
Abstract
In recent years, our understanding of the physiology of the aldosterone-sensitive distal nephron (ASDN) has greatly advanced thanks to the discovery of the complex with-no-lysine kinase (WNK) signaling and the molecular characterization of the epithelial sodium channel (ENaC). A series of studies, initially focused on rare tubulopathies such as Gordon and Liddle syndromes, eventually led to a partial elucidation of the so-called “aldosterone paradox”, the traditional explanation of the physiology of such disparate conditions such as hyperkalemia and low effective arterial blood volume. The physiology of the ASDN is herein illustrated in light of the novel acquisitions in an easy-to-understand fashion, with the aim of giving the practicing nephrologist a solid “first glance” into this exciting but challenging field. Focus is on ion channels and transporters, their regulation by key hormones such as aldosterone and angiotensin II, and dietary implications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the last 30 years, two intertwined fields of research in renal physiology have greatly expanded our knowledge of potassium and sodium handling by the kidney. These novel insights arose from seminal observations, respectively in the early 1990s and the early 2000s: on the one hand, the discovery of the epithelial sodium channel (ENaC) molecular composition by the groups of Rossier, Lifton [1, 2] and Lingueglia [3], on the other hand the identification of with no lysine kinases (WNKs) mutations leading to an extremely rare Mendelian tubulopathy characterized by volume-dependent hypertension and hyperkalemic metabolic acidosis (pseudohypoaldosteronism type-2 or Gordon’s syndrome) [4]. These acquisitions and the ones that followed helped to fully or partially explain not only the pathophysiology of other rare monogenic tubulopathies, but also of common pathologic conditions as well as the physiology of sodium and potassium handling in the distal nephron. Altogether, these insights helped overcome the traditional view that held aldosterone as the sole responsible of potassium secretion and sodium reabsorption in the aldosterone-sensitive distal nephron (ASDN), a view that could not explain the different handling of potassium and sodium in high aldosterone states as diverse as hypovolemia and hyperkalemia, the so-called “aldosterone paradox”.
In this review, modern views of potassium and sodium renal pathophysiology will be examined, encompassing the molecular mechanisms in the kidney and the gut, the gut–kidney axis, and the circadian rhythm of kaliuresis, along with dietary implications of these new concepts.
The aldosterone paradox
Aldosterone has traditionally been appreciated as the key regulator of the physiologic response to two extremely different conditions: hypovolemia and hyperkalemia. Indeed, both conditions stimulate aldosterone secretion. However, the response to hypovolemia is salt retention without increased potassium excretion, while the response to hyperkalemia is increased potassium excretion without salt retention. This is commonly referred to as “the aldosterone paradox”, an expression which assumes that the same hormone mediates different effects in two different, irreconcilable scenarios. Thus, a single hormone would be responsible of sodium reabsorption and potassium secretion, although variations of sodium and potassium balance can still occur independently. The first objection to this view is that during hypovolemia, angiotensin II (AII) levels are also elevated, which is not the case of hyperkalemia, thus highlighting the possibility of a direct action of AII in blunting aldosterone-dependent potassium excretion while maintaining sodium retention. The second objection is that aldosterone-mediated potassium-lowering actions have a slow onset of action and would not prevent a normal-high dietary potassium load from causing hyperkalemia if aldosterone were the only regulator at play [5].
The discovery of the differential expression of sodium and potassium channels along the distal nephron and the modulation of complex kinase pathways signaling by different stimuli shed light on this paradox. Before offering an integrative view of sodium and potassium handling in different physiological/pathophysiological conditions and dietary patterns, the distribution and regulation of sodium and potassium channels in the distal nephron will be reviewed.
The aldosterone-sensitive distal nephron
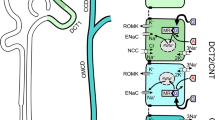
The distal nephron is comprised of the distal convoluted tubule (DCT), the connecting tubule (CNT) and the collecting duct (CD). Only principal-like cells in the late DCT, i.e. the DCT2 cells, the CNT and the cortical collecting duct (CCD) are aldosterone-sensitive, thereof the definition of ASDN for this particular portion of the nephron (Fig. 1). The aldosterone-sensitivity is determined by the cellular expression of both the mineralocorticoid receptor (MR) and the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11BHSD2), which converts cortisol to cortisone, thus preventing cortisol from occupying MR [6, 7].

Roadmap of the distal convoluted tubule and of the aldosterone-sensitive distal nephron. In the upper part of the figure, distal tubular segments are shown. A distinction is made between the early portion of the distal convoluted tubule, the late portion, the connecting tubule and the cortical collecting duct, since the former is not aldosterone-sensitive. In the middle part of the figure, cells responsible for electroneutral sodium and electrogenic sodium are indicated. In the lower part of the figure, the different cell types and the different channels they express are shown, from the distal convoluted tubule towards the collecting duct. DCT1 early portion of the distal convoluted tubule, DCT2 late portion of the distal convoluted tubule, CNT connecting tubule, CCD cortical collecting duct, ASDN aldosterone-sensitive distal nephron, NCC sodium–chloride cotransporter, ENaC epithelial sodium channel, ROMK renal outer medullary potassium channel, BK big potassium channel, Na/K-ATPase sodium–potassium ATPase, Kir4.1/5.1 inward rectifier potassium channel 4.1/5.1
Sodium channels: distribution, regulation and activity
Six to 10% of filtered sodium is reabsorbed in the DCT and CNT, while the CD handles about 1% [8]. In the early portion of DCT, i.e. DCT1, which is not aldosterone-sensitive, sodium is reabsorbed via the sodium chloride cotransporter (NCC), which is thiazide-sensitive [9]. In the principal-like cells in the DCT2, sodium is reabsorbed via both NCC and ENaC, whereas principal cells in the CNT and CCD only express ENaC (Fig. 1) [10]. In type B intercalated cells (DCT2, CNT and CCD) other two transporters are expressed: the sodium-driven chloride bicarbonate exchanger (NDCBE), and pendrins (chloride–bicarbonate exchangers), which work in tandem to reabsorb sodium electroneutrally. NDCBE is also thiazide-sensitive [10].
NCC
NCC mediates the electroneutral reabsorption of sodium chloride (i.e., it reabsorbs one sodium and one chloride ion). The activity of NCC is regulated by three main mechanisms: (1) an increase in sodium delivery to the distal tubule leads to an increase in NCC sodium transport capacity (this is the reason behind hypertrophic changes in DCT cells during chronic treatment with loop diuretics); (2) phosphorylation of NCC by a complex system of kinases (and prevention of these kinases’ degradation) following a series of stimuli causes its activation; (3) aldosterone, insulin and vasopressin increase NCC apical abundance via yet unidentified molecular mechanisms [9].
NCC is phosphoactivated by Ste20-like proline–alanine rich kinase (SPAK)–oxidative stress responsive kinase 1 (OSR1). In order to phosphorylate NCC, SPAK–OSR1 must be phosphorylated by WNKs. WNKs are activated by autophosphorylation, a phenomenon that depends on intracellular chloride concentration [11]. A low chloride concentration leads to WNK autophosphorylation, thus switching on the SPAK–OSR1 signaling pathway. WNK1, WNK3 and WNK4 have all been shown to phosphoactivate NCC. They differ, however, in terms of intracellular chloride concentration sensitivity, WNK4 being the most and WNK3 being the least chloride-sensitive. Moreover, WNK4 has an inhibitory effect in basal conditions: it complexes with WNK1 or WNK3, thus preventing their activation. The higher chloride-sensitivity and its inhibitory role under basal conditions explain why WNK4 among all WNKs is the key regulator of NCC: its activity can be modulated. Since intracellular chloride concentration directly depends on membrane potential, hyperpolarization or depolarization of DCT cells leads to WNK4 activation or inactivation, respectively. Through these mechanisms, low plasma potassium activates NCC whereas high plasma potassium blunts them. Kelch-like 3 (KLHL3) and cullin 3 (CUL3) form a RING-type E3-ubiquitin ligase complex that recruits WNKs for ubiquitination and promotes their proteasomal degradation. AII-induced signaling both prevents WNK4 degradation and promotes WNK4 phosphorylation [12,13,14].
Magnesium restriction has been recently shown to downregulate NCC in mice via neural precursor cell expressed developmentally down-regulated protein 4-2 (Nedd4-2), which ubiquitinates NCC tagging it for proteasomal degradation; interestingly, if potassium intake is also restricted, the effect of magnesium restriction prevails, i.e. NCC is not upregulated by potassium restriction, thus partly explaining the sustained hypokalemia observed in states of hypomagnesemia (see also the effect of magnesium on renal outer medullary potassium channels (ROMK) in the following section) [15]. Finally, there is experimental evidence in animals that zinc deficiency leads to hypertension via NCC upregulation [16].
NDCBE/pendrins
NDCBE works in tandem with pendrins to reabsorb sodium chloride electroneutrally in type B intercalated cells. Two cycles of pendrin-mediated bicarbonate/chloride exchange and one cycle of NDBCE-mediated chloride/sodium bicarbonate exchange result in the net reabsorption of one molecule of sodium chloride (Fig. 2). The chloride ions recycle across the apical membrane. Chloride exits the cell via CLCK2 [17]. Sodium exits the cell via the basolateral anion exchanger 4, together with bicarbonate. Intracellular bicarbonate ions derive from the activity of a carbonic anhydrase. While in principal cells the gradient driving sodium reabsorption is due to the activity of the sodium–potassium ATPase, in intercalated cells the activity of the latter is low. Instead, the necessary gradient is generated by a two-step process driven by the basolateral hydrogen ATPase. Hydrogen is pumped out of the cell, thus favoring bicarbonate generation by an intracellular carbonic anhydrase (step one). The intracellular accumulation of bicarbonate in turn drives pendrin activity, with accumulation of intracellular chloride. Bicarbonate and chloride in turn drive NDCBE activity and thus sodium reabsorption (step two). The generation of bicarbonate and its excretion via pendrins is particularly important as its intraluminal concentration is low in this segment, and would not be sufficient to drive NDBCE. Thus, sodium reabsorption by intercalated cells is driven by a bicarbonate (firstly) and chloride (secondly) gradient generated by the hydrogen ATPase, in contrast with the sodium gradient generated by sodium–potassium ATPase in principal cells [18]. As stated, like NCC, NDCBE is thiazide-sensitive. Experimental studies showed that the integrity of this system (i.e. the complex of NDCBE and pendrin) is required for sodium chloride reabsorption: genetic ablation of pendrin blunts thiazide-sensitive sodium reabsorption in this segment [18, 19]. Pendrin appears to be of critical importance in extracellular volume (and thus blood pressure) conservation during salt restriction: pendrin knockout mice waste salt and become hypovolemic and hypotensive during salt restriction, despite appropriately high aldosterone levels [20].

Electroneutral sodium chloride reabsorption in the cortical collecting duct via sodium-driven chloride–bicarbonate exchanger. The process of electroneutral sodium reabsorption via the combined actions of sodium-driven chloride–bicarbonate exchanger (NDCBE) and pendrin is shown. NDCBE activity results in the electroneutral exchange of one intracellular chloride for two bicarbonate and one sodium ions; pendrin mediates the also electroneutral exchange of one intracellular bicarbonate for one chloride ion. Thus, two cycles of transport via pendrin, when coordinated with one cycle of NDCBE, result in the net uptake of one sodium and one chloride. NDCBE sodium-driven chloride–bicarbonate exchanger, H+-ATPase hydrogen ATPase, AE4 anion exchanger 4, CLCK2 chloride channel kidney type 2
ENaC
ENaC is also called amiloride-sensitive sodium channel, since amiloride (as well as triamterene) specifically blocks the channel, which reabsorbs sodium ions, thus creating a lumen-negative transepithelial gradient (electrogenic sodium reabsorption). The latter in turn drives paracellular chloride reabsorption. The activity of ENaC is mainly modulated by aldosterone, which is antagonized by MR-antagonists (spironolactone, eplerenone, finerenone). Other relevant mediators that up-regulate ENaC are insulin, which also stimulates sodium reabsorption via sodium–hydrogen antiporter 3 in the proximal tubule, and vasopressin. Insulin has vasodilatory effects that are blunted in insulin-resistant states. Thus, insulin-resistance might directly contribute to the development of salt-sensitive hypertension in affected patients. Surprisingly, vasopressin has a stimulatory effect on ENaC. A possible explanation is that in states of low-water and low-sodium intake vasopressin-mediated ENaC stimulation helps maintain central volume. Moreover, vasopressin-mediated ENaC stimulation may contribute, through incompletely elucidated mechanisms, to the increased urinary concentration that leads to hyponatremia in high vasopressin states, suggesting a mechanistic shift of the role of ENaC from plasma volume to plasma osmolarity regulation. Indeed, adrenalectomized mice with markedly elevated vasopressin levels have increased ENaC activity in the absence of aldosterone or other steroids, thus facilitating urinary concentration and the development of hyponatremia, providing an adjunctive explanation for hyponatremia of adrenal insufficiency to already established mechanisms (e.g., the lack of cortisol-mediated inhibition of vasopressin secretion, the corticotropin releasing hormone-mediated stimulation of vasopressin secretion, the hypovolemic baroceptor-mediated vasopressin secretion and the decreased fluid delivery to the diluting segments of the nephron) [9, 21, 22].
The afore-mentioned paracellular chloride absorption is mediated by anion transporters located in the tight junctions between tubular epithelial cells, called claudins. Several claudins have been identified in the CD but current evidence mainly regards claudin 4 and 8. The presence of claudins confers anion-specificity to paracellular transport (in this specific case, to chloride ions), presumably because of intrinsic structural features. Claudin 4 and 8 knockout rodents develop hypochloremia, hypovolemia, hypotension and metabolic alkalosis from profound chloride depletion, especially after restriction of dietary salt intake. The drop in blood pressure is more marked in claudin 8 knockout animal models, which additionally develop hypokalemia, probably due to the exaggerated lumen-negative transepithelial gradient which favors potassium secretion. Claudin 8 is removed by the proteasome after ubiquitination by KLHL3, while claudin 4 is downregulated by channel-activating protease 1, whose expression is induced by aldosterone. While from a mechanistic standpoint claudin 8 regulation by KLHL3 remains quite obscure, the actions of aldosterone, i.e. upregulating both ENaC and claudin 4 degradation, seem to serve the purpose of switching from sodium reabsorption to potassium secretion: by increasing sodium reabsorption and reducing paracellular chloride reabsorption, the transepithelial lumen-negative gradient is maximized, favoring potassium secretion [23,24,25].
Potassium channels: distribution, regulation and activity
The proximal tubule and the loop of Henle mediate the reabsorption of the most part of filtered potassium, so that less than 10% of the latter reaches the distal tubule [26].
Three are the main potassium channels in the ASDN, two apical and one basolateral: on the one hand, ROMK and big K channels (BK), on the other hand Kir4.1/5.1 [12].
Kir4.1/5.1
Inward-rectifier potassium channel 4.1/5.1 (Kir4.1/5.1) is a basolateral secretory potassium channel. It is found all along the ASDN, but its role has been well-established in the DCT. Potassium exit via Kir4.1/5.1 leads to cell hyperpolarization and subsequent chloride exit through chloride channel Kb (CLCNKB), thus lowering chloride intracellular content. A low intracellular chloride concentration “switches on” WNK4, which phosphorylates SPAK–OSR1, that in turn phosphoactivates NCC. When potassium cannot exit via Kir4.1/5.1, the cell membrane potential decreases as there is a pump-leak uncoupling (sodium–potassium ATPase keeps pumping in potassium ions that cannot exit the cell). Depolarization blunts chloride exit through CLCNKB. A higher intracellular chloride concentration halts the WNK4-SPAK/OSR1 pathway, the end result being the lack of NCC activation (Fig. 3). Ki4.1/5.1 two main regulators are: (1) extracellular potassium concentration [27], (2) AII. Low extracellular potassium concentration leads to DCT cells hyperpolarization (i.e., more potassium is able to exit the cell via Kir4.1/5.1), whereas an elevation of extracellular potassium levels (as in hyperkalemia or mild elevation due to dietary loading) causes depolarization (i.e., less potassium can leave the cell via Kir4.1/5.1). AII stimulates Kir4.1/5.1 activity, thus leading to cell hyperpolarization [12]. Norepinephrine has been shown to increase sodium reabsorption through NCC by increasing potassium current via Kir4.1/5.1 [28].

Differential handling of sodium and potassium during high-aldosterone states. In the figure, a DCT1 cell is exemplified. a High-aldosterone state of volume depletion. During volume depletion, angiotensin II and aldosterone are both secreted. Angiotensin II, through its AT1 receptor, promotes WNKs activity via direct and indirect mechanisms. One key indirect mechanism is the promotion of Kir4.1/5.1 activity, a basolateral potassium channel. Potassium exits the cell, leading to hyperpolarization that in turn drives chloride exit via CLCKB, a basolateral chloride channel. Lower chloride intracellular concentration promotes WNKs activation and finally NCC activation and sodium chloride reabsorption. b High-aldosterone state of hyperkalemia. During hyperkalemia, aldosterone is secreted whereas angiotensin II is not. In this scenario, hyperkalemia per se blunts Kir4.1/5.1 activity since there is an unfavorable electrochemical gradient for potassium secretion. Depolarization leads to increased intracellular chloride concentration, which prevents WNKs from activating NCC. Blunting sodium chloride reabsorption allows for increased sodium delivery to DCT2 and CNT/CCD cells, i.e. the aldosterone-sensitive distal nephron, so that sodium can be exchanged with potassium by the combined actions of ENaC, ROMK and BK (not shown). NCC sodium–chloride cotransporter, SPAK Ste20-like proline–alanine rich kinase, WNKs with-no-lysine kinases, Kir4.1/5.1 inward rectifier potassium channel 4.1/5.1, Na/K-ATPase sodium–potassium ATPase, AII angiotensin II, AT1R angiotensin II receptor type 1, CLCKB chloride channel kidney type B, DCT1 early portion of the distal convoluted tubule, CNT connecting tubule, CCD cortical collecting duct, ENaC epithelial sodium channel, ROMK renal outer medullary potassium channel, BK big potassium channel
ROMK and BK
Two conditions are needed for potassium secretion in the ASDN: (1) a transepithelial lumen-negative gradient, which is provided by electrogenic sodium reabsorption via ENaC (in contrast with electroneutral sodium reabsorption via NCC, where chloride reabsorption is coupled with that of sodium), and (2) the presence of a sufficient number of open potassium channels. A third element, tubular flow, determines the activity of a particular type of potassium channels, the BKs [12, 29].
ROMK is a secretory potassium channel with a high basal open probability. It is found in principal-like and principal cells in the DCT, CNT and CCD. ROMK regulation is the result of a balance between opposing signals driving either the transport of newly synthesized channels to the apical membrane or their clathrin-dependent internalization. Potassium depletion/low plasma potassium via WNKs signaling and AII via angiotensin II receptor type 1 (AT1R) are inhibitory signals, while aldosterone, via serum and glucocorticoid-regulated kinase 1 (SGK-1), and hyperkalemia/dietary potassium load are stimulatory signals once again via WNKs signaling. Kir4.1/5.1 might regulate ROMK trafficking by modulating WNKs activity as with NCC phosphoactivation. By either depolarizing or hyperpolarizing the cell membrane, changes in potassium flow through Kir4.1/5.1 cause either a decrease or an increase of intracellular chloride concentration leading to either activation/inactivation of WNKs, which in turn promote either endocytosis or synthesis of ROMKs. Thus, high serum potassium leads to ROMK translocation to the apical membrane while low serum potassium leads to its internalization, which is opposite and complementary to what happens to NCC (Fig. 4) [12].

Regulation of ROMK activity during high-aldosterone states and during potassium depletion. A cell from the aldosterone-sensitive distal nephron is shown. Three main scenarios are depicted. (1) In response to potassium excess (either hyperkalemia or a dietary load), which is “sensed” by Kir4.1/5.1, the latter promotes depolarization, which in turn leads to an increase in intracellular chloride concentration that blunts WNKs, which would otherwise promote ROMK degradation. At the same time aldosterone, secreted in response to potassium excess, blocks WNKs via SGK-1. Kaliuretic factors might lead to the same end result through yet to be clarified mechanisms. (2) In response to potassium depletion, the opposite occurs. (3) During volume contraction, aldosterone promotes ROMK activity via the inhibition of WNKs. This signal is mediated by SGK-1. To prevent potassium loss during volume contraction, AII, through its AT1 receptor, promotes ROMK degradation. Green and red arrows stand, respectively, for activating and inhibitory signals. ROMK renal outer medullary potassium channel, WNKs with-no-lysine kinases, SGK-1 serum and glucocorticoid-regulated kinase 1, Aldo aldosterone, Kir4.1/5.1 inward rectifying potassium channel 4.1/5.1, AngII angiotensin II, AT1R angiotensin II receptor type 1 (color figure online)
Difficult-to-treat hypokalemia in the presence of hypomagnesemia is a common clinical scenario. Under physiological conditions, magnesium has an inhibitory effect on potassium secretion via ROMKs. During states of potassium wasting, concomitant low magnesium levels lead to reduced intracellular magnesium, removing its inhibitory effect on ROMKs. This might explain why correction of hypokalemia requires both potassium and magnesium supplementation, if hypomagnesemia coexists [30].
BKs are potassium channels possibly found both in intercalated and principal-like/principal cells of the CNT and CCD, although current evidence based on functional data seem to support their selective localization in intercalated cells. Interestingly, intercalated cells lack basolateral sodium–potassium ATPase and instead rely on NKCC1 to sustain potassium secretion. BKs are responsible for what is called flow-induced potassium secretion (FIKS). In states of potassium excess, sodium reabsorption in upwards tubular segments is decreased, leading to an increase in tubular flow rate in the distal nephron, which in turn bends primary cilia in principal cells. The bending of cilia leads to an increase in intracellular calcium which, in turn, activates BKs (Fig. 5) in intercalated cells via paracrine signals. Thus, primary stimulatory signals for BKs activity are tubular flow rate (any condition that increases tubular flow rate, e.g. osmotic diuresis) and serum potassium via WNKs, which have a coordinated effect on ROMKs and BKs, inhibiting the latter presumably via an ubiquitin-related degradation pathway [12, 31].

Flow-induced potassium secretion in the cortical collecting duct. Flow-induced bending of cilia in principal cells leads to an increase in cellular calcium concentration, in turn leading to the secretion of potassium via big potassium channels (BK) in intercalated cells via paracrine signals. Green and red arrows stand, respectively, for activating and inhibitory signals. BK big potassium channel, Na/K-ATPase sodium–potassium ATPase, H+-ATPase hydrogen ATPase, WNKs with-no-lysine kinases (color figure online)
Aldosterone actions on the ASDN
Aldosterone increases the number of functional ENaC units via their translocation on the apical membrane of ASDN epithelial cells [32]. This is the most rapid effect of aldosterone on the kidney (less than 2 h) and is partly mediated by SGK-1, which also induces other early-onset aldosterone effects: indeed, it enhances ENaC activity both by stimulating the channel via the phosphorylation of WNK4 [33] and by inhibiting the ubiquitin ligase Nedd4-2 [34], which would otherwise promote ENaC degradation. SGK-1 also enhances basolateral sodium–potassium ATPase activity [35].
Slower aldosterone-mediated actions include: (1) the stimulation of the basolateral sodium–potassium ATPase, both indirectly by the means of an increase in sodium entrance via ENaC and directly by the means of increased recruiting from the intracellular pool and/or increased expression of the pump; (2) the increased synthesis of ENaC.
Moreover, aldosterone increases ROMK apical abundance through SGK-1- and protein kinase A-mediated phosphorylation [12]. Altogether, these aldosterone-mediated effects facilitate ROMK activity, since the final effect is an increase in the transepithelial lumen-negative gradient, which is favorable for potassium secretion. Thus, aldosterone is necessary to maximize ROMK secretory activity. However, it is not sufficient. Indeed, in an experimental model, aldosterone levels comparable to those observed after a high dietary potassium load were not sufficient to stimulate ROMK [36]. This implies that other kaliuretic factors, particularly potassium serum levels per se and a gut–kidney axis, are necessary to rapidly stimulate ROMK in an aldosterone-independent fashion in order to prevent hyperkalemia [37].
Finally, there is experimental evidence that in the presence of aldosterone, insulin or vasopressin, the apical abundance of phosphorylated NCC is also increased. However, whether these stimuli act through SGK1 and/or WNKs is yet to be clarified [9]. Aldosterone also increases NCC activity (via phosphorylation) within minutes in mice as shown in both ex vivo and in vivo models [38]. A more definite role of AII in NCC activation has recently emerged, and current evidence will be reviewed in the next section.
Pendrin expression is upregulated by aldosterone, i.e. the plasma membrane-enriched fraction is increased in high aldosterone states [39]. This effect appears to be mediated by MR. However, additional mechanisms are at play. The phosphorylated or dephosphorylated status of a specific site in the ligand-binding domain of the MR determines if the aldosterone-MR interaction actually leads to pendrin upregulation. In more detail, hyperkalemia leads to phosphorylation of the site therefore shutting down the effects of aldosterone, whereas AII leads to its dephosphorylation, thereby allowing the aldosterone-MR interaction to be effective and thus upregulate pendrin expression. Interestingly, while MR can be phosphorylated in multiple sites, the phosphorylation of the specific site involved in pendrin upregulation is almost exclusively observed in intercalated cells [40]. Hypokalemia appears to have similar effects to aldosterone, but only in the presence of the latter [39].
Angiotensin II actions on the ASDN
From a distal nephron perspective of sodium and potassium handling, AII is responsible of three key effects: NCC activation, Kir4.1/5.1 stimulation and ROMK inhibition.
There are two proposed molecular mechanisms of NCC activation by AII. By binding to AT1R on DCT cells, AII might: (1) prevent WNK4 from interacting with WNK1 and with WNK3, therefore letting WNK4 phosphorylate SPAK–OSR1; (2) activate free inactive WNK4 [13].
AII, through AT1R, stimulates Kir4.1/5.1 to hyperpolarize DCT cells, thus driving chloride basolateral exit, the end result being NCC activation. Moreover, AII-induced signaling activates protein kinase C, which phosphorylates KLHL3, thus preventing it from binding WNK4 and the subsequent proteasomal degradation of the latter, the end result being an increase in WNK4 and therefore increased NCC activity [14].
AII, again via AT1R, leads to ROMK clathrin-dependent internalization by the means of a Src family of protein tyrosine kinases signaling pathway, an effect that is opposite to that of aldosterone. AII receptor type 2 mediates opposite effects, as it stimulates ROMK through a protein kinase A-signaling pathway [12].
The relationship between the calcium-sensing receptor, NCC and WNKs in the ASDN
Thiazide and thiazide-like diuretics have been traditionally used in calcium stone formers to prevent stone recurrence as they lower urinary calcium excretion [41]. On the other hand, loop diuretics increase calciuria and this is the rationale for their use in the treatment of hypercalcemia [42]. Two hereditary tubulopathies, Bartter syndrome (BS) and Gitelman syndrome (GS), which emulate the effects of loop and thiazide diuretics, also induce hyper- and hypocalciuria, respectively. It has long been appreciated that while in the thick ascending limb of the loop of Henle (TAL) calcium and sodium chloride are transported in the same direction (either both reabsorbed or both excreted), in DCT they go opposite ways (when one is reabsorbed, the other is excreted and vice versa). In TAL, sodium chloride enters the cell via apical sodium–potassium–chloride cotransporter 2 (NKCC2) and exits the cell via basolateral sodium–potassium ATPase and CLCNKB. Potassium is recycled in the tubular lumen, i.e. it enters the cell via NKCC2 and exits the cell via ROMK, thus providing a lumen-positive gradient that drives paracellular calcium (and magnesium) reabsorption. The calcium-sensing receptor (CaSR) is located in the basolateral membrane in TAL; when calcium levels in the interstitial fluid are high (or when calcimimetics are administered), CaSR blunts NKCC2 and ROMK, thus favoring calcium excretion and sodium chloride distal tubular delivery. In DCT, there is an inverse relationship between salt reabsorption via NCC and calcium excretion via transient receptor potential cation channel subfamily V (TRPV5), a relationship which is still largely unclear. CaSR is found both on the apical and basolateral membrane of DCT cells. When interstitial calcium levels are high (or when a calcimimetic is administered) and/or distal calcium delivery is increased (e.g., during hypercalcemia for the afore-mentioned consequences on TAL, or secondarily to loop diuretics or BS), basolateral and/or apical DCT CaSRs activate (Fig. 6). This, in turn, leads to WNK4 phosphoactivation via the WNK4-SPAK pathway and increased NCC activity. The mechanistic interpretation is that in order to excrete calcium, TAL wastes sodium chloride that must be reabsorbed in the DCT, the two actions being coordinated by CaSR [43].

Relationship between calcium and sodium handling in the thick ascending limb of the loop of Henle and in the distal convoluted tubule. Green and red arrows stand for activating or inhibitory signals. NKCC sodium–potassium–chloride cotransporter, ROMK renal outer medullary potassium channel, CLCKB chloride channel kidney type B, Ca-mim calcimimetics, CaSR calcium-sensing receptor, Na/K-ATPase sodium–potassium ATPase, NCC sodium–chloride cotransporter, TRPV5 transient receptor potential cation channel subfamily V (color figure online)
It has long been appreciated that impaired urine concentration in the CD is responsible for hypercalcemia-associated polyuria. It appears that CaSR in the CD blunts vasopressin-induced aquaporin insertion, thus allowing a dilution of calcium ions, presumably in order to prevent their precipitation, orchestrating a complex mechanism not dissimilar to the one we described in the DCT [44]. As a final note, it is worth reporting that both acute and chronic furosemide administration have been found to induce an increase of apical calcium transporters abundance in the DCT, in a similar fashion to what happens to sodium transporters (chronic loop diuretic use induces hypertrophic changes in DCT cells) [42].
Intestinal potassium handling: a brief overview
On a normal Western diet, 100 mmol of potassium are ingested daily, with 90% being excreted by the kidneys, the rest (10 mmol) with feces. Normal stool volume is about 100 ml, which means that fecal potassium concentration would be 100 mmol per liter, that is more than 28 times normal serum potassium concentration. The daily excretion of potassium with stools ranges between 2 and 20 mmol, reflecting oral intake, which means that in normal (100 ml) stool volume, the fecal potassium concentration can rise up to 50 times the serum potassium concentration. On the other hand, on a potassium restriction diet, fecal potassium excretion can drop to as low as to 3.5 mmol/day, a value corresponding to the obligatory excretion rate. Since kidneys remove the major share of ingested potassium, a reduced intestinal potassium excretion does not cause hyperkalemia per se. On the contrary, severe intestinal potassium wasting (e.g. due to diarrhea) is a relevant cause of hypokalemia, which can sometimes be hidden since metabolic acidosis and/or hypovolemia (with hemoconcentration) frequently coexist. On a brighter note, intestinal potassium excretion appears to be increased in oligo-anuric patients with end-stage renal disease, thus partially replacing the impaired kidney potassium excretory functions. In the small intestine, 90% of ingested potassium is absorbed passively. On the contrary, in the colon, passive absorption in proximal and distal segments, and active absorption and secretion of potassium in all segments are observed. The active absorption is mediated by an apical hydrogen-potassium ATPase, similar to the gastric transporter but ouabain- and not omeprazole-sensitive. Potassium secretion in the colon follows the pump-leak dynamics observed in other tissues: it enters the cell via basolateral sodium–potassium ATPase (directly or in a “secondarily active” fashion via basolateral NKCC1) and it exits the cell via apical potassium channels (presumably BK). Passive potassium secretion is paracellular, driven by the lumen negativity. A recent hypothesis postulates that distal colon potassium secretion and absorption are mediated by separate cells, respectively the crypt and surface enterocytes, both expressing BKs. On surface enterocytes, ENaC is also expressed.
During a low sodium diet, aldosterone stimulates both potassium secretion (presumably through BKs, as mentioned) and potassium absorption via hydrogen-potassium ATPase; however, experimental models showed that no potassium fecal wasting is observed in mice fed with a low sodium diet despite high aldosterone levels, the mechanistic interpretation being that during a low sodium diet the “unwanted” potassium loss is balanced by colonic potassium absorption, since the primary aim of aldosterone secretion is to keep volume (sodium chloride) balance. During a low potassium diet, hydrogen-potassium ATPase is upregulated by different (yet to be clarified) means, as obviously aldosterone is not at play. Finally, during a high potassium diet/potassium loading, BKs on crypt (secretory) enterocytes are upregulated. We mentioned that surface (absorptive) enterocytes express both ENaC and BK. It appears that in high aldosterone states (sodium restriction or potassium loading), surface cells contribute to active potassium secretion following an ENaC-dependent mechanism which recapitulates that observed in the ASDN: potassium is excreted since sodium reabsorption via ENaC promotes a lumen-negative electrical gradient [45].
Aldosterone-independent kaliuresis, the gut–kidney axis, the kaliuresis circadian rhythm
The intestine absorbs virtually the whole bulk of ingested potassium. If all the potassium content of a potassium-rich meal were to reach the extracellular fluid, fatal cardiac arrhythmias would inevitably ensue, as serum potassium would rapidly rise towards life-threatening levels. Luckily this is not the case, but the mechanisms that allow us to “survive” potassium-rich meals are still largely obscure.
The physiological mechanisms dealing with a potassium-rich meal (and with its opposite, a potassium-deficient meal) can be distinguished in five “blocks”: a feedback system regulating kaliuresis; potassium transcellular shifts; a feed-forward control of kaliuresis; circadian clock-driven kaliuresis; non-renal (i.e., mainly colonic) potassium handling.
A very small rise in serum potassium (0.1 mmol/l) is sufficient to induce depolarization of adrenal cells in the zona glomerulosa, causing aldosterone secretion [46]. As explained, aldosterone mediates both rapid- and slow-onset actions on the ASDN, the rapid ones (onset in less than 2 h) mainly involving ENaC translocation to the apical membrane [32]. Aldosterone facilitates ROMK to maximize potassium secretory activity, but it would not be sufficient alone to prevent hyperkalemia due to a dietary potassium load, since aldosterone-induced kaliuresis has a slow onset of action [37]. One fundamental kaliuretic factor is serum potassium concentration per se: serum potassium levels are sensed by Kir4.1/5.1 in the ASDN and potassium secretion through ROMKs follows, as we previously mentioned [12]. Although this feedback loop is rapid enough, it requires an “error” to start, that is, an increase in potassium concentration in peritubular capillaries. This is also the case for potassium transcellular shifts, as they happen only after sensing a change in serum potassium concentration: in response to a potassium-rich meal that includes glucose, pancreatic insulin secretion causes skeletal muscle and liver sodium–potassium ATPase activation, so that potassium is pumped from the plasma to the intracellular fluid, minimizing the postprandial increase in serum potassium concentration; subsequent muscle activity causes potassium release into plasma; in order to maintain balance, the amount of potassium ingested (minus the minimal amount lost in the feces) must eventually be secreted in urine. These mechanisms are blunted in case of hypokalemia, i.e. sodium-potassium ATPase is down-regulated in the liver and in skeletal muscle, allowing a “leak” of potassium from the intracellular to the extracellular fluid [47].
It has long been appreciated, however, that variations in potassium dietary intake too little to cause changes in serum potassium concentration, and thus directly or indirectly (through aldosterone secretion) stimulate kaliuresis through the feedback system, did indeed alter kaliuresis in sheep. Moreover, sheep fed with a potassium-rich meal, that is, a meal that could alter serum potassium concentration, were shown to increase kaliuresis in 20 min, well before any change in serum potassium concentration and aldosterone secretion might have occurred [47]. These observations led to the hypothesis of a feed-forward control of kaliuresis. Unlike feedback systems, a feed-forward control does not need an “error” to start. Instead, it starts when a signal that could lead to an “error” is sensed, i.e. it is a predictive system, whereas a feedback system is reactive. Instead of hyperkalemia (the “error” in the feedback system), the signal in the feed-forward system is the amount of potassium ingested. Even though the sensors that detect the signal and the mediators that transmit it to the kidneys to promote kaliuresis are still unknown, there is evidence that a gut–kidney kaliuretic axis actually exists in humans [48]. Preston et al. in their seminal work performed a series of experiments in healthy humans to assess the existence of an aldosterone-independent gut–kidney axis. A potassium load without glucose led to an increase in venous potassium concentration, activating the feedback system and leading to aldosterone-dependent kaliuresis. A potassium load with concomitant glucose-derived meal did not alter venous potassium concentration, presumably because insulin led to a transcellular potassium shift, but there was a small increase in aldosterone concentration, and increased kaliuresis was observed, whether due to aldosterone or unknown “gut factors” cannot be ascertained. In the third intervention, a potassium load with concomitant glucose-derived meal was administered to subjects who had been given eplerenone, an MR antagonist. As expected, venous potassium concentration did not change, but even though aldosterone-dependent signals were blunted, kaliuresis was nonetheless observed, corroborating the hypothesis that an aldosterone-independent gut–kidney axis actually exists. These experiments, however, prove more than this: (1) they do not rule out the possibility that the unknown “gut factor” is indeed once again potassium concentration itself (via hyperpolarization of DCT cells, ensuing downregulation of NCC and increased sodium delivery to the CCD with increased sodium/potassium exchange via the coordinated activity of ENaC and ROMK), however not the venous but the arterial potassium concentration, similarly to the wide variations in arterial sodium concentration with small or no variation in sodium venous concentration, demonstrated by other investigators in other conditions, such as water loading [49]; (2) an arterial potassium concentration variation might be responsible for the early aldosterone secretion observed, which occurred as early as 15 min after the intervention, an observation that conflicts with the traditional view of a slow (hours) feedback kaliuresis versus a fast (minutes) feedforward kaliuresis, suggesting instead that aldosterone might be an early actor and that the two systems act simultaneously. It must be observed, however, that a large body of evidence showed that aldosterone alone fails to promote significant kaliuresis in the absence of increased serum potassium concentration [48, 50].
Nonetheless, in adrenalectomized animal models dietary potassium intake increases apical sodium and potassium transport in the CCD despite aldosterone deficiency [51, 52]. However, up until recently there were concerns that adrenalectomy in animal models might not have been complete and that a residual local production of aldosterone (e.g. in the kidney itself) might persist [53, 54]. A mouse model of complete aldosterone deficiency, relying on targeted inactivation of aldosterone synthase rather than adrenalectomy, has been effectively used to explore potassium homeostasis in this setting: investigators showed that despite complete aldosterone deficiency, dietary potassium intake resulted in increased sodium and potassium urinary excretion, mainly via upregulation of ROMK; moreover, sodium and thus volume balance was partly maintained due to AII-induced upregulation of ENaC [55]. Altogether, these experiments show that the aldosterone-independent kaliuresis, including the mechanisms at play in the gut–kidney axis illustrated by Preston et al., might indeed be mediated by potassium per se and/or AII, at least in part.
Underlying these mechanisms, there is an ever-active clock dictating the circadian rhythm of potassium urinary excretion. Indeed, a central clock in the suprachiasmatic nucleus of the brain and peripheral clocks in renal cells regulate the circadian rhythm of kaliuresis. The circadian rhythm of potassium excretion is so important that the ablation of the suprachiasmatic nucleus, which is responsible for the disruption of many other circadian rhythms, does not affect it, presumably for the preserved activity of renal cell clocks. This rhythm is preserved even after adrenalectomy. The central circadian oscillator in the suprachiasmatic nucleus responds to the ambient light–dark cycle. Over a 24-h period, the clock dictates the kaliuresis rhythm so that the maximum potassium excretion is at noon, while the minimum is at midnight. If the potassium content of single meals is fixed and meals are evenly distributed across the day (e.g., every 6 h), the kaliuresis can increase from nadir to maximum (e.g., midnight–noon) by a factor of 2–4 in normal potassium intake (70–100 mmol per day), or to 1.6 in high potassium diets (400 mmol per day). In any case, the circadian periodicity is always retained. Long distance travels with changes in day–night cycle cause the rhythm to slowly adjust (over several days) to the new ambient light–dark cycle. The role of hormones, particularly cortisol and aldosterone, in relation to kaliuresis circadian rhythm has been partly unveiled. Glucocorticoids promote potassium excretion and have been shown to act on the phase of circadian clock gene expression both centrally (the suprachiasmatic nucleus) and peripherally (the kidneys), thus acting to coordinate, or “synchronize” the clocks. Aldosterone has been shown to modestly increase potassium excretion in the afternoon, with no effect in the morning or at night. On the other hand, it promotes the transcription of circadian clock genes which, in turn, promote the expression of the ENaC alfa subunit, with the final effect of enhancing sodium retention [56].
The “aldosterone paradox” reconciled
In two different high aldosterone states, namely hyperkalemia and hypovolemia, sodium and potassium are handled independently. Neither hyperkalemia causes sodium retention, nor does hypovolemia cause potassium wasting. The novel acquisitions we outlined have led to a partial reconciliation of this paradox. In this section we will briefly review how recent insights in renal physiology led to a better understanding of sodium and potassium handling in high aldosterone states.
During hypovolemia, both AII and aldosterone levels are elevated. AII promotes sodium retention in the ASDN by enhancing NCC activity, while aldosterone mediates sodium retention by enhancing ENaC activity. The increased sodium reabsorption through NCC reduces sodium delivery to the CCD, thus preventing potassium excretion, both by impairing FIKS and by decreasing the amount of sodium which can be “exchanged” with potassium by the means of the already-described electrogenic sodium reabsorption/potassium excretion through ENaC and ROMK. In addition, AII inhibits ROMK, further limiting potassium secretion.
Hyperkalemia increases aldosterone secretion, without changing circulating AII levels, since it does not affect renin release. Hyperkalemia per se inhibits NCC, thus increasing sodium delivery to the CCD, allowing ENaC to reabsorb sodium in so promoting a negative tubular lumen, which in turn allows potassium excretion through ROMK. Moreover, the rise in aldosterone level increases the expression and activity of ROMK. At the same time, the increased sodium delivery to the CCD promotes FIKS.
The unfortunate coexistence of hypovolemia and hyperkalemia poses a problem, as the maintenance of volume is the priority, preventing the correction of hyperkalemia until an euvolemic state is restored. On the other hand, a high dietary potassium load leads to increased sodium excretion.
In the opposite condition of potassium depletion (e.g. due to low dietary potassium intake), hypokalemia lowers aldosterone secretion, which results in reduced activity of both ENaC and ROMKs, so diminishing potassium excretion. At the same time, hypokalemia-induced hyperpolarization in the DCT activates NCC via WNKs, which contributes to limit the potassium excretion, but at the cost of sodium retention and blood pressure increase. The dietary implications of these conditions will be reviewed in the next section.
Dietary implications
Over million years, renal physiology evolved to adapt to a low sodium high potassium diet, i.e. to retain sodium and excrete potassium. Agriculture and industrialization, however, led to a drop in dietary potassium intake and a rise in sodium consumption, which is associated with a variety of unfavourable outcomes. Low potassium intake leads to principal-like and principal cells hyperpolarization in DCT, causing the activation of the ASDN sodium-retaining mechanisms we already outlined [57]. An explanation of the mechanisms at play during a high-potassium and low-sodium diet, irrespective of WNKs signaling, is available in a recent, in-depth review, to which the reader is referred [58]. Among the unfavourable outcomes associated with a low-potassium and high-sodium diet, are disorders such as hypertension, obesity, nephrolithiasis and osteoporosis. In recent years, the return to a “paleolithic diet” was proposed as a way to reduce the risk of developing the afore-mentioned conditions. The peculiarity of the paleolithic diet lies not only in the high potassium but also in the high alkaline content [59].
Potassium intake is inversely related to the prevalence of hypertension and the risk of stroke. Blood pressure decreases in hypertensive patients put on a high potassium diet. Two large scale studies, NHANES III and DASH, found that increased dietary potassium consumption was associated with lower blood pressure [60, 61].
The Western diet tends to be poor in alkali, too (fruits, vegetables). If bicarbonate ions are insufficient to maintain arterial blood pH within normal ranges, alkaline calcium salts are mobilized from bones, leading to calcium loss and eventually osteoporosis. There is experimental evidence that supplementation of potassium chloride to potassium depleted mice reduces bone resorption. Studies in postmenopausal women either taking potassium bicarbonate supplements or adhering to a diet rich in potassium-containing fruits and vegetables showed they had less bone resorption and higher bone densities, respectively, compared to controls [59].
Postulated mechanisms include: (1) a thiazide-like effect of potassium loading, which by inhibiting NCC through already-discussed mechanisms leads to hypocalciuria, (2) increased urinary pH with subsequent increased citraturia (the higher the urinary pH the higher the fraction of trivalent citrate will be, and only the divalent form of citrate is reabsorbed by the proximal convoluted tubule) [29]. Potassium loading presumably leads to increased urinary citrate excretion as a consequence of proximal intracellular alkalosis due to transcellular potassium shifts (exchanged with hydrogen), which cause a decrease in both ammoniagenesis and proximal bicarbonate reabsorption. Moreover, the increased intracellular pH inhibits citrate metabolism by m-aconitase [62]. Another mechanism involves metabolic acidosis and hypokalemia, both leading to increased proximal citrate reabsorption via NaDC1 [63]. Therefore, a paleolithic diet, thanks to its high content of potassium and alkali, promotes hypocalciuria and increases citraturia, both beneficial for the prevention of calcium stone recurrence [59].
Abbreviations
- ENaC:
-
Epithelial sodium channel
- WNK:
-
With no lysine kinase
- ASDN:
-
Aldosterone-sensitive distal nephron
- AII:
-
Angiotensin II
- DCT:
-
Distal convoluted tubule
- CNT:
-
Connecting tubule
- CD:
-
Collecting duct
- DCT2:
-
The late portion of the distal convoluted tubule
- CCD:
-
Cortical collecting duct
- MR:
-
Mineralocorticoid receptor
- 11BHSD2:
-
11β-Hydroxysteroid dehydrogenase type 2
- DCT1:
-
The early portion of the distal convoluted tubule
- NCC:
-
Sodium chloride cotransporter
- NDCBE:
-
Sodium-driven chloride bicarbonate exchanger
- SPAK:
-
Ste20-like proline–alanine rich kinase
- OSR1:
-
Oxidative stress responsive kinase 1
- KLHL3:
-
Kelch-like 3
- CUL3:
-
Cullin 3
- Nedd4-2:
-
Neural precursor cell expressed developmentally down-regulated protein 4-2
- ROMK:
-
Renal outer medullary potassium channel
- BK:
-
Big potassium channels
- Kir4.1/5.1:
-
Inward-rectifier potassium channel 4.1/5.1
- CLCNKB:
-
Chloride channel, kidney b
- AT1R:
-
Angiotensin II receptor type 1
- SGK-1:
-
Serum and glucocorticoid-regulated kinase 1
- FIKS:
-
Flow-induced potassium secretion
- BS:
-
Bartter syndrome
- GS:
-
Gitelman syndrome
- TAL:
-
Thick ascending limb of the loop of Henle
- NKCC2:
-
Sodium–potassium–chloride cotransporter 2
- CaSR:
-
Calcium-sensing receptor
- TRPV5:
-
Transient receptor potential cation channel subfamily V
References
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367(6462):463–467. https://doi.org/10.1038/367463a0
Canessa CM, Horisberger JD, Rossier BC (1993) Epithelial sodium channel related to proteins involved in neurodegeneration. Nature 361(6411):467–470. https://doi.org/10.1038/361467a0
Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P (1993) Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett 318(1):95–99
Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP (2001) Human hypertension caused by mutations in WNK kinases. Science 293(5532):1107–1112. https://doi.org/10.1126/science.1062844
Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G (2011) Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology (Bethesda) 26(2):115–123. https://doi.org/10.1152/physiol.00049.2010
Bostanjoglo M, Reeves WB, Reilly RF, Velazquez H, Robertson N, Litwack G, Morsing P, Dorup J, Bachmann S, Ellison DH (1998) 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na–Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9(8):1347–1358
Funder JW (2013) Mineralocorticoid receptor antagonists: emerging roles in cardiovascular medicine. Integr Blood Press Control 6:129–138. https://doi.org/10.2147/IBPC.S13783
Palmer LG, Schnermann J (2015) Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 10(4):676–687. https://doi.org/10.2215/CJN.12391213
Subramanya AR, Ellison DH (2014) Distal convoluted tubule. Clin J Am Soc Nephrol 9(12):2147–2163. https://doi.org/10.2215/CJN.05920613
Roy A, Al-bataineh MM, Pastor-Soler NM (2015) Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10(2):305–324. https://doi.org/10.2215/CJN.08880914
Chen JC, Lo YF, Lin YW, Lin SH, Huang CL, Cheng CJ (2019) WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1817220116
Welling PA (2016) Roles and regulation of renal K channels. Annu Rev Physiol 78:415–435. https://doi.org/10.1146/annurev-physiol-021115-105423
Hadchouel J, Ellison DH, Gamba G (2016) Regulation of renal electrolyte transport by WNK and SPAK–OSR1 kinases. Annu Rev Physiol 78:367–389. https://doi.org/10.1146/annurev-physiol-021115-105431
Shibata S, Arroyo JP, Castaneda-Bueno M, Puthumana J, Zhang J, Uchida S, Stone KL, Lam TT, Lifton RP (2014) Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci USA 111(43):15556–15561. https://doi.org/10.1073/pnas.1418342111
Ferdaus MZ, Mukherjee A, Nelson JW, Blatt PJ, Miller LN, Terker A, Staub O, Lin DH, McCormick JA (2019) Mg2+ restriction downregulates NCC through NEDD4-2 and prevents its activation by hypokalemia. Am J Physiol Ren Physiol. https://doi.org/10.1152/ajprenal.00216.2019
Williams CR, Mistry M, Cheriyan AM, Williams JM, Naraine MK, Ellis CL, Mallick R, Mistry AC, Gooch JL, Ko B, Cai H, Hoover RS (2019) Zinc deficiency induces hypertension by promoting renal Na+ reabsorption. Am J Physiol Ren Physiol 316(4):F646–F653. https://doi.org/10.1152/ajprenal.00487.2018
Chambrey R, Trepiccione F (2015) Relative roles of principal and intercalated cells in the regulation of sodium balance and blood pressure. Curr Hypertens Rep 17(4):538. https://doi.org/10.1007/s11906-015-0538-0
Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D (2010) The Na+-dependent chloride–bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Investig 120(5):1627–1635. https://doi.org/10.1172/JCI40145
Eladari D, Chambrey R, Peti-Peterdi J (2012) A new look at electrolyte transport in the distal tubule. Annu Rev Physiol 74:325–349. https://doi.org/10.1146/annurev-physiol-020911-153225
Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW (2004) NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl-conservation. Hypertension 44(6):982–987. https://doi.org/10.1161/01.HYP.0000145863.96091.89
Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD (2012) Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci USA 109(25):10095–10100. https://doi.org/10.1073/pnas.1201978109
Raff H (1987) Glucocorticoid inhibition of neurohypophysial vasopressin secretion. Am J Physiol Regul Integr Comp Physiol 252(4 Pt 2):R635–R644. https://doi.org/10.1152/ajpregu.1987.252.4.R635
Hou J (2016) Paracellular transport in the collecting duct. Curr Opin Nephrol Hypertens 25(5):424–428. https://doi.org/10.1097/MNH.0000000000000253
Gong Y, Yu M, Yang J, Gonzales E, Perez R, Hou M, Tripathi P, Hering-Smith KS, Hamm LL, Hou J (2014) The Cap1–claudin-4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proc Natl Acad Sci USA 111(36):E3766–E3774. https://doi.org/10.1073/pnas.1406741111
Gong Y, Wang J, Yang J, Gonzales E, Perez R, Hou J (2015) KLHL3 regulates paracellular chloride transport in the kidney by ubiquitination of claudin-8. Proc Natl Acad Sci USA 112(14):4340–4345. https://doi.org/10.1073/pnas.1421441112
Palmer BF (2015) Regulation of potassium homeostasis. Clin J Am Soc Nephrol 10(6):1050–1060. https://doi.org/10.2215/CJN.08580813
Su XT, Ellison DH, Wang WH (2019) Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K+ excretion. Am J Physiol Ren Physiol 316(3):F582–F586. https://doi.org/10.1152/ajprenal.00412.2018
Duan XP, Gu L, Xiao Y, Gao ZX, Wu P, Zhang YH, Meng XX, Wang JL, Zhang DD, Lin DH, Wang WH, Gu R (2019) Norepinephrine-induced stimulation of Kir4.1/Kir5.1 is required for the activation of NaCl transporter in distal convoluted tubule. Hypertension 73(1):112–120. https://doi.org/10.1161/hypertensionaha.118.11621
Kamel KS, Schreiber M, Halperin ML (2018) Renal potassium physiology: integration of the renal response to dietary potassium depletion. Kidney Int 93(1):41–53. https://doi.org/10.1016/j.kint.2017.08.018
Huang CL, Kuo E (2007) Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 18(10):2649–2652. https://doi.org/10.1681/ASN.2007070792
Carrisoza-Gaytan R, Carattino MD, Kleyman TR, Satlin LM (2016) An unexpected journey: conceptual evolution of mechanoregulated potassium transport in the distal nephron. Am J Physiol Cell Physiol 310(4):C243–C259. https://doi.org/10.1152/ajpcell.00328.2015
Loffing J, Zecevic M, Feraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F (2001) Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Ren Physiol 280(4):F675–F682. https://doi.org/10.1152/ajprenal.2001.280.4.F675
Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP (2007) An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA 104(10):4025–4029. https://doi.org/10.1073/pnas.0611728104
Alvarez de la Rosa D, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, Canessa CM (1999) The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem 274(53):37834–37839
de la Alvarez Rosa D, Gimenez I, Forbush B, Canessa CM (2006) SGK1 activates Na+–K+-ATPase in amphibian renal epithelial cells. Am J Physiol Cell Physiol 290(2):C492–C498. https://doi.org/10.1152/ajpcell.00556.2004
Palmer LG, Frindt G (2000) Aldosterone and potassium secretion by the cortical collecting duct. Kidney Int 57(4):1324–1328. https://doi.org/10.1046/j.1523-1755.2000.00970.x
Welling PA (2013) Regulation of renal potassium secretion: molecular mechanisms. Semin Nephrol 33(3):215–228. https://doi.org/10.1016/j.semnephrol.2013.04.002
Cheng L, Poulsen SB, Wu Q, Esteva-Font C, Olesen ETB, Peng L, Olde B, Leeb-Lundberg LMF, Pisitkun T, Rieg T, Dimke H, Fenton RA (2019) Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 30(8):1454–1470. https://doi.org/10.1681/ASN.2018101025
Xu N, Hirohama D, Ishizawa K, Chang WX, Shimosawa T, Fujita T, Uchida S, Shibata S (2017) Hypokalemia and pendrin induction by aldosterone. Hypertension 69(5):855–862. https://doi.org/10.1161/HYPERTENSIONAHA.116.08519
Hirohama D, Ayuzawa N, Ueda K, Nishimoto M, Kawarazaki W, Watanabe A, Shimosawa T, Marumo T, Shibata S, Fujita T (2018) Aldosterone is essential for angiotensin II-induced upregulation of pendrin. J Am Soc Nephrol 29(1):57–68. https://doi.org/10.1681/ASN.2017030243
Reilly RF, Peixoto AJ, Desir GV (2010) The evidence-based use of thiazide diuretics in hypertension and nephrolithiasis. Clin J Am Soc Nephrol 5(10):1893–1903. https://doi.org/10.2215/CJN.04670510
Lee CT, Chen HC, Lai LW, Yong KC, Lien YH (2007) Effects of furosemide on renal calcium handling. Am J Physiol Ren Physiol 293(4):F1231–F1237. https://doi.org/10.1152/ajprenal.00038.2007
Bazua-Valenti S, Rojas-Vega L, Castaneda-Bueno M, Barrera-Chimal J, Bautista R, Cervantes-Perez LG, Vazquez N, Plata C, Murillo-de-Ozores AR, Gonzalez-Mariscal L, Ellison DH, Riccardi D, Bobadilla NA, Gamba G (2018) The calcium-sensing receptor increases activity of the renal NCC through the WNK4-SPAK pathway. J Am Soc Nephrol 29(7):1838–1848. https://doi.org/10.1681/ASN.2017111155
Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW (1997) Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Investig 99(6):1399–1405. https://doi.org/10.1172/JCI119299
Sorensen MV, Matos JE, Praetorius HA, Leipziger J (2010) Colonic potassium handling. Pflugers Arch 459(5):645–656. https://doi.org/10.1007/s00424-009-0781-9
Guagliardo NA, Yao J, Hu C, Barrett PQ (2012) Minireview: aldosterone biosynthesis: electrically gated for our protection. Endocrinology 153(8):3579–3586. https://doi.org/10.1210/en.2012-1339
Greenlee M, Wingo CS, McDonough AA, Youn JH, Kone BC (2009) Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med 150(9):619–625
Preston RA, Afshartous D, Rodco R, Alonso AB, Garg D (2015) Evidence for a gastrointestinal–renal kaliuretic signaling axis in humans. Kidney Int 88(6):1383–1391. https://doi.org/10.1038/ki.2015.243
Shafiee MA, Charest AF, Cheema-Dhadli S, Glick DN, Napolova O, Roozbeh J, Semenova E, Sharman A, Halperin ML (2005) Defining conditions that lead to the retention of water: the importance of the arterial sodium concentration. Kidney Int 67(2):613–621. https://doi.org/10.1111/j.1523-1755.2005.67117.x
Hoorn EJ, Zietse R (2015) Gut–kidney kaliuretic signaling: looking forward to feeding. Kidney Int 88(6):1230–1232. https://doi.org/10.1038/ki.2015.272
Palmer LG, Antonian L, Frindt G (1994) Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol 104(4):693–710. https://doi.org/10.1085/jgp.104.4.693
Stanton B, Pan L, Deetjen H, Guckian V, Giebisch G (1987) Independent effects of aldosterone and potassium on induction of potassium adaptation in rat kidney. J Clin Investig 79(1):198–206. https://doi.org/10.1172/JCI112783
Xue C, Siragy HM (2005) Local renal aldosterone system and its regulation by salt, diabetes, and angiotensin II type 1 receptor. Hypertension 46(3):584–590. https://doi.org/10.1161/01.HYP.0000175814.18550.c0
Kobayashi M, Yasuoka Y, Sato Y, Zhou M, Abe H, Kawahara K, Okamoto H (2011) Upregulation of calbindin D28k in the late distal tubules in the potassium-loaded adrenalectomized mouse kidney. Clin Exp Nephrol 15(3):355–362. https://doi.org/10.1007/s10157-011-0414-4
Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, Makhanova N, Korbmacher C, Wagner CA, Loffing J (2015) Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26(2):425–438. https://doi.org/10.1681/ASN.2013111156
Gumz ML, Rabinowitz L, Wingo CS (2015) An integrated view of potassium homeostasis. N Engl J Med 373(1):60–72. https://doi.org/10.1056/NEJMra1313341
Wu P, Gao ZX, Su XT, Wang MX, Wang WH, Lin DH (2019) Kir4.1/Kir5.1 activity is essential for dietary sodium intake-induced modulation of Na–Cl cotransporter. J Am Soc Nephrol 30(2):216–227. https://doi.org/10.1681/asn.2018080799
Cornelius RJ, Wang B, Wang-France J, Sansom SC (2016) Maintaining K+ balance on the low-Na+, high-K+ diet. Am J Physiol Ren Physiol 310(7):F581–F595. https://doi.org/10.1152/ajprenal.00330.2015
Palmer BF, Clegg DJ (2016) Achieving the benefits of a high-potassium, paleolithic diet, without the toxicity. Mayo Clin Proc 91(4):496–508. https://doi.org/10.1016/j.mayocp.2016.01.012
Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, Sacks FM, Bray GA, Vogt TM, Cutler JA, Windhauser MM, Lin PH, Karanja N, DASH Collaborative Research Group (1997) A clinical trial of the effects of dietary patterns on blood pressure. N Engl J Med 336(16):1117–1124. https://doi.org/10.1056/nejm199704173361601
Hajjar IM, Grim CE, George V, Kotchen TA (2001) Impact of diet on blood pressure and age-related changes in blood pressure in the US population: analysis of NHANES III. Arch Intern Med 161(4):589–593
Zuckerman JM, Assimos DG (2009) Hypocitraturia: pathophysiology and medical management. Rev Urol 11(3):134–144
Osis G, Webster KL, Harris AN, Lee HW, Chen C, Fang L, Romero MF, Khattri RB, Merritt ME, Verlander JW, Weiner ID (2019) Regulation of renal NaDC1 expression and citrate excretion by NBCe1-A. Am J Physiol Ren Physiol. https://doi.org/10.1152/ajprenal.00015.2019
Acknowledgements
The authors would like to thank Prof. G. Capasso and F. Trepiccione for their useful comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
This article does not contain any studies with animals or human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rossi, G.M., Regolisti, G., Peyronel, F. et al. Recent insights into sodium and potassium handling by the aldosterone-sensitive distal nephron: a review of the relevant physiology. J Nephrol 33, 431–445 (2020). https://doi.org/10.1007/s40620-019-00684-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-019-00684-1