluated the relation between left ventricular diastolic dysfunction (LVDD) and OSA (Table 1). While one early study did not find an association between OSA and diastolic dysfunction [10], several others have correlated echocardiographic LV diastolic dysfunction with OSA [11, 13,14,15, 17,18,19,20]. The prevalence of LV diastolic dysfunction varied from 23 [14] to 56% [13] in moderate to severe OSA patients who did not have underlying cardiovascular disease (CVD). In the large RICCADSA trial of revascularized patients with coronary artery disease, OSA was associated with worse diastolic function (odds ratio (OR) 1.9), whereas older age, obesity, and current smoking status were not significant predictors [19]. Contrarily, in a separate study, older age (OR 3.29) and mean nocturnal oxygen saturation < 92% (OR 2.76) were associated with LV diastolic dysfunction [14]. Nadir oxygen saturation, urine epinephrine, and percentage of sleep time spent with < 90% SpO2 were also predictors of LV diastolic dysfunction [11]. Additionally, the severity of apnea hypopnea index (number of apneas and hypopneas per hour (h) sleep, and a measure of OSA severity) [15, 16] and AHI during REM sleep [17] were other important significant predictors. Moreover, in 2058 subjects from the Sleep Heart Health Study (SHHS), severe OSA (AHI ≥ 30/h) was associated with a higher risk of having concomitant LV hypertrophy (OR 1.78) independent of age, sex, BMI, hypertension, and cardiovascular disease.
OSA may also be associated with right ventricular (RV) remodeling and dysfunction in the absence of LV dysfunction. However, the data supporting this notion are conflicting. A few studies had elevated mean pulmonary artery pressure, suggesting that PH from other causes may have been the precursor for RV dysfunction [21]. Evidence from the Framingham Heart Study suggested that RV dysfunction was present in OSA independent of other factors. RV wall thickness, as assessed by echocardiogram, was significantly greater after multivariable adjustment in patients with OSA compared to those without (0.78 vs. 0.68 ± 0.02 cm respectively, p = 0.005) [22].
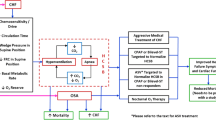
Pathophysiologic Mechanisms (Fig. 1)
The mechanisms which are responsible for cardiac dysfunction in patients with OSA are numerous and intertwined. This section delineates the pathophysiological mechanisms in patients with sleep-disordered breathing which are responsible for right heart dysfunction due to PH as well as LVDD due to mechanisms including systemic hypertension.

Figure demonstrates the potential pathophysiology linking LV diastolic function and PH to SDB. These include intermittent hypoxia, obesity, negative intrathoracic pressure, and/or increased sympathetic activity. The figure also depicts the potential mechanisms underlying these pathways
The etiology of diastolic dysfunction in patients with OSA is mainly due to alterations in LV morphology with subsequent impairment in LV relaxation. This has been demonstrated in both animal and human studies.
Artificially induced OSA in canine models have shown that LV afterload increases with each hypoxic episode with resultant systolic dysfunction after 3 months [23]. In humans, the same phenomenon occurs, with diastolic function being affected first; however systolic function was only affected after approximately 10 years [12].
LV remodeling in OSAS is due to a number of mechanisms including repetitive disturbances in respiratory patterns and subsequent hypoxia, hypercapnia, increased negative intrathoracic pressure, and microarousals. Due to these disturbances, patients with OSA experience variation in their hemodynamics leading to inappropriate activation of the sympathetic nervous system during sleep [24]. Sympathetic overactivity is more pronounced during sleep; however, even after awakening and respiration returning to normal levels, the effect persists [25]. This is in part due to maladaptation to inappropriate sympathetic activity leading to overactivation of the hindbrain regions controlling sympathetic outflow in addition to heightened synaptic signaling from the forebrain through the paraventricular nucleus [26]. These persistent pathophysiologic mechanisms cause peripheral vascular remodeling, increase in vascular resistance, subsequent increase in cardiac afterload and LV hypertrophy [24, 27, 28].
Repetitive episodes of hypoxia and subsequent re-oxygenation are another cause of LVDD due to a number of mechanisms. One of which is resultant oxidative stress causing production of reactive oxygen radicals [29]. This oxidative stress then leads to endothelial dysfunction as well as ischemia-reperfusion, ventricular remodeling, and eventually increase in LV mass [30]. In addition, intermittent hypoxia also leads to neurohormonal disturbances through activation of the renin-aldosterone-angiotensin system due to augmented carotid chemoreceptor activity and inflammation of the carotid body [20, 31]. This mechanism, in addition to overstimulation of the sympathetic nervous system, further exacerbates elevated nocturnal and diurnal mean arterial pressures.
Obesity, a known risk factor for developing OSA [32], further potentiates increase in LV mass and LV remodeling. The incidence of obesity among patients discharged with a diagnosis of HF with preserved ejection fraction was reported to be 41% in a retrospective study of over 4000 HF patients in Minnesota [33]. Various metabolic abnormalities associated with obesity, such as hyperlipidemia and hyperinsulinemia contribute to LV remodeling, which in turn leads to LV dysfunction and changes in LV morphology [34].
Inevitably, LV hypertrophy develops as a result of both direct and indirect etiologies. It is directly affected by remodeling and increased LV mass due to oxidative stress and metabolic derangements associated with obesity [32,33,34]. Indirect effects include persistently elevated MAP due to various etiologies mentioned above, as well as peripheral vascular remodeling leading to increase in cardiac afterload and resultant LV hypertrophy [24, 30].
Mechanical alterations in normal physiology also contribute, however, to a much lesser degree. Recurrent attempts at inspiration against a closed glottis during apnea results in disproportionate negative intrathoracic pressure. This in turn causes increased venous return to the right heart causing the interventricular septum to flatten resulting in decreased LV end diastolic volume [35]. Moreover, OSA is associated with increased arterial stiffness and heart remodeling, and the magnitude of arterial stiffness and the coexistence of OSA and treated hypertension are associated with additive effects on arterial stiffness as well as heart remodeling [36]. The increased episodes of negative intrathoracic pressure during apneic events also contribute to LVDD, increasing LV wall tension and afterload [37].
Diastolic Dysfunction and OHS
Cross-Sectional Associations in OHS
OHS is defined as daytime hypercapnia and hypoxemia (PaCO2 > 45 mmHg and PaO2 < 70 mmHg at sea level) in an obese patient (body mass index ≥ 30 kg/m2) with OSA in the absence of any other cause of hypoventilation. Data to support diastolic dysfunction in OHS is much less robust when compared with that of OSA. Observational retrospective as well as prospective studies suggest that diastolic dysfunction is common among patients with OHS and accounts for left heart pathology [9, 38•]. In a recent prospective observational study of 113 patients with OHS, prevalence of LVDD in OHS patients with mild to moderate OSA (defined as AHI < 30) was significant at 71%. This suggests that LVDD may be due to factors other than repeated apneic events. In this study, OHS patients with higher BMI and presence of hypertension were more likely to have LVDD [38•]. Moreover, in the largest RCT of patients (N = 221) undergoing PAP therapy for OHS, 55% of patients had PH and 51% had evidence of LV hypertrophy at baseline [39•].
Pathophysiologic Mechanisms
Given that increased BMI and hypertension are known risk factors for LVDD in OSA patients, the pathophysiologic mechanisms remain the same. These include metabolic derangements due to obesity, oxidative stress, and neurohormonal disturbances due to intermittent hypoxia and overstimulation of the sympathetic nervous system. All of which eventually lead to LVH, either directly via remodeling and increased LV mass, or indirectly via increased cardiac afterload.
However, the presence of hypercapnia in OHS has unique downstream effects on LV function. The changes in hemodynamics caused by hypercapnia affect myocardial oxygen consumption and supply. Hypercapnia and resultant respiratory acidosis dilate the coronary vessels, and unless the coronary perfusion pressure falls, it will actually increase coronary blood flow. However, hypercapnia also activates the sympathetic nervous system causing increase in cardiac oxygen consumption due to abnormal inotropic and chronotropic effects. The resultant shortening of diastolic filling time offsets any favorable effect of increased diastolic coronary perfusion pressures [40]. Additionally, coexisting hypoxia and obesity may compound the negative cardiac consequences. A rat study showed cardiac Fas receptor- and mitochondrial-dependent apoptotic pathways were more activated in obesity with coexistent nocturnal sustained hypoxia, which may represent one possible apoptotic mechanism for the development of HF in obesity with nocturnal sustained hypoxia [41].
Pulmonary Hypertension and OSA
Cross-Sectional Associations
The prevalence of sleep-disordered breathing in PH patients is higher than in the general population. The prevalence in the different studies has varied from 16 to 67% (Table 2). This is because the studies did not use a standard definition to define PH, included selected sleep clinic or PH clinic populations, did not exclude other respiratory and cardiac conditions that cause PH and/or used variable methods of estimating pulmonary pressures. After excluding relevant confounders, including underlying obstructive or restrictive lung disease or LV heart disease and vasodilator use, the prevalence of PH has varied from 17 to 42% [45, 46, 48,49,50, 52, 53] in selected obese sleep clinic populations with moderate-severe OSA (Table 2). In OSA, PH is mild with mean pulmonary artery pressure (mPpa) ranging from 21.9 to 40.3 mmHg. PH, after excluding underlying other lung or cardiac disease, seemed to be related to the severity of daytime PaO2 and/or duration of nocturnal hypoxia (Table 2). Hypercapnia may be a contributing factor too. The mPpa correlated with weight or body mass index (BMI) in a few studies [48, 50]. The severity of OSA (based on AHI) was not a significant determinant of mPpa in most studies [45, 47, 48]. The mPpa was also found to be state specific with higher values of mPpa in rapid-eye movement (REM) sleep than in non-REM sleep and was also higher in phasic REM compared with tonic REM sleep, independent of the degree of hypoxia [54].
Pathophysiologic Mechanisms (Fig. 1)
In addition to effects on cardiovascular effects, OSA also affects the pulmonary circulation. The pathophysiologic mechanisms that increase mPpa in OSA patients are multiple and in many cases additive. This is thought to be primarily due to downstream effects of intermittent apneas and hypoxia. The effects of sleep on alveolar ventilation pulmonary arterial pressure have been measured in both normal subjects and those with OSA [55]. The mPpa rises and alveolar ventilation decreases in sleep compared to wakefulness in normal subjects, however the changes in mPpa and alveolar ventilation did not change during the different stages of sleep [56]. Contrast this with OSA patients, in which mPpa rises and alveolar ventilation falls during sleep, but mPpa continues to rise and alveolar ventilation continues to fall in an incremental fashion with each sleep stage, peaking during REM sleep [56]. The direct effects of OSA on the change in mPpa and alveolar ventilation were further supported by the fact that normalization of alveolar ventilation and decrease in mPpa occurred in OSA patients after tracheostomy [57].
In this review, we differentiate between precapillary and postcapillary etiologies of PH and the multiple mechanisms that are involved.
Pre-capillary Mechanisms of PH in Patients with OSA
During apneic events, acute pulmonary vasoconstriction occurs as a response to alveolar hypoxia which in turn increases pre-capillary pulmonary artery (PA) pressure [58]. In addition, irreparable changes in the pulmonary vasculature are caused by chronic intermittent hypoxia due to pulmonary arterial smooth muscle hypertrophy and development of muscularized blood vessels [59, 60].
The direct association between hypoxia alone and development of structural remodeling of the pulmonary arteries leading to PH is evident in high altitude population studies. It has been observed that populations living at high altitude have chronic elevation of PA pressures, which is largely resistant to reversal after oxygen administration [61].
This chronic elevation of pulmonary arterial pressures is mainly due to remodeling of the pulmonary vasculature. Hyperplasia and hypertrophy of endothelial cells cause increase in thickness of the subendothelial region of larger, more proximal pulmonary arteries [62]. The activation and exact mechanism of proliferation of cells within the walls of pulmonary vessels has yet to be described. It has been suggested certain populations of cells in the medial layer with membrane-bound receptors are sensitive to hypoxic activation and engage in intracellular signaling pathways leading to proliferation [59].
In animal models, it has been shown that increases in collagen and elastin accumulation in proximal pulmonary arterial vessels in response to exposure to hypoxia further contribute to thickening of the medial and adventitial layers of the vessel wall [63, 64]. In vivo and in vitro studies have shown that hypoxia directly increases collagen synthesis via expression and production of transforming growth factor-β which regulates collagen synthesis in fibroblasts [64,65,66]. It was recently shown that rats with a dominant negative mutation in TGF-β receptor did not develop PH due to chronic hypoxia [67].
Remodeling of the proximal vessels and resultant stiffness of these vessels causes failure of the conductance vessels to store and deliver the stroke volume of the right ventricle and eventually loss of pulmonary flow during diastole [59].
In addition to remodeling of the pulmonary arterial vasculature, pulmonary arterial circulation is further impaired by dysfunction of a number of cellular mechanisms responsible for vasodilation in patients with OSA. Due to aforementioned remodeling of the vasculature and stiffness of vessels resulting in impaired pulmonary vascular flow, there is likely a drop in nitric oxide (NO) production in distal PA endothelial cells, which requires continual pulsatile flow for optimal production of NO [59, 68]. This causes impairment of vasodilation as NO is a known mediator of pulmonary vascular resistance [69]. Another cellular change in OSA patients is elevation of endothelin (ET)-1, a long-acting peptide synthesized in the endothelium that causes vasoconstriction [70]. It has been suggested that intermittent hypoxia increases production of reactive oxygen radicals, which then increase synthesis of ET-1 [71]. The downstream effects cause increased sensitivity of calcium response and increased reactivity of vasoconstrictors [72]. Mice models have suggested that ET-1 is also involved in vascular remodeling. The mechanism by which ET-1 causes early inflammatory remodeling is through hypoxia-inducible factor (HIF)-1 and subsequent activation of nuclear factor (NF)-κB causing increased intima-media thickness. This is supported by the fact that mice partially deficient for HIF-1 gene (HIF-1α+/−) did not demonstrate any increase in intima-media thickness when exposed to the same intermittent hypoxic conditions [73]. The magnitude of response to hypoxia among individuals with OSA may be variable. In patients with OSA, the mean increase of Ppa (pulmonary artery pressure) was 8 ± 1 mmHg to eucapnic hypoxia, and the response to hypercapnic hypoxia was higher at 10 ± 1 mmHg. Ppa response to hypoxia was augmented (> 10 mmHg) by hypercapnia in only four of 20 patients with OSA [53], suggesting inherent variability to hypoxic responsiveness, the underlying mechanisms of which remain unknown.
Post-capillary Mechanisms of PH in Patients with OSA
Pulmonary venous pressure or post-capillary pressures in patients with OSA are affected mostly by the mechanical effects of hemodynamic dysfunction within the cardio-pulmonary circulation. Patients with OSA have an abnormally high threshold of arousal during NREM sleep due to impairment of upper airway mechanoreceptors [74]. It has been shown that negative intrathoracic pressures as high as negative 40–80 cm H2O are required for arousal [75] as compared to normal control subjects who required much lower negative intrathoracic pressures ranging from negative 20–30 cm H2O [76]. This in turn leads to downstream effects causing an increase in post-capillary pulmonary pressures. As mentioned in mechanisms of diastolic dysfunction, increased venous return due to increased negative intrathoracic pressure shifts the interventricular septum to the left causing decrease of diastolic filling volume, which in turn causes an increase in post-capillary pulmonary pressures [35]. Additionally, the increase in negative intrathoracic pressure also increases the transmural pressure of all intra-thoracic structures. This includes not only the ventricles but also the pulmonary vascular beds, atria, and intrathoracic aorta [77]. This cumulative effect causes increase in LV afterload and decreased stroke volume which contributes further to pulmonary venous hypertension [78].
Central Sleep Apnea and Diastolic Dysfunction
The literature is sparse on interactions between central sleep apnea (CSA) and diastolic heart function. Central sleep apnea is the repetitive cessation of airflow in the absence of respiratory effort. Patients with HF and CSA tend to have an exaggerated respiratory response to carbon dioxide, associated with excess sympathetic nervous activity, resulting in inappropriate cyclic hyperventilation and central apneas [79]. While those with CSA do not experience marked episodes of negative intrathoracic pressure as seen in OSA, diurnal sympathetic activation is higher in those with HF and CSA than in matched controls with HF only [80]. Patients with CSA had more advanced left ventricular systolic (lower LVEF) and diastolic dysfunction (higher E/e’) and larger heart chambers [81]. Moreover, CSA with Cheyne-Stokes Respiration may contribute to increased pulmonary artery pressure and right chamber remodeling in HF, independently of the severity of LV systolic and diastolic dysfunction, likely via recurrent hypoxia/hypercapnia cycles and chemoreflex-mediated adrenergic discharge [82]. In one large prospective cohort study of patients receiving guideline-recommended treatment for HF (n = 525), episodes of central apnea were associated with neurohormonal activation, ventricular arrhythmic burden, and systolic and diastolic dysfunction [83]. Patients with severe central sleep apnea during nighttime, daytime, and throughout the 24 h were more frequently males, had a more severe diastolic dysfunction, showed higher neurohormonal activation and ventricular arrhythmic burden, and experienced a higher burden of desaturation [83]. In contrast, in patients without HF (n = 16), the central apnea length and ventilatory cycle length were shorter and majority (80%) did not have diastolic dysfunction [84] or PH. As already mentioned, LV diastolic dysfunction may result from overstimulation of the sympathetic nervous system [12, 24, 26, 27]. Similarly, the hypoxemia caused by CSA may induce activation of the sympathetic nervous system [80]. This hyper-adrenergic state is evidenced by studies showing increased urine and plasma metanephrine levels in patients with CSA [85]. This is further confirmed by studies which also show decrease in urine and plasma metanephrine levels when CSA is treated with nocturnal CPAP [85, 86].
Pulmonary Hypertension and OHS
It is estimated that the prevalence of PH is approximately 50% in patients with OHS in contrast to 20% in patients with OSA alone [87, 88]. In a prospective cross-sectional study, the patient parameters associated with presence of PH or increase in severity of PH were BMI, low DLCO, and lack of consistent noninvasive positive pressure ventilation (NIPPV) use [89].
The etiology of PH in OHS relates primarily due to precapillary PH [88]. This is likely secondary to the known effects of hypoxemia on the pulmonary vasculature including both direct vasoconstriction and vascular remodeling [58, 59, 68,69,70]. It has been shown, however, that PH in OHS patients substantially worsens during exercise and is related to a combination of pre- and postcapillary PH as observed by elevated pulmonary artery occlusion pressures during exercise when compared to rest [48]. The mechanism behind this observation is the diastolic dysfunction of the left atrium, which is commonly associated with obesity [90].
It likely that alveolar hypoventilation in combination with high prevalence of diastolic dysfunction in obese patients, account for the higher prevalence of PH in patients with OHS when compared to OSA. Whether or not hypercapnia has a direct role in the development of PH, whether pre or postcapillary, has yet to be established.
Implications of Therapy
Studies demonstrating reversibility of LV diastolic dysfunction or PH following adequate therapy of OSA provide supportive evidence of the link between these clinical conditions. However, the data are conflicting and only a few randomized clinical trials (RCTs) have studied the impact of OSA therapy on LV diastolic dysfunction or PH. The data on OHS is even more limited. The handful of RCTs and several small observational prospective and retrospective studies provide additional supportive evidence of the direction of associations between the disease entities.
Oxygen Therapy
PH
Hypoxia due to obstructive apneas, may lead to a steady increase in PAP, detectable both at the beginning and at the end of the episodes [91]. Supplemental oxygen to eliminate hypoxemia blunted pulmonary artery pressure (PAP) elevation in dog models of artificially induced recurrent obstructive apnea [92]. However, in humans, oxygen attenuated the falls and oscillations on oximetry but did not effectively reduce transmural PAP nor the amplitude of its variations induced by obstructive apneas [91].
There are no clinical studies of the impact of oxygen on diastolic dysfunction.
PAP Therapy
LV Diastolic Dysfunction
Given that HF concomitant with moderate-severe SDB is associated with increased mortality than in patients with HF without severe SDB [4, 93], it is undoubtedly crucial to treat the SDB prior to the development of overt HF. However, the impact of continuous positive airway pressure (CPAP) on LV diastolic function in patients with OSA is unclear. In a small study of newly diagnosed patients with moderate- severe OSA, there was improved LV systolic and diastolic function after 6 months of CPAP therapy [94]. In another study, patients with moderate or severe OSA (AHI ≥ 20), treatment with CPAP for 6 months was associated with improvement in diastolic function as well as in measures of LA function compared to sham CPAP [95]. Noninvasive ventilation for 6 months also improved measures of diastolic function [96]. Moreover, compared with sham, 3 months of CPAP was associated with improvements in e′ velocity and arterial stiffness [97]. A 5-year uncontrolled prospective observational trial of CPAP in nonhypertensive patients with OSA revealed a significant increase in the acceleration time(AT), Em/Am ratio and ejection time (AT: p = 0.04; Em/Am ratio p = 0.03 ET: p = 0.04) while a significant decrease was observed on deceleration time, isovolumetric relaxation time, and myocardial performance index in all subjects [98]. However, a large RCT trial of 171 patients with CAD found CPAP had no significant impact on diastolic dysfunction (enlarged left atrium, decreased mean é tissue velocity, or increased E/é filling index) in the intention-to-treat population, but on-treatment analysis revealed a significant increase in diastolic relaxation velocity in patients using CPAP for ≥ 4 h/night (OR 2.3, 95% confidence interval 1.0–4.9; p = 0.039) after adjustment for age, sex, body mass index, and left atrium diameter at baseline [99•].
OHS
There are very few studies evaluating whether PAP has an impact on diastolic function in patients with OHS. In one study, 6 weeks of PAP (CPAP or bilevel PAP) treatment reduced nocturnal beat-to-beat BP surges but not the daytime BP in 17 OHS patients with OSA, and this improvement in nocturnal BP regulation was greater in patients with higher PAP adherence [100]. There were no ECHO measures of diastolic heart function on this study.
In the largest multicenter randomized controlled trial of OHS (Pickwick project, n = 221) the effect of 2 months of NIPPV, CPAP or lifestyle modification (control) was studied in patients with severe OSA with baseline hypertension in 55% and LV hypertrophy in 51% patient. The authors found that treatment with NIPPV but not CPAP, lowered systolic pulmonary artery pressure (−3.4 mmHg, adjusted p = 0.025 vs control and P = 0.033 vs CPAP). The degree of improvement in systolic pulmonary artery pressure was greater in patients treated with NIPPV who had pulmonary hypertension at baseline (−6.4 mmHg) [39•]. NIPPV therapy also decreased LV hypertrophy with a significant reduction in LV mass index. Whether PAP therapy has an impact on HF and associated mortality in OHS populations is not known.
PH
Five years of treatment of severe OSA with CPAP (mean use 5.2 h/night) did not change daytime arterial oxygen tension or the pulmonary artery pressure [101]. The pulmonary artery wedge pressure increased significantly from 6 ± 2 to 9 ± 3 mmHg. However, this study may have been underpowered for patients with PH. Conversely, a meta-analysis of seven studies in patients with isolated OSA (n = 222, 77% men, age 52.5 years, AHI 58 events/h) and mPpa of 39.3 ± 6.3 mmHg, CPAP therapy for 3 to 70 months, was associated with a decrease in mPpa of 13.3 mmHg [102]. In one of the randomized crossover trials, CPAP therapy in comparison to sham CPAP for 4 months in patients with OSA and PH reduced pulmonary systolic pressure (28.9 ± 8.6 to 24.0 ± 5.8 mmHg, p < 0.0001). The reduction was greatest in patients with either PH or LVDD at baseline [103, 104]. Similarly, CPAP therapy for 4 months and 6 months decreased mPpa from 16.8 ± 1.2 to 13.9 ± 0.6 mmHg, p < 0.05, and from 25.6 ± 4.0 to 19.5 ± 1.5 mmHg, p < 0.001), respectively, with a reduction in the total pulmonary vascular resistance [105]. However, there are no randomized controlled studies of whether treatment of SDB in PH reduces mortality or hospitalizations.
Prognosis
LV diastolic dysfunction in OSA and OSA coexisting with PH increase the risk for HF, morbidity, and mortality [51, 106]. In a community sample, diastolic dysfunction was common, without recognized HF, with a 20.8%, 6.6%, and 0.7% of mild, moderate, and severe diastolic dysfunction, respectively, with normal EF [106]. Moreover, diastolic dysfunction was associated with marked increases in all-cause mortality [106]. In patients with PH and OSA, survival at 1, 4, and 8 years of follow-up period was 93%, 75%, and 43% compared to 100%, 90%, and 76% for patients without PH, respectively [51]. Patients with severe PH had more nocturnal desaturations, worse pulmonary hemodynamics, and greater mortality (37%) than the groups with mild or moderate PH (16%) or no PH (16%). Although the recent multicenter randomized controlled trials, SAVE and RICCADSA [107, 108], did not find a mortality benefit with CPAP therapy in patients with moderate-severe OSA and cardiovascular diseases, a firm conclusion cannot drawn on the effect of PAP on CV mortality because these studies were limited by inadequate adherence to PAP therapy. Subsequent subgroup analysis demonstrated that patients with CPAP use > 4 h per night did have reduced mortality risk [108]. For systolic HF patients with CSA, the CANPAP trial did not demonstrate an overall survival or hospitalization advantage with CPAP therapy [109], even though CPAP improved surrogate markers of cardiovascular outcome, including LV ejection fraction, 6-min walk test distance, and reduced plasma noradrenaline concentrations.
Conclusion
Moderate to severe OSA leads to serious adverse cardiovascular sequelae, including HF and PH via multiple pathways. The limitations of the enumerated studies were that most were of cross-sectional design, conducted in small clinic-based populations that did not adjust for relevant confounding factors. Overall, the data support an association between OSA, diastolic dysfunction, systemic hypertension and PH. However, there is limited evidence regarding the pathways that link OHS, diastolic function, and PH, and comprehensive mechanistic studies are required in OHS populations. Effect of CSA on these parameters needs to be delineated further. Given that SDB coexisting with PH and LVDD increase the risk for HF, morbidity, and mortality, future prospective therapeutic studies should target the putative pathophysiologic mechanisms to alleviate the serious negative cardiovascular consequences of SDB.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. Heart disease and stroke Statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–360.
Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933–44.
Javaheri S, Barbe F, Campos-Rodriguez F, Dempsey JA, Khayat R, Javaheri S, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69(7):841–58.
Khayat R, Jarjoura D, Porter K, Sow A, Wannemacher J, Dohar R, et al. Sleep disordered breathing and post-discharge mortality in patients with acute heart failure. Eur Heart J. 2015;36(23):1463–9.
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328(17):1230–5.
Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–14.
Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of Obesity Among Adults and Youth: United States. NCHS Data Brief. 2015-2016;2017(288):1–8.
Mokhlesi B. Obesity hypoventilation syndrome: a state-of-the-art review. Respir Care. 2010;55(10):1347–62 discussion 63-5.
Alawami M, Mustafa A, Whyte K, Alkhater M, Bhikoo Z, Pemberton J. Echocardiographic and electrocardiographic findings in patients with obesity hypoventilation syndrome. Intern Med J. 2015;45(1):68–73.
Niroumand M, Kuperstein R, Sasson Z, Hanly PJ. Impact of obstructive sleep apnea on left ventricular mass and diastolic function. Am J Respir Crit Care Med. 2001;163(7):1632–6.
Kraiczi H, Caidahl K, Samuelsson A, Peker Y, Hedner J. Impairment of vascular endothelial function and left ventricular filling : association with the severity of apnea-induced hypoxemia during sleep. Chest. 2001;119(4):1085–91.
Fung JWH, Li TST, Choy DKL, Yip GWK, Ko FWS, Sanderson JE, et al. Severe obstructive sleep apnea is associated with left ventricular diastolic dysfunction. Chest. 2002;121(2):422–9.
Arias MA, GarcíA-RíO F, Alonso-FernáNdez A, Mediano O, MartíNez I, Villamor J. Obstructive sleep apnea syndrome affects left ventricular diastolic function. Circulation. 2005;112(3):375–83.
Baguet JP, Barone-Rochette G, Levy P, Vautrin E, Pierre H, Ormezzano O, et al. Left ventricular diastolic dysfunction is linked to severity of obstructive sleep apnoea. Eur Respir J. 2010;36(6):1323–9.
Wachter R, Lüthje L, Klemmstein D, Lüers C, Stahrenberg R, Edelmann F, et al. Impact of obstructive sleep apnoea on diastolic function. Eur Respir J. 2013;41(2):376–83.
Usui Y, Takata Y, Inoue Y, Tomiyama H, Kurohane S, Hashimura Y, et al. Severe obstructive sleep apnea impairs left ventricular diastolic function in non-obese men. Sleep Med. 2013;14(2):155–9.
Chen YL, Su MC, Liu WH, Wang CC, Lin MC, Chen MC. Influence and predicting variables of obstructive sleep apnea on cardiac function and remodeling in patients without congestive heart failure. J Clin Sleep Med. 2014;10(1):57–64.
Bodez D, Lang S, Meuleman C, Boyer-Chatenet L, Nguyen XL, Soulat-Dufour L, et al. Left ventricular diastolic dysfunction in obstructive sleep apnoea syndrome by an echocardiographic standardized approach: an observational study. Arch Cardiovasc Dis. 2015;108(10):480–90.
Glantz H, Thunstrom E, Johansson MC, Wallentin Guron C, Uzel H, Ejdeback J, et al. Obstructive sleep apnea is independently associated with worse diastolic function in coronary artery disease. Sleep Med. 2015;16(1):160–7.
Fung ML, Tipoe GL, Leung PS. Mechanisms of maladaptive responses of peripheral chemoreceptors to intermittent hypoxia in sleep-disordered breathing. Sheng Li Xue Bao. 2014;66(1):23–9.
Araz O, Yilmazel Ucar E, Degirmenci H, Pulur D, Acemoglu H, Bayram E, et al. The correlation of ECHO findings of right cardiac pathologies with BNP, uric acid, and CRP in OSAS. Turk J Med Sci. 2014;44(5):832–8.
Guidry UC, Mendes LA, Evans JC, Levy D, O'Connor GT, Larson MG, et al. Echocardiographic features of the right heart in sleep-disordered breathing. Am J Respir Crit Care Med. 2001;164(6):933–8.
Parker JD, Brooks D, Kozar LF, Render-Teixeira CL, Horner RL, Douglas Bradley T, et al. Acute and chronic effects of airway obstruction on canine left ventricular performance. Am J Respir Crit Care Med. 1999;160(6):1888–96.
Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103(6):1763–8.
Narkiewicz K, Somers VK. The sympathetic nervous system and obstructive sleep apnea: implications for hypertension. J Hypertens. 1997;15(12 Pt 2):1613–9.
Shell B, Faulk K, Cunningham JT. Neural Control of Blood Pressure in Chronic Intermittent Hypoxia. 2016;18(3).
Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96(4):1897–904.
Schulz R, Hummel C, Heinemann S, Seeger W, Grimminger F. Serum levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea and severe nighttime hypoxia. Am J Respir Crit Care Med. 2002;165(1):67–70.
Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, et al. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Am J Respir Crit Care Med. 2000;162(2):566–70.
Nieto FJ, Herrington DM, Redline S, Benjamin EJ, Robbins JA. Sleep apnea and markers of vascular endothelial function in a large community sample of older adults. Am J Respir Crit Care Med. 2004;169(3):354–60.
Fung ML. The role of local renin-angiotensin system in arterial chemoreceptors in sleep-breathing disorders. Front Physiol 2014;5.
Newman AB. Progression and regression of sleep-disordered breathing with changes in weight. Arch Intern Med. 2005;165(20):2408–13.
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251–9.
Rayner JJ, Banerjee R, Holloway CJ, Lewis AJM, Peterzan MA, Francis JM, et al. The relative contribution of metabolic and structural abnormalities to diastolic dysfunction in obesity. Int J Obes. 2018;42(3):441–7.
Shiomi T, Guilleminault C, Stoohs R, Schnittger I. Leftward shift of the interventricular septum and pulsus paradoxus in obstructive sleep apnea syndrome. Chest. 1991;100(4):894–902.
Drager LF, Bortolotto LA, Figueiredo AC, Silva BC, Krieger EM, Lorenzi-Filho G. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest. 2007;131(5):1379–86.
Virolainen J, Ventila M, Turto H, Kupari M. Effect of negative intrathoracic pressure on left ventricular pressure dynamics and relaxation. J Appl Physiol (1985). 1995;79(2):455–60.
• Al Otair HA, Elshaer F, Elgishy A, Nashwan SZ, Almeneessier AS, Olaish AH, et al. Left ventricular diastolic dysfunction in patients with obesity hypoventilation syndrome. Journal Thorac Dis. 2018;10(10):5747–54 Study showed that diastolic left ventricular dysfunction was prevalent among OHS patients even in the absence of severe OSA.
• Corral J, Mogollon MV, Sanchez-Quiroga MA, Gomez de Terreros J, Romero A, Caballero C, et al. Echocardiographic changes with non-invasive ventilation and CPAP in obesity hypoventilation syndrome. Thorax. 2018;73(4):361–8 Study presents the effect of PAP therapy on echocardiographic findings in OHS.
Rasmussen JP. Cardiac Function and Hypercarbia. Arch Surg. 1978;113(10):1196.
Lee SD, Kuo WW, Bau DT, Ko FY, Wu FL, Kuo CH, et al. The coexistence of nocturnal sustained hypoxia and obesity additively increases cardiac apoptosis. J Appl Physiol (1985). 2008;104(4):1144–53.
Tilkian AG. Hemodynamics in sleep-induced apnea. Ann Intern Med. 1976;85(6):714–9.
Podszus T, Bauer W, Mayer J, Penzel T, Peter JH, von Wichert P. Sleep apnea and pulmonary hypertension. Klin Wochenschr. 1986;64(3):131–4.
Fletcher EC, Schaaf JW, Miller J, Fletcher JG. Long-term cardiopulmonary sequelae in patients with sleep apnea and chronic lung disease. Am Rev Respir Dis. 1987;135(3):525–33.
Weitzenblum E, Krieger J, Apprill M, Vallee E, Ehrhart M, Ratomaharo J, et al. Daytime pulmonary hypertension in patients with obstructive sleep apnea syndrome. Am Rev Respir Dis. 1988;138(2):345–9.
Krieger J, Sforza E, Apprill M, Lantpert E, Weitzenblum E, Ratomaharv J. Pulmonary hypertension, hypoxemia, and hypercapnia in obstructive sleep apnea patients. Chest. 1989;96(4):729–37.
Laks L, Lehrhaft B, Grunstein RR, Sullivan CE. Pulmonary hypertension in obstructive sleep apnoea. Eur Respir J. 1995;8(4):537–41.
Chaouat A, Weitzenblum E, Krieger J, Oswald M, Kessler R. Pulmonary hemodynamics in the obstructive sleep apnea syndrome: results in 220 consecutive patients. Chest. 1996;109(2):380–6.
Sajkov D, Cowie RJ, Thornton AT, Espinoza HA, McEvoy RD. Pulmonary hypertension and hypoxemia in obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 1994;149(2):416–22.
Bady E, Achkar A, Pascal S, Orvoen-Frija E, Laaban JP. Pulmonary arterial hypertension in patients with sleep apnoea syndrome. Thorax. 2000;55(11):934–9.
Minai OA, Ricaurte B, Kaw R, Hammel J, Mansour M, McCarthy K, et al. Frequency and impact of pulmonary hypertension in patients with obstructive sleep apnea syndrome. Am J Cardiol. 2009;104(9):1300–6.
Sanner BM, Doberauer C, Konermann M, Sturm A, Zidek W. Pulmonary hypertension in patients with obstructive sleep apnea syndrome. Arch Intern Med. 1997;157(21):2483–7.
Laks L, Lehrhaft B, Grunstein RR, Sullivan CE. Pulmonary artery pressure response to hypoxia in sleep apnea. Am J Respir Crit Care Med. 1997;155(1):193–8.
Niijima M, Kimura H, Edo H, Shinozaki T, Kang J, Masuyama S, et al. Manifestation of pulmonary hypertension during REM sleep in obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 1999;159(6):1766–72.
Coccagna G, Lugaresi E. Haemodynamics during sleep: old results and new perspectives. J Sleep Res. 1995;4:2–7.
Coccagna G, Mantovani M, Brignani F, Parchi C, Lugaresi E. Continuous recording of the pulmonary and systemic arterial pressure during sleep in syndromes of hypersomnia with periodic breathing. Bull Physiopathol Respir (Nancy). 1972;8(5):1159–72.
Coccagna G, Mantovani M, Brignani F, Parchi C, Lugaresi E. Tracheostomy in hypersomnia with periodic breathing. Bull Physiopathol Respir (Nancy). 1972;8(5):1217–27.
Schneider H, Schaub CD, Chen CA, Andreoni KA, Schwartz AR, Smith PL, et al. Neural and local effects of hypoxia on cardiovascular responses to obstructive apnea. J Appl Physiol (1985). 2000;88(3):1093–102.
Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling. Circ Res. 2006;99(7):675–91.
Kay JM, Suyama KL, Keane PM. Effect of intermittent normoxia on muscularization of pulmonary arterioles induced by chronic hypoxia in rats. Am Rev Respir Dis. 1981;123(4 Pt 1):454–8.
Arias-Stella J, SaldañA M. The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation. 1963;28(5):915–25.
Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med (Berl). 2013;91(3):297–309.
Kobs RW, Chesler NC. The mechanobiology of pulmonary vascular remodeling in the congenital absence of eNOS. Biomech Model Mechanobiol. 2006;5(4):217–25.
Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodeling in mouse pulmonary arteries with hypoxia-induced hypertension. Am J Phys Heart Circ Phys. 2005;288(3):H1209–H17.
Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, et al. Transforming growth factor-signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L86–95.
Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol. 1997;59(1):89–144.
Chen Y-F, Feng J-A, Li P, Xing D, Zhang Y, Serra R, et al. Dominant negative mutation of the TGF-β receptor blocks hypoxia-induced pulmonary vascular remodeling. J Appl Physiol. 2006;100(2):564–71.
Weiss JW, Liu Y, Li X, Ji E-S. Nitric oxide and obstructive sleep apnea. Respir Physiol Neurobiol. 2012;184(2):192–6.
Cooper CJ, Landzberg MJ, Anderson TJ, Charbonneau F, Creager MA, Ganz P, et al. Role of nitric oxide in the local regulation of pulmonary vascular resistance in humans. Circulation. 1996;93(2):266–71.
Phillips BG, Narkiewicz K, Pesek CA, Haynes WG, Dyken ME, Somers VK. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J Hypertens. 1999;17(1):61–6.
Friedman JK, Nitta CH, Henderson KM, Codianni SJ, Sanchez L, Ramiro-Diaz JM, et al. Intermittent hypoxia-induced increases in reactive oxygen species activate NFATc3 increasing endothelin-1 vasoconstrictor reactivity. Vascul Pharmacol. 2014;60(1):17–24.
Kanagy NL. Vascular effects of intermittent hypoxia. ILAR J. 2009;50(3):282–8.
Gras E, Belaidi E, Briançon-Marjollet A, Pépin J-L, Arnaud C, Godin-Ribuot D. Endothelin-1 mediates intermittent hypoxia-induced inflammatory vascular remodeling through HIF-1 activation. J Appl Physiol. 2016;120(4):437–43.
Orr RS, Jordan AS, Catcheside P, Saunders NA, McEvoy RD. Sustained isocapnic hypoxia suppresses the perception of the magnitude of inspiratory resistive loads. J Appl Physiol (1985). 2000;89(1):47–55.
Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med. 2004;169(5):623–33.
Issa FG, Sullivan CE. Arousal and breathing responses to airway occlusion in healthy sleeping adults. J Appl Physiol. 1983;55(4):1113–9.
Javaheri S, Barbe F, Campos-Rodriguez F, Dempsey JA, Khayat R, Javaheri S, et al. Sleep Apnea. J Am Coll Cardiol. 2017;69(7):841–58.
Buda AJ, Pinsky MR, Ingels NB Jr, Daughters GT 2nd, Stinson EB, Alderman EL. Effect of intrathoracic pressure on left ventricular performance. N Engl J Med. 1979;301(9):453–9.
Javaheri S. A mechanism of central sleep apnea in patients with heart failure. N Engl J Med. 1999;341(13):949–54.
Spaak J, Egri ZJ, Kubo T, Yu E, Ando S, Kaneko Y, et al. Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension. 2005;46(6):1327–32.
Kazimierczak A, Krzesiński P, Gielerak G, Uziębło-Życzkowska B, Smurzyński P, Ryczek R, et al. Association of central sleep apnea with impaired heart structure and cardiovascular hemodynamics in patients with chronic heart failure. Med Sci Monit. 2016;22:2989–98.
Giannoni A, Raglianti V, Mirizzi G, Taddei C, Del Franco A, Iudice G, et al. Influence of central apneas and chemoreflex activation on pulmonary artery pressure in chronic heart failure. Int J Cardiol. 2016;202:200–6.
Emdin M, Mirizzi G, Giannoni A, Poletti R, Iudice G, Bramanti F, et al. Prognostic significance of central apneas throughout a 24-hour period in patients with heart failure. J Am Coll Cardiol. 2017;70(11):1351–64.
Solin P, Jackson DM, Roebuck T, Naughton MT. Cardiac diastolic function and hypercapnic ventilatory responses in central sleep apnoea. Eur Respir J. 2002;20(3):717–23.
Naughton MT, Benard DC, Liu PP, Rutherford R, Rankin F, Bradley TD. Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1995;152(2):473–9.
Naughton MT, Benard DC, Rutherford R, Bradley TD. Effect of continuous positive airway pressure on central sleep apnea and nocturnal PCO2 in heart failure. Am J Respir Crit Care Med. 1994;150(6 Pt 1):1598–604.
Atwood CW, McCrory D, Garcia JGN, Abman SH, Ahearn GS. Pulmonary artery hypertension and sleep-disordered breathing. Chest. 2004;126(1):72S–7S.
Kessler R, Chaouat A, Weitzenblum E, Oswald M, Ehrhart M, Apprill M, et al. Pulmonary hypertension in the obstructive sleep apnoea syndrome: prevalence, causes and therapeutic consequences. Eur Respir J. 1996;9(4):787–94.
Kauppert CA, Dvorak I, Kollert F, Heinemann F, Jörres RA, Pfeifer M, et al. Pulmonary hypertension in obesity-hypoventilation syndrome. Respir Med. 2013;107(12):2061–70.
Rochester DF, Enson Y. Current concepts in the pathogenesis of the obesity-hypoventilation syndrome. Mechanical and circulatory factors. Am J Med. 1974;57(3):402–20.
Marrone O, Bellia V, Pieri D, Salvaggio A, Bonsignore G. Acute effects of oxygen administration on transmural pulmonary artery pressure in obstructive sleep apnea. Chest. 1992;101(4):1023–7.
Iwase N, Kikuchi Y, Hida W, Miki H, Taguchi O, Satoh M, et al. Effects of repetitive airway obstruction on O2 saturation and systemic and pulmonary arterial pressure in anesthetized dogs. Am Rev Respir Dis. 1992;146(6):1402–10.
Jilek C, Krenn M, Sebah D, Obermeier R, Braune A, Kehl V, et al. Prognostic impact of sleep disordered breathing and its treatment in heart failure: an observational study. Eur J Heart Fail. 2011;13(1):68–75.
Akar Bayram N, Ciftci B, Durmaz T, Keles T, Yeter E, Akcay M, et al. Effects of continuous positive airway pressure therapy on left ventricular function assessed by tissue Doppler imaging in patients with obstructive sleep apnoea syndrome. Eur J Echocardiogr. 2009;10(3):376–82.
Oliveira W, Campos O, Cintra F, Matos L, Vieira ML, Rollim B, et al. Impact of continuous positive airway pressure treatment on left atrial volume and function in patients with obstructive sleep apnoea assessed by real-time three-dimensional echocardiography. Heart. 2009;95(22):1872–8.
D'Andrea A, Martone F, Liccardo B, Mazza M, Annunziata A, Di Palma E, et al. Acute and chronic effects of noninvasive ventilation on left and right myocardial function in patients with obstructive sleep apnea syndrome: a speckle tracking echocardiographic study. Echocardiography. 2016;33(8):1144–55.
Shim CY, Kim D, Park S, Lee CJ, Cho HJ, Ha JW, et al. Effects of continuous positive airway pressure therapy on left ventricular diastolic function: a randomised, sham-controlled clinical trial. Eur Respir J. 2018;51(2).
Bilge AR, Yavuz V, Cetin N, Dalgic O, Kum G, Yilmaz H, et al. The effect of long-term continuous positive airway pressure treatment on systolic and diastolic function in patients with obstructive sleep apnoea syndrome: a five year observational study. Anadolu Kardiyol Derg. 2014;14(3):265–71.
• Glantz H, Johansson MC, Thunstrom E, Guron CW, Uzel H, Saygin M, et al. Effect of CPAP on diastolic function in coronary artery disease patients with nonsleepy obstructive sleep apnea: a randomized controlled trial. Int J Cardiol. 2017;241:12–8 One of the few studies that described the effects of PAP therapy on LV diastolic function in OSA patients.
Carter JR, Fonkoue IT, Grimaldi D, Emami L, Gozal D, Sullivan CE, et al. Positive airway pressure improves nocturnal beat-to-beat blood pressure surges in obesity hypoventilation syndrome with obstructive sleep apnea. Am J Phys Regul Integr Comp Phys. 2016;310(7):R602–R11.
Chaouat A, Weitzenblum E, Kessler R, Oswald M, Sforza E, Liegeon MN, et al. Five-year effects of nasal continuous positive airway pressure in obstructive sleep apnoea syndrome. Eur Respir J. 1997;10(11):2578–82.
Imran TF, Ghazipura M, Liu S, Hossain T, Ashtyani H, Kim B, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev. 2016;21(5):591–8.
Arias MA, García-Río F, Alonso-Fernández A, Martínez I, Villamor J. Pulmonary hypertension in obstructive sleep apnoea: effects of continuous positive airway pressure. Eur Heart J. 2006;27(9):1106–13.
Sajkov D, Wang T, Saunders NA, Bune AJ, Douglas MR. Continuous positive airway pressure treatment improves pulmonary hemodynamics in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;165(2):152–8.
Alchanatis M, Tourkohoriti G, Kakouros S, Kosmas E, Podaras S, Jordanoglou JB. Daytime pulmonary hypertension in patients with obstructive sleep apnea: the effect of continuous positive airway pressure on pulmonary hemodynamics. Respiration. 2001;68(6):566–72.
Redfield MM, Jacobsen SJ, Burnett JJC, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community. JAMA. 2003;289(2):194–202.
McEvoy RD, Antic NA, Heeley E, Luo Y, Ou Q, Zhang X, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375(10):919–31.
Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunstrom E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med. 2016;194(5):613–20.
Bradley TD, Logan AG, Kimoff RJ, Sériès F, Morrison D, Ferguson K, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2005;353(19):2025–33.
Dumitrascu R, Tiede H, Eckermann J, Mayer K, Reichenberger F, Ghofrani HA, et al. Sleep apnea in precapillary pulmonary hypertension. Sleep Med. 2013;14(3):247–51.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Chowdhuri, Dr. Venkat, and Dr. Abbas each declare no conflicts of interest.
Human and Animal Rights Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Heart Disease and Sleep Disturbances
Rights and permissions
About this article

Cite this article
Venkat, D., Abbas, H. & Chowdhuri, S. Sleep-Disordered Breathing and Diastolic Heart Disease. Curr Sleep Medicine Rep 5, 243–254 (2019). https://doi.org/10.1007/s40675-019-00160-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40675-019-00160-z