
Abstract
Background
Vitamin D deficiency is associated with increased risks of mortality in people with chronic kidney disease. The benefits and harm of vitamin D supplementation on cardiovascular outcomes and mortality are unknown. We aimed to assess the effectiveness of calcifediol in reducing mortality in patients with vitamin D insufficiency on hemodialysis compared to no additional therapy.
Methods
A phase III, multicenter, randomized, open-label trial was conducted including 284 adults with vitamin D insufficiency undergoing hemodialysis who were randomly assigned to receive oral calcifediol or standard care for 24 months.
Results
Two hundred eighty-four participants were enrolled (143 assigned to the calcifediol group and 141 to the no additional therapy group). The primary outcome (mortality) occurred in 34 and 31 participants in the calcifediol and control group, respectively [hazard ratio (HR) 1.03; 95% confidence interval (CI) 0.63–1.67]. Calcifediol had no detectable effects on cardiovascular death (HR 1.06; 95% CI 0.41–2.74), non-cardiovascular death (HR 1.13; 95% CI 0.62–2.04), nonfatal myocardial infarction (HR 0.20; 95% CI 0.02–1.67) or nonfatal stroke (HR could not be estimated). The incidence of hypercalcemia and hyperphosphatemia was similar between groups. None of the participants underwent parathyroidectomy.
Conclusions
In adults treated with hemodialysis and who had vitamin D insufficiency, calcifediol supplementation for 24 months had inconclusive effects on mortality and cardiovascular outcomes.
Trial registration number
NCT01457001
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Circulating levels of 25-hydroxy vitamin D (calcifediol), a pre-hormone produced in the liver from vitamin D3 (cholecalciferol) and then converted within kidney tissue into active vitamin D (1,25 dihydroxy vitamin D or calcitriol), are deficient (< 10 ng/ml) in over 80% of patients with severe chronic kidney disease [1]. Vitamin D deficiency is associated with increased risks of premature mortality in people with chronic kidney disease, although it is not clear whether vitamin D insufficiency has a causative role in cardiovascular events [2]. Kidney failure leads to impaired enzymatic conversion of calcifediol into calcitriol. Because of this, trials evaluating vitamin D therapy have focused on the efficacy of calcitriol and vitamin D analogs to treat the clinical consequences of dysregulated vitamin D metabolism in chronic kidney disease. To date, these agents have not been conclusively shown to prevent mortality and cardiovascular complications, while incurring hypercalcemia and accelerated cardiovascular calcification [3].
Although the kidney is the primary site for vitamin D activation, the enzyme is present also in extra renal sites including the parathyroid gland [4]. Thus, despite the loss of renal function with progressive chronic kidney disease, supplementation with nutritional vitamin D (such as cholecalciferol and calcifediol) has been shown to increase calcitriol levels without increasing the risk of hypercalcemia or hyperphosphatemia [5, 6]. Cholecalciferol and calcifediol are also associated with favorable changes in intermediary pathways related to left ventricular hypertrophy, microalbuminuria, insulin-resistance, vascular function, calcification and injury [7, 8]. However, evidence of any beneficial impact of supplementation on cardiovascular and mortality endpoints and potential treatment harm in patients with chronic kidney disease requires clarification [5, 9, 10].
This randomized controlled trial was designed to test the hypothesis that calcifediol supplementation reduces cardiovascular events in adults with end-stage kidney disease treated with hemodialysis and 25-hydroxy vitamin D insufficiency.
Methods
Study design
This investigator-initiated, phase III, multicenter, randomized, open-label trial was conducted by members of the Italian Society of Nephrology. The trial protocol is available in Item S1. The trial was registered prior to enrollment at http://www.clinicaltrials.gov, registration number NCT01457001.
Setting and participants
Adults who were 18 years of age or older, on long term hemodialysis for 90 days or more, with serum parathyroid hormone (PTH) levels 2- to 9-fold above the upper limit of normal [11] and vitamin D insufficiency (circulating levels of 25-hydroxy vitamin D less than 30 ng/ml, including patients with vitamin D deficiency-levels < 15 ng/ml) were eligible to participate in this trial. Participants with secondary hyperparathyroidism under treatment and therefore under control were eligible. Key exclusion criteria were prior kidney transplant or peritoneal dialysis within the previous 3 years, or pregnancy or lactating.
Randomization and masking
Eligible patients were randomly assigned to treatment with calcifediol or standard care without calcifediol in a 1:1 ratio. The sequence generation for randomization was performed using an electronically generated list created by an independent statistician. Randomization was stratified by center and in randomly permuted blocks. Randomization was performed through telephone contact with staff at the study coordination center after verification of the inclusion and exclusion criteria. Participants and trial investigators, including physicians and study nurses, were not blinded to treatment assignment.
Intervention and concomitant therapy
Participants assigned to the calcifediol group were prescribed 40 mcg administered by dialysis staff at the end of each dialysis session three times per week. Calcifediol supplement was discontinued by the treating physician if stable hypercalcemia occurred (persistent serum calcium ≥ 11.5 mg/dl despite calcium salt withdrawal, dietary modification, calcium reduction in dialysate and calcimimetic administration), or if 25-OH-vitD serum levels rose above 100 ng/ml or serious adverse events occurred that were considered attributable to the assigned treatment. Participants could resume treatment after the serum calcium or 25-OH-vitD levels dropped again. Those who discontinued therapy, as well as patients undergoing parathyroidectomy, renal transplantation or transfer to peritoneal dialysis during the study follow-up (for whom treatment with 25-OH-vitD became optional and entrusted to the clinical context and the policy of the study site) were asked to continue with trial assessments and outcome ascertainment. During the follow-up period, participants were assessed by their treating team every 3 months after randomization unless they died, withdrew consent or were not contactable for follow up. Participants in both treatment groups could be administered active vitamin D (paricalcitol or calcitriol) and/or a calcimimetic agent by their treating physician in order to obtain the parathyroid hormone target level recommended by the Kidney Disease Improving Global Outcomes (KDIGO) clinical practice guidelines. Phosphate binders could be administered to achieve guideline recommended levels of serum calcium and phosphate [12]. Dialysate calcium concentrations were maintained at 1.25–1.50 mmol/l throughout the intervention period.
Outcomes
The primary outcome was a composite of nonfatal myocardial infarction, nonfatal stroke, and death from any cause excluding trauma. The secondary outcomes were the individual components of the primary composite outcome, cardiovascular and non-cardiovascular mortality, fatal myocardial infarction, fatal stroke, normalization of circulating levels of calcifediol (at least one measurement > 30 ng/ml), circulating calcifediol > 100 ng/ml, dose reduction of active vitamin D, calcimimetic and phosphate binder therapy, and parathyroidectomy. Safety outcomes included hypercalcemia (serum calcium > 10.5 mg/dl), hyperphosphatemia (serum phosphate > 5.5 mg/dl), and adverse events.
Ethics and oversight
The study received Institutional Review Board approval from the Ethics Committee of the “Azienda Ospedaliera G. Rummo” of Benevento (deliberation n. 294, 13/04/2012) and was monitored by an external data and safety monitoring board that regularly reviewed safety parameters and study conduct. The study was conducted in compliance with the principles of the Declaration of Helsinki (1964) and in accordance with Good Clinical Practice Guidelines. All participants gave written informed consent prior to inclusion into the study.
Statistical analysis
The analyses were based on the intention-to-treat principle. All participants who were randomly assigned to treatment were included in analyses according to their assigned treatment group. The trial was designed to enroll 524 participants with 262 assigned to each treatment group to provide 80% power to detect an effect size of 0.66 for the primary composite outcome. The power calculation assumed a proportion surviving of 0.54 in the control group at three years (based an annual mortality of 15%) [13] and a withdrawal of 10% and two-sided α = 0.05.
The study was prematurely discontinued after inclusion of 284 participants due to the bankruptcy of the clinical research organization Consorzio Mario Negri Sud which occurred in May 2015. Participant recruitment was stopped and follow-up of participants was completed through December 2015. The Clinical Research Organisation (CRO) role was reassigned to the Istituto di Ricerche Farmacologiche Mario Negri to complete the study closure. Clinical centers were requested to provide all follow-up data. A revised power calculation indicated that the power of the study with 284 participants for the composite endpoint with 24 months of follow-up provided 28% power to detect a risk reduction of 34% in the primary outcome between the study groups.
The composite outcome and its individual components were analyzed using a time-to-event approach. Time-to-event data for each group were compared using the Cox proportional hazards model and expressed as hazard ratio (HR) with 95% confidence intervals (CIs). The proportions of the following secondary outcomes were analyzed by means of a chi-square test or Fisher's Exact test, as appropriate: hypercalcemia (serum calcium levels > 10.5 mg/dl), hyperphosphatemia (phosphate level > 5.5 mg/dl), normalization of circulating levels of 25-OH-vitD (> 30 ng/ml), increase in circulating levels of 25-OH-vitD above 100 ng/ml, down titration of therapy with calcitriol or paricalcitol, calcimimetic or phosphate binder therapy, parathyroidectomy and adverse events. Incidence rate ratio for episodes of circulating levels of calcifediol > 30 ng/ml, hypercalcemia and hyperphosphatemia were obtained using ‘stptime’ command in STATA version 15.
Pre-specified subgroup analyses of the primary outcome were conducted using the same methods. The following baseline characteristics formed pre-specified subgroups: age (above or below 65 years of age), prior calcifediol treatment, and concurrent hemodialysis treatment type (hemodialysis or hemodiafiltration).
Results
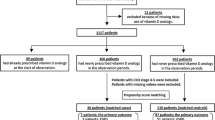
Between 8th November, 2012 and 11th November, 2014, 284 patients in 28 dialysis centers around Italy (Item S2) were randomly assigned to calcifediol (n = 143) or standard care (n = 141) (Fig. 1) and were assessed for the primary outcome.

CONSORT (Consolidated Standards of Reporting Trials) flow diagram of the trial. Adverse events were: in the calcifediol group, central vascular catheter infection (n = 1), sepsis (n = 2), pulmonary infection (n = 1), erythroderma (n = 1), pneumonia (n = 1), hemorrhage from epigastric artery (n = 1), cognitive impairment (n = 1), anemia (n = 1); in the control group, cachexia (n = 3), phlegmon (n = 1), respiratory insufficiency (n = 1), gallstones (n = 1), septic shock (n = 1), percutaneous transluminal coronary angioplasty (n = 1)
Baseline characteristics were similar between treatment groups except for sex (Table 1). There was a higher proportion of men in the calcifediol group than in the control group (65% vs 55%). The mean age was 66.1 ± 12.9 years. Diabetes was the cause of kidney disease in 66 (23%) participants. The mean serum calcifediol level was 12.3 ± 6.7 ng/ml. One hundred ninety-six (69%) participants showed vitamin D deficiency (< 15 ng/ml). Thirty-nine (14%) participants had circulating levels of calcifediol < 5 ng/ml, without showing any concomitant clinical patterns or fractures, and the median serum parathyroid hormone level was 241 (interquartile range [IQR] 167–327) pg/ml. Most patients received hemodialysis with a dialysate calcium concentration of 1.5 mmol/l. At baseline, 170 (60%) participants were prescribed concomitant calcitriol or paricalcitol, 55 (19%) were prescribed cinacalcet and 225 (79%) were prescribed a phosphate binder.
Primary composite outcome
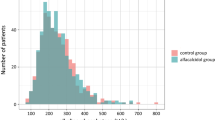
During a median follow-up of 24 months (IQR 12–30 months), 34 participants in the calcifediol group and 31 participants in the control group experienced the primary composite endpoint of nonfatal myocardial infarction, nonfatal stroke, and death from any cause excluding trauma [hazard ratio (HR) 1.03; 95% confidence interval (CI) 0.63–1.67] (Table 2 and Fig. 2). In subgroup analysis, there was no evidence of different intervention effects on the primary composite outcome based on age, prior treatment with calcifediol, or modality of hemodialysis (Table S1).

Kaplan–Meier estimates of primary composite outcome according to treatment allocation
Secondary outcomes
There was no difference in the risks of cardiovascular mortality (HR 1.06; 95% CI 0.41–2.74), non-cardiovascular mortality (HR 1.13; 95% CI 0.62–2.04), nonfatal myocardial infarction (HR 0.20; 95% CI 0.02–1.67) or nonfatal stroke (could not be estimated) (Table 2). A significantly greater proportion of patients in the calcifediol group (N = 52, 36.4%) achieved normalization of serum 25-hydroxy vitamin D compared with the control group (N = 15, 10.6%): incidence rates were 35.1 (95% CI 26.7–46.1) per 100 patient-years in the calcifediol group and 8.9 (95% CI 5.4–14.8) per 100 patient-years in the control group (incidence rate ratio: 3.9, 95% CI 2.2–7.5, p < 0.0001). Two and one patients alone in the control and calcifediol group, respectively, achieved vitamin D sufficiency (serum 25-hydroxy vitamin D > 100 ng/ml). During treatment, the dose of activated vitamin D was reduced in 126 (74%) participants, 28 (51%) had dose reductions in cinacalcet therapy, and 33 (15%) received dose reductions in their phosphate binder therapy. Rates of reduced doses in concomitant medications were statistically similar between groups (Table S2).
Safety outcomes
Serious adverse events occurred at a similar frequency in the treatment groups, 45 (32%) participants in the calcifediol group compared to 39 (27%) participants in the control group (p = 0.48) (Table 3). Two participants in the calcifediol and eight participants in the control group experienced hypercalcemia requiring withdrawal of therapy. Hyperphosphatemia (> 5.5 mg/ml) occurred in 28 participants in the calcifediol group and in 32 participants in the control group. The risk of other adverse events was similar between treatment groups (Table S3). Similar calcium, phosphate and parathyroid hormone levels between groups were reported during the study period (Table S4). No difference was reported between treatment groups.
Discussion
In adults treated with long-term hemodialysis who had circulating 25-hydroxy vitamin D insufficiency, supplementation with calcifediol did not prevent mortality or cardiovascular events compared with standard care. Restoration of serum total 25-hydtoxy vitamin D levels to > 30 mg/ml was higher in the supplementation group compared with standard care, although it was low overall (36% versus 11%). There did not appear to be different rates of adverse events including hypercalcemia or hyperphosphatemia in patients receiving supplementation. These findings support existing evidence that calcifediol therapy may restore insufficient circulating levels of 25-hydroxy vitamin D without incurring adverse effects on serum calcium and phosphate levels [14], although there is little evidence that calcifediol treatment prevents mortality or cardiovascular outcomes in this population.
The inconclusive effects of calcifediol supplementation on cardiovascular events and all-cause mortality is in contrast with the available observational data and has a number of possible explanations. First, vitamin D normalization was achieved only in a small portion of patients (36%). This could be due to limited enzymatic conversion of calcifediol to calcitriol relying mostly on extra renal tissues, and it might prevent the detection of any beneficial effect with calcifediol. Second, vitamin D supplementation may reduce blood pressure, proteinuria, cardiac myocyte hypertrophy and vascular calcification [15, 16]. However, cardiovascular pathobiology for hemodialysis is very complex and involves multiple pathways including oxidative stress, endothelial dysfunction, malnutrition and inflammation [17]. Therefore, the physiological changes associated with vitamin D supplementation may not suffice to reduce mortality due to competing biological mechanisms. Third, adverse vascular processes leading to cardiovascular complications in patients with advanced kidney disease may be so advanced and thus unmodifiable by vitamin D supplementation. This phenomenon has been demonstrated in relation to other cardiovascular interventions (such as antihypertensive drugs or statins) of proven benefit for other high cardiovascular risk populations [18, 19]. Fourth, participants assigned to the calcifediol group were treated with adequate doses of Vitamin D Receptor Activators (either paracalcitriol or calcitriol), which may have higher Vitamin D Receptor affinity compared to calcifediol thereby limiting the evaluation of the effects of the intervention on cardiovascular outcomes. Fifth, the statistical power and duration of the present study might have been insufficient to observe any survival benefit in hemodialysis patients receiving vitamin D compared with placebo. Finally, the positive findings of the observational study could be biased by residual confounding, and the true link between vitamin D insufficiency and clinical outcomes could be via unrelated pathways.
Our findings are consistent with results from a previous randomized controlled trial in 105 long-term dialysis patients with vitamin D deficiency showing that nutritional vitamin D (50,000 IU of ergocalciferol weekly or monthly) corrects 25-hydroxy vitamin D levels but does not lower mortality and cardiovascular events compared with placebo [10]. Our results are also in agreement with randomized controlled trials of cholecalciferol and calcifediol in the general population. While the present study was underpowered for the primary outcome, the results are concordant with a trial among 25,781 older men and women in whom cholecalciferol 2,000 IU per day had no effect on the risk of a major cardiovascular event (myocardial infarction, stroke, or death from cardiovascular causes) both in vitamin D-deficient and not deficient participants [20]. Similarly, in a placebo-controlled randomized controlled trial of 5,108 older men and women, monthly vitamin D3 treatment had no effect on cardiovascular disease incidence over 3.3 years regardless of the presence of vitamin D deficiency or previous cardiovascular disease [15]. In addition, no beneficial effect of vitamin D supplementation on other clinical outcomes such as incidence of falls, fracture and cancer were found in this same study [21].
The current evidence in dialysis, combined with the lack of efficacy shown in large trials in the broader population, indicates that nutritional vitamin D agents may make little or no difference to clinical outcomes in patients with end-stage kidney disease. The ongoing pragmatic, registry-based SIMPLIFIED trial comparing cholecalciferol (60,000 IU fortnightly) to standard care in 4,200 dialysis patients is likely to provide definitive evidence for treatment on survival, cardiovascular events, quality of life, cancer and cost-effectiveness [22].
The strengths of the present trial include investigation of mortality and cardiovascular disease as core outcomes identified by patients, caregivers and clinicians within the Standardized Outcomes in NephroloGy (SONG) initiative [23], a multicenter design having supervised administration of the medication and two years of treatment duration. The limitations include the early termination of the trial due to closure of the coordinating institution, leading to an inadequate number of participants enrolled and a small number of events for efficacy and safety outcomes which may have led to ascertainment bias [23]. Serum calcitriol and 24-metabolite levels were not measured, therefore we were not able to evaluate whether the hydroxylation pathways (such as via 1-alpha-hydroxylation and 24-hydroxylation) were influenced by calcifediol, thus leading to inconclusive effects on mortality and cardiovascular outcomes.
In conclusion, calcifediol supplementation for 2 years did not appear to prevent mortality and improve cardiovascular outcomes in hemodialysis patients with 25-hydroxy vitamin D insufficiency.
Data availability statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. Authors confirmed that the results presented in this paper have not been published previously in whole or part, except in abstract format.
Code availability
Not applicable.
References
LaClair RE, Hellman RN, Karp SL et al (2005) Prevalence of calcidiol deficiency in CKD: a cross-sectional study across latitudes in the United States. Am J Kidney Dis 45(6):1026–1033
Barreto DV, Barreto FC, Liabeuf S et al (2009) Vitamin D affects survival independently of vascular calcification in chronic kidney disease. Clin J Am Soc Nephrol 4(6):1128–1135
Palmer SC, McGregor DO, Macaskill P, Craig JC, Elder GJ, Strippoli GF (2007) Meta-analysis: vitamin D compounds in chronic kidney disease. Ann Intern Med 147(12):840–853
Dusso A, Lopez-Hilker S, Rapp N, Slatopolsky E (1988) Extra-renal production of calcitriol in chronic renal failure. Kidney Int 34(3):368–375
Kandula P, Dobre M, Schold JD, Schreiber MJ Jr, Mehrotra R, Navaneethan SD (2011) Vitamin D supplementation in chronic kidney disease: a systematic review and meta-analysis of observational studies and randomized controlled trials. Clin J Am Soc Nephrol 6(1):50–62
Hewitt NA, O’Connor AA, O’Shaughnessy DV, Elder GJ (2013) Effects of cholecalciferol on functional, biochemical, vascular, and quality of life outcomes in hemodialysis patients. Clin J Am Soc Nephrol 8(7):1143–1149
Kumar V, Yadav AK, Lal A et al (2017) A randomized trial of vitamin D supplementation on vascular function in CKD. J Am Soc Nephrol 28(10):3100–3108
Galassi A, Bellasi A, Auricchio S, Papagni S, Cozzolino M (2013) Which vitamin D in CKD-MBD? The time of burning questions. Biomed Res Int 2013:864012
Cardoso MP, Pereira LAL (2019) Native vitamin D in pre-dialysis chronic kidney disease. Nefrologia 39(1):18–28
Bhan I, Dobens D, Tamez H et al (2015) Nutritional vitamin D supplementation in dialysis: a randomized trial. Clin J Am Soc Nephrol 10(4):611–619
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO (2017) Clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl 2017(7):1–59
Kidney Disease: Improving global outcomes (KDIGO) CKD-MBD working group (2009). KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl 113:S1–S130
https://ridt.sinitaly.org/2017/03/18/2008-6/. Accessed 31 Jan 2021
Shieh A, Ma C, Chun RF et al (2017) Effects of cholecalciferol vs calcifediol on total and free 25-hydroxyvitamin D and parathyroid hormone. J Clin Endocrinol Metab 102(4):1133–1140
Scragg R, Stewart AW, Waayer D et al (2017) Effect of monthly high-dose vitamin d supplementation on cardiovascular disease in the vitamin D assessment study : a randomized clinical trial. JAMA Cardiol 2(6):608–616
de Zeeuw D, Agarwal R, Amdahl M et al (2010) Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet 376(9752):1543–1551
Stenvinkel PHC (2014) Cardiovascular disease in chronic kidney disease. In: Johnson RJ, Feehally J, Floege J (eds) Comprehensive clinical nephrology. Elsevier, Philadelphia, pp 949–966
Palmer SC, Craig JC, Navaneethan SD, Tonelli M, Pellegrini F, Strippoli GF (2012) Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med 157(4):263–275
Palmer SC, Di Micco L, Razavian M et al (2012) Effects of antiplatelet therapy on mortality and cardiovascular and bleeding outcomes in persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med 156(6):445–459
Manson JE, Cook NR, Lee IM et al (2019) Vitamin D supplements and prevention of cancer and cardiovascular disease. N Engl J Med 380(1):33–44
Scragg RKR (2019) Overview of results from the vitamin D assessment (ViDA) study. J Endocrinol Investig 42(12):1391–1399
Eudra CT (2016) Natural vitamin D (cholecalciferol) versus standard care in patients receiving dialysis. https://doi.org/10.1186/ISRCTN15087616
Tong A, Gill J, Budde K et al (2017) Toward establishing core outcome domains for trials in kidney transplantation: report of the standardized outcomes in nephrology-kidney transplantation consensus workshops. Transplantation 101(8):1887–1896
Acknowledgements
We acknowledge Dr. Boris Biclow for his support with the statistical analyses.
Mineral Metabolism Study Group of the Italian Society of Nephrology: Carlo Massimetti, Fabio Pennacchiotti, Antonio Mannarino, Cristina Grimaldi, Vincenzo Savica, Onofrio Schillaci, Olga Credentino, Maria Domenica Casu, Carlo Lomonte, Valentina Vigo, Giuseppe Grandaliano, Stefano Netti, Filippo Aucella, Massimo Morosetti, Roberto Boero, Francesco Soleti, Efstratios Fasianos, Maria Polidoro, Domenico Santoro, Alessandra Perna, Fabio Malberti, Ludovica d'Apice, Romano Musacchio, Maria Carla Porcu, Giuseppe Cianciolo, Silverio Rotondi e Maria Luisa Muci.
Funding
This work was a no-profit trial supported by the Italian Society of Nephrology. Grant/award number: not applicable.
Author information
Authors and Affiliations
Consortia
Contributions
Conception and design: LM, GS; data acquisition: LM, GC, AC, BDI, MDL, SM, AP; data analysis: AP; data interpretation: LM, SP, VS, AP, GS. Each author contributed important intellectual content during manuscript drafting or revision, accepts personal accountability for the author’s own contributions, and agrees to ensure that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. The study received Institutional Review Board approval from the Ethics Committee of the “Azienda Ospedaliera G. Rummo” of Benevento (deliberation n. 294, 13/04/2012).
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The members of the Mineral Metabolism Study Group of the Italian Society of Nephrology are listed in Acknowledgements.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Morrone, L., Palmer, S.C., Saglimbene, V.M. et al. Calcifediol supplementation in adults on hemodialysis: a randomized controlled trial. J Nephrol 35, 517–525 (2022). https://doi.org/10.1007/s40620-021-01104-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-021-01104-z