
Abstract
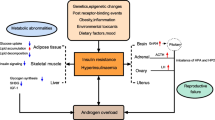
PCOS is a common and heterogeneous endocrine disorder in women of reproductive age, frequently associated with metabolic abnormalities. It was estimated that about 75% of these subjects have an impairment of insulin action, as measured by gold standard methods. While the relationship between insulin resistance and PCOS is consistently shown by a number of studies, the mechanisms underlying its primary origin still remains an unsolved issue. Insulin resistance and the associated hyperinsulinemia can induce both the endocrine and reproductive traits of PCOS. However, androgen excess, in turn, can impair insulin action, directly and/or through several changes occurring in different tissues. Body fat excess, which is another common feature in these women, can contribute to worsening the whole picture. Nevertheless, insulin resistance may also be found in many normal-weight individuals. Endocrine and metabolic abnormalities can develop in different moments, and probably there is fetal programming of these alterations. However, a number of vicious circles, with bidirectional relationships between androgen excess and insulin resistance, and with the contribution of several other factors, make it extremely difficult to understand where this process really originates. This review summarizes available evidence on this topic, in order to better understand the complex relationships linking hyperandrogenism and impaired insulin action in women with PCOS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycystic ovary syndrome (PCOS) is a very common and heterogeneous endocrine disorder in women of reproductive age, with several relevant clinical implications. Based on the generally adopted Rotterdam diagnostic criteria, established in a workshop consensus of the American Society for Reproductive Medicine (ASRM) and the European Society of Human Reproduction and Embriology (ESHRE) [1], it was estimated that its prevalence is at least 10% [2].
In its typical expression, PCOS is characterized by hyperandrogenism and evidence of ovarian dysfunction, such as chronic oligoanovulation and/or micropolycystic morphology of the ovary. However, there are other frequent abnormalities in these women, which are not included among the diagnostic features. In particular, insulin resistance is a very common finding in subjects with PCOS. By using the hyperinsulinemic euglycemic clamp, the gold standard methodology for measuring in vivo insulin action on glucose metabolism, and the WHO criteria for defining insulin resistance (i.e. glucose uptake in the lowest quartile for the background population under hyperinsulinemic euglycemic clamp conditions [3]), about 75% of these women have impaired insulin sensitivity [4]. Interestingly, highlighting the clinical impact of this phenomenon, in a cohort of 137 consecutive PCOS women, with a mean age of just 23 years and a mean BMI in the overweight range, about one third of subjects had metabolic syndrome [5]. These data, obtained in Caucasian women, fit well with those obtained in a multi-ethnic American sample, despite a substantially higher degree of obesity [6], suggesting that PCOS per se plays a major role in these alterations.
While the relationship between insulin resistance and PCOS is consistently shown by several studies which used the glucose clamp methodology [7], what the cause is and what the effect is in this dangerous liaison still remain unsolved issues.
This question prompts a number of additional queries, which are relevant in order to try to answer the first one: is insulin resistance in these women accounted for by any PCOS-specific molecular abnormalities? If so, are these abnormalities due to intrinsic defects or are they rather induced by PCOS-specific environmental factors? And again, if the latter hypothesis is correct, when do these factors come into play during the lifetime of PCOS women? Unfortunately, we have only vague ideas about these crucial aspects.
Molecular defects in insulin resistance associated with PCOS
A key point is to understand whether specific mechanisms underlie insulin resistance associated with PCOS. In this regard, about 25 years ago, in cultured cells from a sample of 14 insulin resistant women with PCOS, Dunaif et al. reported abnormally increased serine phosphorylation and reduced tyrosine phosphorylation of the insulin receptor and its substrate IRS-1, with impaired downstream metabolic insulin signalling [8]. Nevertheless, this unique abnormality was not observed in 50% of examined patients, although they too were insulin resistant, suggesting heterogeneity in the mechanisms responsible for this phenomenon. Subsequent research by the same group showed that, unlike metabolic signalling, mitogenic signalling of insulin was activated in cultured cells from PCOS women [9]. These findings suggested that either this pathway, or other pathways which are not defective and remain sensitive to insulin, may account for increased insulin action—driven by insulin resistance-induced hyperinsulinemia, on non-metabolic pathways, specifically androgen production in the ovary [10].
However, confirmation of these molecular findings has been elusive. Indeed, other studies have suggested that molecular defects underlying insulin resistance in PCOS may not be related to alterations in the proximal portion of the insulin signalling cascade [11,12,13], and that an impaired rather than stimulated mitogenic pathway, with reduced levels of activated MEK1/2 and ERK1/2, can be associated with increased androgen production in the ovary, independent of insulin action [14]. In a recent study, carried out in muscle biopsies of lean women with PCOS, who had whole body insulin resistance and impaired insulin-induced glucose oxidation in euglycemic clamp studies, proximal insulin signalling cascade did not show any alterations [13]. Interestingly, in this study, reduced expression and activity of AMP-activated protein kinase (AMPK) was found, associated with increased testosterone and lowered adiponectin levels. AMPK is an enzyme that plays a key role in cellular energy homeostasis, activating substrate uptake and oxidation when cellular energy is low. Based on these findings, direct or indirect effects of androgen excess on energy sensing signalling were hypothesized in subjects with PCOS. Yet, another study, carried out in skeletal muscle of obese insulin-resistant PCOS women, failed to detect any alterations in the AMPK pathway [12]. As a whole, available data on the molecular defects underlying insulin resistance in these women are inconsistent, suggesting possible heterogeneity in the mechanisms involved.
Hindrances in assessing what comes first in the relationship between insulin resistance and PCOS
Unfortunately, a number of factors limit most existing information that is needed to establish which comes first in the association between PCOS and insulin resistance. An important point is that clear identification of actors playing in the scene is often imprecise. PCOS is a heterogeneous condition, and its diagnosis is based on unspecific features which are generally measured by inaccurate methods, especially in terms of serum androgen levels. And also insulin resistance is rarely measured by appropriate techniques, being generally estimated by rough surrogate indexes. Moreover, in order to answer the question, we would need to understand the chronological sequence of events in the process. However, while there is evidence of potential fetal programming of the syndrome [15], PCOS can be diagnosed only in reproductive age, and there is a lack of prospective studies. Finally, there are interesting animal models of PCOS, but they do not completely mirror the human condition. So, transferring their results to humans requires caution.
As regards heterogeneity of PCOS, a contribution came from the adoption of current diagnostic criteria, established in the Rotterdam workshop consensus [1]. According to these criteria, diagnosis is based on any combination of at least two of three clinical features, i.e. hyperandrogenism (clinical and/or biochemical), chronic oligoanovulation, and micropolycystic ovarian morphology at ultrasound examination. However, other disorders that can potentially cause these alterations, in particular other hyperandrogenic conditions, must be excluded before diagnosis. Adoption of these criteria means that, under the general term of PCOS, there are women with different clinical phenotypes. In particular, there are women with hyperandrogenism plus oligoanovulation, the so-called classic phenotype of PCOS, but also women who have only one of these two main abnormalities, hyperandrogenism or oligoanovulation, combined with the micropolycystic aspect of the ovary, from which the ovulatory and the normoandrogenic phenotypes originate, respectively. There is also the possibility that all three clinical features coexist, but this complete phenotype is barely distinguishable from the classic one. It is noteworthy that the 2012 NIH workshop on PCOS, while confirming the Rotterdam criteria for diagnosis, has recommended distinguishing women with these phenotypes [16]. Actually, it is important to make a distinction between the different phenotypes of PCOS, as they differ in several ways, including in insulin resistance. A comparison of insulin-induced glucose utilization in euglycemic clamp studies has shown that this figure is strikingly reduced in PCOS women with the classic/complete phenotype, but it is normal in those with the normoandrogenic phenotype, while showing intermediate values in subjects with the ovulatory phenotype (Fig. 1) [5].

Reproduced with permission from Moghetti et al. [5]
Insulin-induced glucose utilization during the hyperinsulinemic euglycemic clamp in women with the different clinical phenotypes of PCOS and in control subjects.
Another obvious influential issue is the amount of body fat, which is increased in many of these women. In our cohort of patients, about two thirds among 375 subjects with PCOS, referred to the outpatient clinics of the Verona City Hospital for hirsutism and/or menstrual dysfunction, were overweight or obese [4]. In this sample, as expected, frequency of insulin resistance, as measured by the hyperinsulinemic euglycemic clamp, was higher in patients with body fat excess. However, insulin resistance was also present in many lean individuals. In this cohort about 60% of normal weight subjects showed impaired insulin sensitivity (Fig. 2). Remarkably, there is evidence that the effect of increased BMI in worsening insulin resistance may be proportionally more evident in PCOS women, as compared with control women [17, 18]. This could possibly be due to a preferential visceral distribution of fat in these subjects, even in normal weight individuals [19, 20], and/or to the presence of dysfunctional adipocytes [21]. Notably, the association between body fat and hyperandrogenism, in these subjects, seems to be to a large extent explained by insulin resistance [22]. However, there is evidence of a bidirectional link between obesity and PCOS, as suggested by genetic, epidemiologic, and experimental data [23].

Frequency of insulin resistance, measured by the hyperinsulinemic euglycemic clamp technique, in women with PCOS of the Verona 3P Study cohort subdivided according to BMI categories
Some studies have suggested that the association between obesity and PCOS may be inflated by significant referral bias [24]. Nevertheless, a couple of recent studies, which used bidirectional Mendelian randomization analysis to explore the causal relationships between obesity and PCOS, in large cohorts of subjects of European and Chinese origin, respectively, have found that known single nucleotide polymorphisms associated with BMI predicted increased risk of PCOS [25, 26]. In these analyses, the reverse was not true. However, as regards this latter aspect, it should not be forgotten that our knowledge of the genetic basis of PCOS is still limited. In another recent study, that used polygenic risk prediction to analyse the phenome-wide comorbidity patterns in a very large cohort of subjects across different genetic origins, a high level of shared biology was identified between PCOS and a range of metabolic outcomes, in particular obesity and diabetes [27].
Interestingly, PCOS is clearly associated with an increased risk of diabetes, especially type 2 diabetes, as indicated by a number of population and case–control studies, both cross-sectional and prospective [28,29,30,31]. However, studies carried out in premenopausal women with overt type 2 diabetes did not clearly support this association. While some small studies reported consistent findings, others reported increased frequency of polycystic ovarian morphology, but not of PCOS [32,33,34,35]. It was argued that these discordant findings may dispute the role of insulin resistance, which is a typical characteristic of type 2 diabetes, as a primary actor in the pathogenesis of PCOS. However, other potential explanations can be hypothesized. Indeed, many physicians do not assess glucose tolerance in young women with normal fasting glucose. As a consequence, type 2 diabetes may be underdiagnosed in women with this condition, limiting the apparent frequency of PCOS among diabetic subjects. In addition, this inconsistency may also reflect the reduction of insulin levels typically found in subjects with overt diabetes, due to pancreatic beta-cell glucotoxicity and other mechanisms [36].
Current diagnostic criteria of PCOS do not distinguish between clinical and biochemical hyperandrogenism. However, it was recently proposed that the definition of hyperandrogenism should consider androgen excess and hirsutism separately, because their pathophysiological mechanisms may differ [37]. Unfortunately, assessment of biochemical hyperandrogenism, which is a key aspect of the syndrome and is fundamental in understanding the pathophysiology of PCOS, is often biased by major problems. Methods generally available in routine clinical laboratories for assaying serum androgens are both inaccurate and imprecise, and when using these assays there is a striking overestimation of hormone concentrations, as compared with measures obtained by gold standard methods, i.e. liquid chromatography-tandem mass spectrometry and equilibrium dialysis [38]. In particular, the low precision of currently available commercial methods is a major obstacle, from both a clinical and speculative point of view. In actual fact, with routine assays many women may be misclassified in terms of biochemical hyperandrogenism. We have found that about 30% of subjects with PCOS were erroneously classified by these methods as hyperandrogenemic or normoandrogenemic, with the assignment of many patients to the incorrect clinical phenotypes and, thus, also inappropriate estimation of their metabolic risk [38].
Another important point is that, although current diagnostic criteria of PCOS do not clearly discriminate between different hormones, androgens cannot be considered equal in terms of action. Testosterone and its derivative DHT have a much greater affinity with the androgen receptor as compared with other circulating androgens, such as androstenedione and DHEA, which can be considered pro-hormones [39]. Interestingly, inclusion of free testosterone, as measured by gold standard methods, in the panel of assessed serum androgens substantially increases the fraction of PCOS women recognized as hyperandrogenemic, and makes the contribution of other androgens in distinguishing these subjects negligible [38]. Remarkably, in these women, insulin resistance is highly correlated with serum free testosterone (Fig. 3), whereas the relationships with total testosterone or other androgens are weaker.

Reproduced with permission from Tosi et al. [22]
Relationship between serum free testosterone, as measured by liquid chromatography-mass spectrometry and equilibrium dialysis, and glucose utilization during the hyperinsulinemic euglycemic clamp.
An intriguing additional problem arises from the acknowledgement that there are other circulating androgens, apart from the classic androgens that are generally measured. Interestingly, some recent studies showed that serum concentrations of 11-oxygenated androgens, which are mainly produced in the adrenals and converted to active hormones (11-ketotestosterone and 11-ketodihydrotestosterone) in peripheral tissues, are higher in PCOS subjects, as compared with healthy women [40]. This could be biologically relevant, as recent data in cultured cells transfected with the androgen receptor showed that 11-ketotestosterone may effectively bind and activate this receptor, with a potency of action that, in this in vitro model, was about 75% as compared with testosterone [41]. In this regard, it is noteworthy that in women with PCOS serum 11-ketotestosterone was found to be 3–5 times higher than testosterone [40]. Other studies showed that 11-oxygenated androgens may be the predominant circulating androgens also in other conditions, such as during normal and premature adrenarche [41] and in women with 21-hydroxylase deficiency [42]. Moreover, comparison of activated genes after stimulation of cultured cells with either 11-ketotestosterone or testosterone indicated that the biological effects may to some extent differ [41]. Our knowledge on the clinical implications of these aspects is still extremely limited.
Unfortunately, the accurate assessment of insulin resistance is another difficult task. Therefore, in clinical studies surrogate indexes, such as the HOMA index, are generally used. Indeed, in PCOS women, these indexes revealed fair correlations with direct measures of in vivo insulin action [4, 43]. Nevertheless, it is necessary to be aware that using these surrogate indexes to categorize an individual PCOS woman as insulin resistant or insulin sensitive may be misleading and should be avoided. In actual fact, there may be a striking mismatch in this classification when using the glucose clamp and the HOMA index, or other surrogate indexes [4]. Indeed, these indexes have a good positive predictive power, but a poor negative predictive power. In other words, in individual subjects with PCOS these indexes can be used to rule in, but not rule out, insulin resistance.
An interesting point is that insulin levels, which mainly drive these surrogate indexes, depend on several factors, not only insulin sensitivity and glucose levels. In particular, beta-cell function, insulin metabolism and obesity also play a major role. Among these factors, insulin clearance might be an important issue in women with PCOS, which has been little investigated. Indeed, plasma insulin concentrations depend on both the insulin secretion rate from beta-cells and the metabolic clearance rate of insulin from the plasma compartment. Insulin degradation follows the insulin-receptor complex internalization in target cells, and is carried out, in the cytoplasm, by the insulin-degrading enzyme and other lysosomal enzymes [44]. Interestingly, an early study, by Buffington and Kitabchi, showed marked reduction of endogenous insulin clearance in women with PCOS, compared with either lean or obese healthy controls, as estimated by the fasting plasma C-peptide over insulin ratio [45]. In this study, impaired in vitro insulin degradation was also found in cultured lymphocytes from PCOS women. Moreover, in cultured lymphocytes obtained from healthy controls, testosterone showed a biphasic effect on both insulin binding and insulin degradation. In particular, both binding and degradation of insulin were impaired at moderately high concentrations of testosterone, corresponding to those found in hyperandrogenic women, whereas they returned to normal at higher concentrations, corresponding to those found in men. The authors hypothesized that this phenomenon could contribute to the sexual dimorphism in the metabolic effect of androgens.
Consistent results have been reported in a recent study, carried out in 190 women with PCOS of the Verona 3P cohort. In this study, metabolic clearance of insulin was calculated as the insulin infusion rate divided by the serum insulin concentrations during the steady state period of euglycemic hyperinsulinemic clamp studies. Interestingly, insulin clearance proved to be strikingly impaired in PCOS women, as compared with reference values in healthy controls (Fig. 4), and this reduction was confirmed after adjustment for several potentially confounding factors [46]. Moreover, in multivariate analysis, body fat, estimates of insulin secretion and serum androgen levels were independent predictors of insulin clearance, all with negative relationships. However, insulin sensitivity did not show an independent association with insulin clearance. Conversely, age, insulin clearance and insulin sensitivity, with inverse relationships, and body fat, with a direct relationship, were independent predictors of insulin secretion, while serum androgens were not. These data suggest that, in women with PCOS, obesity contributes to hyperinsulinemia by both increasing secretion and lowering metabolism of insulin, whereas age and insulin resistance regulate insulin secretion, and serum androgens specifically regulate insulin clearance. There appears to be also a reciprocal modulation between secretion and degradation of insulin. Thus, androgens could mainly contribute to generating hyperinsulinemia, in women with PCOS, by reducing insulin degradation, although the underlying mechanisms remain unclear.

Reproduced with permission from Tosi et al. [33]
Comparison of metabolic clearance rate of insulin in women with PCOS and healthy controls.
Genetic and epigenetic mechanisms underlying the origin of endocrine and metabolic PCOS traits
Interestingly, several family studies showed that PCOS and its typical traits are frequent in female relatives of PCOS women [47,48,49]. Moreover, metabolic features are frequent in both male and female relatives of these subjects [50], suggesting a common genetic background of these alterations.
A large study in twins estimated that genetic heritability may explain about 80% of the origin of PCOS [51]. These findings prompted a series of genetic studies, initially by analysis of candidate genes and more recently by large-scale genome-wide association studies, carried out in women with either Han Chinese, Korean or European ancestry [52]. In these studies, 19 genetic loci associated with risk of PCOS have been identified, of which several are common to different ancestries. A recent meta-analysis, conducted in women with European ancestry (10,074 PCOS cases and 103,164 controls), confirmed association with 11 previously reported loci and also identified three novel loci. However, it has been estimated that they may explain just 10% of predicted heritability [53]. Although the biological significance of these findings remains little understood, identified variants were found to be associated with hyperandrogenism and gonadotropin regulation in PCOS women. Moreover, linkage disequilibrium score regression analysis showed genetic correlations with obesity, serum insulin and lipid levels, type 2 diabetes and cardiovascular disease, suggesting shared genetic architecture between metabolic traits and PCOS. Interestingly, Mendelian randomization analysis of these data suggested that genetic variants linked to some features typically associated with PCOS, such as body fat excess, hyperinsulinemia, but also late menopause timing and depression, as well as genetic variants linked to male pattern balding, which is a putative male phenotype [54], may be causally linked to the syndrome [53]. These intriguing findings require further functional research.
Nevertheless, association within families of women with this syndrome of both PCOS traits and metabolic abnormalities does not necessarily prove that genetic mechanisms play the major role in the origin of PCOS and the associated insulin resistance. Indeed, epigenetic mechanisms could be of greater relevance in these phenomena. In this regard, an interesting hypothesis is that the intrauterine environment may play a key role in the development of PCOS phenotype, as strongly suggested by studies in animal models in which androgen administration during gestation induced several PCOS traits in female progeny [15], as further discussed below.
MicroRNAs (miRNAs) are short non-coding RNAs which participate in the post-transcriptional regulation of gene expression. Some studies suggested that PCOS women may have alterations in their miRNA pattern, in the blood and in the ovary and other tissues [55]. However, available data are limited and do not allow us to understand whether these changes may be a cause or a consequence of PCOS abnormalities. Interestingly, in a recent study, a number of miRNA transcripts with different expression in PCOS women and controls were identified in skeletal muscle biopsies [56]. While the pathophysiological significance of these alterations remains uncertain, it is noteworthy that, in functional studies, the expression of several of these miRNAs was affected by either insulin or testosterone, suggesting a relationship with the endocrine and metabolic features of PCOS. In another study, which compared the non-targeted circulating miRNA profile in women with PCOS, control women and men, who were similar in terms of BMI, the expression of 38 miRNAs proved to be different between groups [57]. In particular, the differences in 15 miRNAs followed a pattern suggestive of androgenisation, as their expression was similar in PCOS women and men but different from control women. Conversely, the expression of 13 miRNAs was similar in PCOS and control women but different from men, suggesting sexual dimorphism. Finally, 5 miRNAs were expressed differently in women with PCOS as compared with both control women and men, suggesting PCOS-specific abnormalities. Interestingly, the expression levels of many of these circulating miRNAs were associated with hyperandrogenism, body fat excess and metabolic alterations.
Recent studies have shown that circular RNAs, long non-coding RNAs and RNA-binding proteins are other potential intermediate actors in the interaction between androgens and insulin action, possibly modulating the expression of key genes involved in this relationship [58,59,60]. However, these studies are still in the early stages.
Some studies reported altered DNA methylation, another fundamental mechanism of epigenetic control, in blood cells and in some tissues of women with PCOS [61]. However, available data are extremely limited. A study which combined genome-wide DNA methylation analysis and gene expression patterns, in ovarian tissue from a very small sample of PCOS and control women, reported that there were almost 8,000 differentially methylated CpG sites, about 3% of total sites, and several hundreds differential transcripts in PCOS compared to normal ovary [62]. Moreover, in tissue from PCOS subjects, 54 genes showed methylated levels which correlated with gene transcription. Another pilot study, carried out by profiling the methylation levels of the DNA extracted from umbilical cord blood of PCOS and control women submitted to in vitro fertilization, reported a different methylation status, of prevalent hypomethylation, affecting 918 genes. Network analysis showed relationships between these genes and several pathways, including those of glucose and lipid metabolism, and inflammation [63]. These findings suggested that the maternal PCOS environment may affect the epigenetic programming in utero of the developing embryo, inducing changes in genomic regions involved in the PCOS phenotype. The authors hypothesized a key role for this epigenetic signature in perpetuating the maternal endocrine and metabolic alterations, typical of PCOS, in the progeny. More recently, sex-specific differences in the DNA methylation patterns were found in the promoter regions of some reproductive and metabolic genes, selected on the basis of their potential pathophysiological role in PCOS, in blood cells from 2–3 months old sons or daughters of PCOS and control women [64]. Interestingly, metformin treatment of PCOS mothers during pregnancy prevented some of these changes, whereas, in the whole group of daughters, the maternal BMI was associated with the methylation level of one CpG site of the androgen receptor gene promoter. Likewise, studies in animal models strongly support a role for epigenetic modifications in the pathophysiology of PCOS, also providing potential links between endocrine and metabolic alterations [15].
Evidence of a developmental origin of PCOS
While investigations into epigenetic aspects are still in their early stages, there is consistent evidence, especially in animal models, that developmental factors underlie susceptibility to PCOS and to the phenotypic expression of this condition. Indeed, an altered maternal endocrine-metabolic environment could permanently program the physiology of the fetus, with changes that emerge in early reproductive life [15].
Many intriguing results have come from androgen-induced animal models of PCOS, in particular as regards the effect of global or tissue-specific loss of androgen receptor signalling on the development of PCOS traits. In this regard, in rodent, sheep and primate models, administration of testosterone or DHT, either prenatally or postnatally, may induce different reproductive, endocrine and metabolic PCOS traits [65]. Interestingly, in DHT-induced mice models, although global loss of the androgen receptor reversed all the reproductive and metabolic features of PCOS, its selective knockout in theca cells improved the reproductive effects only, whereas in granulosa cells it had minimal effects. However, selective brain loss of androgen receptors totally or partially abolished many of the reproductive and metabolic traits of PCOS, suggesting the importance of neuroendocrine mechanisms [66]. It should be noted that androgen receptors are expressed in several brain areas, especially in the hypothalamus. However, the specific neurons targeted by androgens, and the mechanisms involved in these alterations remain unclear [67].
Some data in animal models also indicate potential sex-specific effects of androgens on beta cells. Interestingly, in female mice, DHT administration induced insulin resistance and hyperinsulinemia, which were reversed in animals with selective loss of the androgen receptor in these cells [68]. Moreover, in isolated islets from both humans and mice, while insulin response to glucose was increased by DHT, this response was abolished by either an androgen receptor antagonist or rapamycin, suggesting involvement of an androgen receptor dependent mTORC pathway [68].
Against this background, another intriguing finding is that serum androgens are higher in PCOS women, as compared to controls, even during pregnancy [69]. Moreover, it was recently reported that serum AMH as well is higher in pregnant PCOS women, especially in those who are lean and hyperandrogenic, than in pregnant healthy controls [70]. These abnormalities may have relevant implications in the neuroendocrine mechanisms underlying the origin of PCOS, as it was demonstrated that both androgens and AMH can persistently alter the intrinsic firing rate of GnRH neurons, favouring LH oversecretion and ovarian androgen production [70, 71].
Of particular interest, in this pathophysiological scenario, are recent findings in animal models in which AMH was administered during pregnancy. In these animals, while the mother developed androgen excess and impaired reproductive capacity, the female offspring showed subtle masculinization at birth and, more importantly, PCOS-like neuroendocrine changes, which persisted in adult life [70]. Remarkably, these effects were prevented by the co-administration of GnRH-antagonists, suggesting that AMH, which cannot cross the placental barrier, exerts its effects on the maternal brain. However, to further complicate the interpretation of the chain of events underlying the relationship between hyperandrogenism and insulin resistance, in an androgen-induced sheep model of PCOS, it was reported that the administration of the insulin sensitizer rosiglitazone may prevent the GnRH-stimulated LH hypersecretion [72].
Pathophysiological links between hyperandrogenism and insulin resistance
Consistent with data indicating that insulin may play a key role in the origin of PCOS, young women who have either congenital or acquired primary severe insulin resistance, such as those with type A or type B insulin resistance syndrome, i.e. with a genetic defect of the insulin receptor or the presence of autoantibodies impairing insulin receptor function, show all the typical symptoms of PCOS, i.e. marked hyperandrogenism, menstrual dysfunction and polycystic ovarian morphology [73]. Moreover, in women with these autoantibodies, immunosuppression induces remission of hyperandrogenism [74].
Interestingly, in a mouse model of diet-induced obesity and hyperinsulinemia, which shows impaired fertility and increased serum testosterone, mimicking human PCOS, selective knockout of the insulin receptor in theca cells showed no effect in lean normoinsulinemic animals. However, it improved the reproductive and endocrine features in obese hyperinsulinemic animals [75], suggesting that direct insulin effects on the ovary play a key role in determining these PCOS-like traits.
These findings are consistent with data in humans. Insulin stimulates androgen secretion in cultured theca cells from healthy subjects, and this phenomenon is strongly enhanced in cells from women with PCOS [76], suggesting intrinsic abnormalities. In vivo, exposure to increased insulin levels, obtained by a prolonged hyperinsulinemic euglycemic clamp, increases androgen response to GnRH agonist stimulation [77]. In addition, adrenal androgen secretion, under ACTH stimulation, is also greater when stimulation is carried out during hyperinsulinemia, induced by the glucose clamp procedure, than at baseline insulin levels [78, 79]. As a whole, these findings suggest that insulin may enhance androgen secretion in both the ovary and the adrenals.
Thus, available data support the conclusion that in women with PCOS insulin excess, secondary to still undetermined defects in metabolic insulin action, acts at several levels, ovary, adrenal, possibly pituitary, liver and also peripheral tissues, increasing androgen production and lowering SHBG synthesis, thus inducing higher free androgen levels. In accordance with this hypothesis there is also consistent evidence that insulin sensitizers, such as metformin, can improve the reproductive aspects of PCOS [80]. However, it should be noted that, among PCOS women, there are responders and non responders to metformin [81], suggesting, once more, potential heterogeneity in underlying pathogenic mechanisms.
Nevertheless, there are also data supporting the hypothesis that insulin resistance may indeed be the effect, and not the cause, in PCOS. In this regard, a couple of studies, carried out using multi-step hyperinsulinemic glucose clamps, showed a rapid onset of insulin resistance after supraphysiological androgen administration in women [82, 83]. Also in this perspective there are some supporting data from intervention studies in PCOS women, although findings are not fully consistent. In a two-step glucose clamp study, as expected, insulin-induced glucose uptake was impaired in hyperandrogenic women as compared to healthy controls. However, interestingly, this alteration improved, at least in part, after different treatments aimed at reducing either androgen levels or androgen action, which supports an effect of hyperandrogenism on insulin sensitivity [84].
Several mechanisms may potentially underlie these effects of androgens. In ovariectomized rats made hyperandrogenemic by testosterone treatment, with serum androgens similar to those found in PCOS women, insulin increased and in vivo insulin action was impaired. Interestingly, in this model, skeletal muscle fibres had shifted toward a less insulin sensitive type 2 phenotype and there was also a reduction in muscle capillary density [85]. These findings are consistent with the observation that cardiorespiratory fitness may be impaired in lean PCOS women, as compared with healthy controls [86]. Notably, in these subjects serum free testosterone, measured by gold standard methods, was an independent predictor of this impairment, after adjusting for insulin sensitivity and other confounding factors, i.e. the higher the serum free testosterone, the lower maximal oxygen consumption during exercise. These results are consistent with the hypothesis that androgens can modify the skeletal muscle fibre phenotype, shifting from the slow-oxidative to the fast-glycolytic phenotype.
Another target of androgen action with relevant potential implications in terms of altered insulin sensitivity is adipose tissue. In cultured subcutaneous adipocytes, insulin-induced glucose uptake was lowered by testosterone in a dose-dependent manner [87]. Notably, this effect was reversed by androgen receptor blockade, proving that this effect of testosterone was mediated by androgen receptor activation. Very interesting data have recently been reported by O’Reilly et al., using the in vivo microdialysis technique in subcutaneous adipose tissue. These authors reported that PCOS women had increased mRNA expression, in this tissue, of AKR1C3, the key enzyme synthesizing testosterone from its precursor androstenedione [88]. Interestingly, PCOS women, as compared to controls, had increased testosterone levels both in serum and, to a greater extent, in adipose tissue. Moreover, in PCOS women, testosterone over androstenedione ratio, index of androgen activation, was strikingly higher in subcutaneous adipocytes than in serum, proving that synthesis of activated androgens occurs in peripheral tissues of these subjects. In addition, this study showed that androgens determined activation of lipogenesis and reduction of lipolysis and fatty acid oxidation in subcutaneous adipocytes, which differs from previous findings in visceral adipocytes [89]. These alterations favour lipid accumulation and increased fat mass, and may potentially contribute to the insulin resistance of PCOS women. Interestingly, insulin excess, in turn, may boost this process, by increasing AKR1C3 expression in adipocytes [88], which is another example of the many vicious circles linking androgen excess and insulin resistance in these women.
Gonzalez and coworkers have carried out, in the last few years, a series of studies on the short-term effects of experimental hyperandrogenism in healthy women. Interestingly, in these subjects, they observed an increase, both at fasting and after oral glucose, of the expression in mononuclear cells of activated NF-kB, a molecule that plays a key role in metabolic inflammation [90]. In accordance with this finding, plasma TNFalpha also increased. These data support the hypothesis that androgen excess may induce inflammation, both directly and by enhancing the effects of nutrients on this phenomenon, providing evidence of another androgen-driven mechanism that can potentially contribute to impairing insulin action.
Another typical finding in subjects with insulin resistance is metabolic inflexibility, i.e. an impairment in the ability of the mitochondria to switch from fat to carbohydrate oxidation, and vice versa, in response to changes in physiological conditions. This phenomenon was also found in women with PCOS, as compared with healthy controls [91], indicating that these subjects have persistent oxidation of a mixture of glucose and fatty acids, regardless of the nutritional condition. This may imply an alteration in energy homeostasis and cellular dysfunction. Notably, in these women metabolic inflexibility was independently associated with serum testosterone, suggesting that androgens may play a role also in this metabolic abnormality.
Interestingly, the hypothesis that androgens may have a primary role in the association with insulin resistance is also supported by recent results obtained using genome-wide association analyses, in 425,097 individuals of European ancestry included in the UK Biobank. This study concluded that genetic determinants of higher testosterone predicted metabolic diseases in women, independent of SHBG [92].
Conclusions
Putting together all the pieces of this complex puzzle, unfortunately we are unable to conclude which is the chicken and which is the egg in the association between insulin resistance and PCOS. This condition is heterogeneous, and the different phenotypes of PCOS could indeed have different origins. We know that insulin resistance and the associated hyperinsulinemia have the capacity to induce both the endocrine and reproductive traits of PCOS. However, androgen excess, in turn, may impair insulin action, directly and/or through several changes occurring in different body sites, especially muscle and adipose tissue. Moreover, body fat excess itself can contribute to worsening the whole picture. These alterations may develop in different moments, and probably there is fetal programming of these alterations, in which androgens and AMH may play a key role. However, there are a number of vicious circles in this process, with bidirectional relationships between androgen excess, adipose dysfunction, insulin resistance, and other actors, such as inflammation and oxidative stress, which crowd the scene and make it extremely difficult to understand where the guilt lies.
References
Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group (2004) Revised 2003 consensus on diagnostic criteria and long term health risks related to polycystic ovary syndrome (PCOS). The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group. Hum Reprod 19:41–47
Lizneva D, Suturina L, Walker W, Brakta S, Gavrilova-Jordan L, Azziz R (2016) Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril 106:6–15
World Health Organization (1999) Definition, Diagnosis and classification of diabetes mellitus and its complications. Report of a WHO consultation. Part 1: diagnosis and classification of diabetes mellitus. World Health Organization, Geneva
Tosi F, Bonora E, Moghetti P (2017) Insulin resistance in a large cohort of women with polycystic ovary syndrome: a comparison between euglycaemic-hyperinsulinaemic clamp and surrogate indexes. Hum Reprod 32:2515–2521
Moghetti P, Tosi F, Bonin C, Di Sarra D, Fiers T, Kaufman JM, Giagulli VA, Signori C, Zambotti F, Dall'Alda M, Spiazzi G, Zanolin ME, Bonora E (2013) Divergences in insulin resistance between the different phenotypes of the polycystic ovary syndrome. J Clin Endocrinol Metab 98:E628–637
Apridonidze T, Essah PA, Iuorno MJ, Nestler JE (2005) Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab 90:1929–1935
Cassar S, Misso ML, Hopkins WG, Shaw CS, Teede HJ, Stepto NK (2016) Insulin resistance in polycystic ovary syndrome: a systematic review and metaanalysis of euglycaemic hyperinsulinaemic clamp studies. Hum Reprod 31:2619–2631
Dunaif A, Xia J, Book CB, Schenker E, Tang Z (1995) Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Investig 96:801–810
Corbould A, Zhao H, Mirzoeva S, Aird F, Dunaif A (2006) Enhanced mitogenic signalling in skeletal muscle of women with polycystic ovary syndrome. Diabetes 55:751–759
Diamanti-Kandarakis E, Dunaif A (2012) Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 33:981–1030
Ciaraldi TP (2000) Molecular defects of insulin action in the polycystic ovary syndrome: possible tissue specificity. J Pediatr Endocrinol Metab 13(Suppl 5):1291–1293
Højlund K, Glintborg D, Andersen NR, Birk JB, Treebak JT, Frøsig C, Beck-Nielsen H, Wojtaszewski JF (2008) Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. Diabetes 57:357–366
Hansen SL, Svendsen PF, Jeppesen JF, Hoeg LD, Andersen NR, Kristensen JM, Nilas L, Lundsgaard AM, Wojtaszewski JFP, Madsbad S, Kiens B (2019) Molecular mechanisms in skeletal muscle underlying insulin resistance in women who are lean with polycystic ovary syndrome. J Clin Endocrinol Metab 104:1841–1854
Nelson-Degrave VL, Wickenheisser JK, Hendricks KL, Asano T, Fujishiro M, Legro RS, Kimball SR, Strauss JF 3rd, McAllister JM (2005) Alterations in mitogen-activated protein kinase kinase and extracellular regulated kinase signaling in theca cells contribute to excessive androgen production in polycystic ovary syndrome. Mol Endocrinol 19:379–390
Dumesic DA, Hoyos LR, Chazenbalk GD, Naik R, Padmanabhan V, Abbott DH (2020) Mechanisms of intergenerational transmission of polycystic ovary syndrome. Reproduction 159:R1–R13
Johnson T, Kaplan L, Ouyang P, Rizza P (2012) National Institutes of Health evidence-based methodology workshop on polycystic ovary syndrome. NIH EbMW reports. National Institutes of Health, Bethesda 1:1–14. https://www.prevention.nih.gov/sites/default/files/2018-06/FinalReport.pdf. Accessed 31 Mar 2020
Holte J, Bergh T, Berne C, Berglund L, Lithell H (1994) Enhanced early insulin response to glucose in relation to insulin resistance in women with polycystic ovary syndrome and normal glucose tolerance. J Clin Endocrinol Metab 78:1052–1058
Stepto NK, Cassar S, Joham AE, Hutchison SK, Harrison CL, Goldstein RF, Teede HJ (2013) Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic–hyperinsulinaemic clamp. Hum Reprod 28:777–784
Carmina E, Bucchieri S, Esposito A, Del Puente A, Mansueto P, Orio F, Di Fede G, Rini G (2007) Abdominal fat quantity and distribution in women with polycystic ovary syndrome and extent of its relation to insulin resistance. J Clin Endocrinol Metab 92:2500–2505
Dumesic DA, Akopians AL, Madrigal VK, Ramirez E, Margolis DJ, Sarma MK, Thomas AM, Grogan TR, Haykal R, Schooler TA, Okeya BL, Abbott DH, Chazenbalk GD (2016) Hyperandrogenism accompanies increased intra-abdominal fat storage in normal weight polycystic ovary syndrome women. J Clin Endocrinol Metab 101:4178–4188
Mannerås-Holm L, Leonhardt H, Kullberg J, Jennische E, Odén A, Holm G, Hellström M, Lönn L, Olivecrona G, Stener-Victorin E, Lönn M (2011) Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab 96:E304–311
Tosi F, Di Sarra D, Kaufman JM, Bonin C, Moretta R, Bonora E, Zanolin E, Moghetti P (2015) Total body fat and central fat mass independently predict insulin resistance but not hyperandrogenemia in women with polycystic ovary syndrome. J Clin Endocrinol Metab 100:661–669
Barber TM, Hanson P, Weickert MO, Franks S (2019) Obesity and polycystic ovary syndrome: implications for pathogenesis and novel management strategies. Clin Med Insights Reprod Health 13:1179558119874042
Lizneva D, Kirubakaran R, Mykhalchenko K, Suturina L, Chernukha G, Diamond MP, Azziz R (2016) Phenotypes and body mass in women with polycystic ovary syndrome identified in referral versus unselected populations: systematic review and meta-analysis. Fertil Steril 106:1510–1520.e2
Brower MA, Hai Y, Jones MR, Guo X, Chen Y-DI, Rotter JI, Krauss RM, Legro RS, Azziz R, Goodarzi MO (2019) Bidirectional Mendelian randomization to explore the causal relationships between body mass index and polycystic ovary syndrome. Hum Reprod 34:127–136
Zhao Y, Xu Y, Wang X, Xu L, Chen J, Gao C et al (2020) Body mass index and polycystic ovary syndrome: a 2-sample bidirectional Mendelian randomization study. J Clin Endocrinol Metab 105:dgaa25
Joo YY, Actkins K, Pacheco JA, Basile AO, Carroll R, Crosslin DR et al (2020) A polygenic and phenotypic risk prediction for polycystic ovary syndrome evaluated by phenome-wide association studies. J Clin Endocrinol Metab 105:dgz326
Moran LJ, Misso ML, Wild RA, Norman RJ (2010) Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update 16:347–363
Gambineri A, Patton L, Altieri P, Pagotto U, Pizzi C, Manzoli L, Pasquali R (2012) Polycystic ovary syndrome is a risk factor for type 2 diabetes: results from a long-term prospective study. Diabetes 61:2369–2374
Glintborg D, Hass Rubin K, Nybo M, Abrahamsen B, Andersen M (2015) Morbidity and medicine prescriptions in a nationwide Danish population of patients diagnosed with polycystic ovary syndrome. Eur J Endocrinol 172:627–638
Kakoly NS, Earnest A, Teede HJ, Moran LJ, Joham AE (2019) The impact of obesity on the incidence of type 2 diabetes among women with polycystic ovary syndrome. Diabetes Care 42:560–567
Conn JJ, Jacobs HS, Conway GS (2000) The prevalence of polycystic ovaries in women with type 2 diabetes mellitus. Clin Endocrinol (Oxf) 52:81–86
Peppard HR, Marfori J, Iuorno MJ, Nestler JE (2001) Prevalence of polycystic ovary syndrome among premenopausal women with type 2 diabetes. Diabetes Care 24:1050–1052
Zargar AH, Gupta VK, Wani AI, Masoodi SR, Bashir MI, Laway BA, Ganie MA, Salahuddin M (2005) Prevalence of ultrasonography proved polycystic ovaries in North Indian women with type 2 diabetes mellitus. Reprod Biol Endocrinol 3:35
Kelestimur F, Unluhizarci K, Baybuga H, Atmaca H, Bayram F, Sahin Y (2006) Prevalence of polycystic ovarian changes and polycystic ovary syndrome in premenopausal women with treated type 2 diabetes mellitus. Fertil Steril 86:405–410
Marchetti P, Bugliani M, Boggi U, Masini M, Marselli L (2012) The pancreatic beta cells in human type 2 diabetes. Adv Exp Med Biol 771:288–309
Pasquali R, Gambineri A (2018) New perspectives on the definition and management of polycystic ovary syndrome. J Endocrinol Investig 41:1123–1135
Tosi F, Fiers T, Kaufman JM, Dall'Alda M, Moretta R, Giagulli VA, Bonora E, Moghetti P (2016) Implications of androgen assay accuracy in the phenotyping of women with polycystic ovary syndrome. J Clin Endocrinol Metab 101:610–618
Mooradian AD, Morley JE, Korenman SG (1987) Biological actions of androgens. Endocr Rev 8:1–28
O'Reilly MW, Kempegowda P, Jenkinson C, Taylor AE, Quanson JL, Storbeck KH, Arlt W (2017) 11-Oxygenated C19 steroids are the predominant androgens in polycystic ovary syndrome. J Clin Endocrinol Metab 102:840–848
Rege J, Turcu AF, Kasa-Vubu JZ, Lerario AM, Auchus GC, Auchus RJ, Smith JM, White PC, Rainey WE (2018) 11-Ketotestosterone is the dominant circulating bioactive androgen during normal and premature adrenarche. J Clin Endocrinol Metab 103:4589–4598
Turcu AF, Nanba AT, Chomic R, Upadhyay SK, Giordano TJ, Shields JJ, Merke DP, Rainey WE, Auchus RJ (2016) Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur J Endocrinol 174:601–609
Dahan MH, Abbasi F, Reaven G (2019) Relationship between surrogate estimates and direct measurement of insulin resistance in women with polycystic ovary syndrome. J Endocrinol Investig 42:987–993
Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19:608–624
Buffington CK, Kitabchi AE (1994) Evidence for a defect in insulin metabolism in hyperandrogenic women with polycystic ovarian syndrome. Metabolism 43:1367–1372
Tosi F, Dal Molin F, Zamboni F, Saggiorato E, Salvagno GL, Fiers T, Kaufman JM, Bonora E, Moghetti P (2020) Serum androgens are independent predictors of insulin clearance but not of insulin secretion in women with PCOS. J Clin Endocrinol Metab 105:dgaa095 (E-pub ahead of print)
Legro RS, Driscoll D, Strauss JF 3rd, Fox J, Dunaif A (1998) Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA 95:14956–14960
Sam S, Legro RS, Essah PA, Apridonidze T, Dunaif A (2006) Evidence for metabolic and reproductive phenotypes in mothers of women with polycystic ovary syndrome. Proc Natl Acad Sci USA 103:7030–7035
Sir-Petermann T, Codner E, Pérez V, Echiburú B, Maliqueo M, Ladrón de Guevara A, Preisler J, Crisosto N, Sánchez F, Cassorla F, Bhasin S (2009) Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 94:1923–1930
Yilmaz B, Vellanki P, Ata B, Yildiz BO (2018) Diabetes mellitus and insulin resistance in mothers, fathers, sisters, and brothers of women with polycystic ovary syndrome: a systematic review and meta-analysis. Fertil Steril 110:523–533
Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI (2006) Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab 91:2100–2104
Hiam D, Moreno-Asso A, Teede HJ, Laven JSE, Stepto NK, Moran LJ, Gibson-Helm M (2019) The genetics of polycystic ovary syndrome: an overview of candidate gene systematic reviews and genome-wide association studies. J Clin Med 8(10):1606
Day F, Karaderi T, Jones MR et al (2018) Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet 14:e1007813
Cannarella R, Condorelli RA, Mongioì LM, La Vignera S, Calogero AE (2018) Does a male polycystic ovarian syndrome equivalent exist? J Endocrinol Investig 41:49–57
Chen B, Xu P, Wang J, Zhang C (2019) The role of MiRNA in polycystic ovary syndrome (PCOS). Gene 706:91–96
Nilsson E, Benrick A, Kokosar M, Krook A, Lindgren E, Källman T, Martis MM, Højlund K, Ling C, Stener-Victorin E (2018) Transcriptional and epigenetic changes influencing skeletal muscle metabolism in women with polycystic ovary syndrome. J Clin Endocrinol Metab 103:4465–4477
Murri M, Insenser M, Fernández-Durán E, San-Millán JL, Luque-Ramírez M, Escobar-Morreale HF (2018) Non-targeted profiling of circulating microRNAs in women with polycystic ovary syndrome (PCOS): effects of obesity and sex hormones. Metabolism 86:49–60
Che Q, Liu M, Zhang D, Lu Y, Xu J, Lu X, Cao X, Liu Y, Dong X, Liu S (2020) Long noncoding RNA HUPCOS promotes follicular fluid androgen excess in PCOS patients via aromatase inhibition. J Clin Endocrinol Metab 105:dgaa060
Vilariño-García T, Pérez-Pérez A, Santamaría-López E, Prados N, Fernández-Sánchez M, Sánchez-Margalet V (2020) Sam68 mediates leptin signaling and action in human granulosa cells: possible role in leptin resistance in PCOS. Endocr Connect 9:479–488
Che Q, Liu M, Xu J, Liu Y, Cao X, Dong X, Liu S (2019) Characterization of circular RNA expression profiles in cumulus cells from patients withpolycystic ovary syndrome. Fertil Steril 111:1243–1251.e1
Vázquez-Martínez ER, Gómez-Viais YI, García-Gómez E, Reyes-Mayoral C, Reyes-Muñoz E, Camacho-Arroyo I, Cerbón M (2019) DNA methylation in the pathogenesis of polycystic ovary syndrome. Reproduction 158:R27–R40
Wang XX, Wei JZ, Jiao J, Jiang SY, Yu DH, Li D (2014) Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget 5:6603–6610
Lambertini L, Saul SR, Copperman AB, Hammerstad SS, Yi Z, Zhang W, Tomer Y, Kase N (2017) Intrauterine reprogramming of the polycystic ovary syndrome: evidence from a pilot study of cord blood global methylation analysis. Front Endocrinol (Lausanne) 8:352
Echiburú B, Milagro F, Crisosto N, Pérez-Bravo F, Flores C, Arpón A, Salas-Pérez F, Recabarren SE, Sir-Petermann T, Maliqueo M (2020) DNA methylation in promoter regions of genes involved in the reproductive and metabolic function of children born to women with PCOS. Epigenetics. https://doi.org/10.1080/15592294.2020.1754674
Abbott DH, Dumesic DA, Levine JE (2019) Hyperandrogenic origins of polycystic ovary syndrome—implications for pathophysiology and therapy. Expert Rev Endocrinol Metab 14:131–143
Walters KA, Gilchrist RB, Ledger WL, Teede HJ, Handelsman DJ, Campbell RE (2018) New perspectives on the pathogenesis of PCOS: neuroendocrine origins. Trends Endocrinol Metab 29:841–852
Morford JJ, Wu S, Mauvais-Jarvis F (2018) The impact of androgen actions in neurons on metabolic health and disease. Mol Cell Endocrinol 465:92–102
Navarro G, Allard C, Morford JJ et al (2018) Androgen excess in pancreatic β cells and neurons predisposes female mice to type 2 diabetes. JCI Insight 3(12):e98607
Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Pérez-Bravo F, Recabarren SE (2002) Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod 17:2573–2579
Tata B, Mimouni NEH, Barbotin AL et al (2018) Elevated prenatal anti-Müllerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med 24:834–846
Dulka EA, Moenter SM (2017) Prepubertal development of gonadotropin-releasing hormone neuron activity is altered by sex, age, and prenatal androgen exposure. Endocrinology 158:3943–3953
Cardoso RC, Burns A, Moeller J, Skinner DC, Padmanabhan V (2016) Developmental programming: insulin sensitizer prevents the GnRH-stimulated LH hypersecretion in a sheep model of PCOS. Endocrinology 157:4641–4653
Bar RS, Muggeo M, Roth J (1979) Insulin resistant syndromes in humans. Adv Exp Med Biol 124:85–104
Malek R, Chong AY, Lupsa BC, Lungu AO, Cochran EK, Soos MA, Semple RK, Balow JE, Gorden P (2010) Treatment of type B insulin resistance: a novel approach to reduce insulin receptor autoantibodies. J Clin Endocrinol Metab 95:3641–3647
Wu S, Divall S, Nwaopara A, Radovick S, Wondisford F, Ko C, Wolfe A (2014) Obesity-induced infertility and hyperandrogenism are corrected by deletion of the insulin receptor in the ovarian theca cell. Diabetes 63:1270–1282
Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F (1998) Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 83:2001–2005
Tosi F, Negri C, Perrone F, Dorizzi R, Castello R, Bonora E, Moghetti P (2012) Hyperinsulinemia amplifies GnRH agonist stimulated ovarian steroid secretion in women with polycystic ovary syndrome. J Clin Endocrinol Metab 97:1712–1719
Moghetti P, Castello R, Negri C, Tosi F, Spiazzi GG, Brun E, Balducci R, Toscano V, Muggeo M (1996) Insulin infusion amplifies 17 alpha-hydroxycorticosteroid intermediates response to adrenocorticotropin in hyperandrogenic women: apparent relative impairment of 17,20-lyase activity. J Clin Endocrinol Metab 81:881–886
Tosi F, Negri C, Brun E, Castello R, Faccini G, Bonora E, Muggeo M, Toscano V, Moghetti P (2011) Insulin enhances ACTH-stimulated androgen and glucocorticoid metabolism in hyperandrogenic women. Eur J Endocrinol 164:197–203
Morley LC, Tang T, Yasmin E, Norman RJ, Balen AH (2017) Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, d-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev 11:CD003053
Moghetti P, Castello R, Negri C, Tosi F, Perrone F, Caputo M, Zanolin E, Muggeo M (2000) Metformin effects on clinical features, endocrine and metabolic profiles, and insulin sensitivity in polycystic ovary syndrome: a randomized, double-blind, placebo-controlled 6-month trial, followed by open, long-term clinical evaluation. J Clin Endocrinol Metab 85:139–146
Polderman KH, Gooren LJ, Asscheman H, Bakker A, Heine RJ (1994) Induction of insulin resistance by androgens and estrogens. J Clin Endocrinol Metab 79:265–271
Diamond MP, Grainger D, Diamond MC, Sherwin RS, Defronzo RA (1998) Effects of methyltestosterone on insulin secretion and sensitivity in women. J Clin Endocrinol Metab 83:4420–4425
Moghetti P, Tosi F, Castello R, Magnani CM, Negri C, Brun E, Furlani L, Caputo M, Muggeo M (1996) The insulin resistance in women with hyperandrogenism is partially reversed by antiandrogen treatment: evidence that androgens impair insulin action in women. J Clin Endocrinol Metab 81:952–960
Holmäng A, Svedberg J, Jennische E, Björntorp P (1990) Effects of testosterone on muscle insulin sensitivity and morphology in female rats. Am J Physiol 259:E555–560
Bacchi E, Negri C, Di Sarra D, Tosi F, Tarperi C, Moretta R, Schena F, Bonora E, Kaufman JM, Moghetti P (2015) Serum testosterone predicts cardiorespiratory fitness impairment in normal-weight women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 83:895–901
Corbould A (2007) Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol 192:585–594
O'Reilly MW, Kempegowda P, Walsh M, Taylor AE, Manolopoulos KN, Allwood JW, Semple RK, Hebenstreit D, Dunn WB, Tomlinson JW, Arlt W (2017) AKR1C3-mediated adipose androgen generation drives lipotoxicity in women with polycystic ovary syndrome. J Clin Endocrinol Metab 102:3327–3339
Ek I, Arner P, Rydén M, Holm C, Thörne A, Hoffstedt J, Wahrenberg H (2002) A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes 51:484–492
González F, Nair KS, Daniels JK, Basal E, Schimke JM (2012) Hyperandrogenism sensitizes mononuclear cells to promote glucose-induced inflammation in lean reproductive-age women. Am J Physiol Endocrinol Metab 302:E297–306
Di Sarra D, Tosi F, Bonin C, Fiers T, Kaufman JM, Signori C, Zambotti F, Dall'Alda M, Caruso B, Zanolin ME, Bonora E, Moghetti P (2013) Metabolic inflexibility is a feature of women with polycystic ovary syndrome and is associated with both insulin resistance and hyperandrogenism. J Clin Endocrinol Metab 98:2581–2588
Ruth KS, Day FR, Tyrrell J et al (2020) Using human genetics to understand the disease impacts of testosterone in men and women. Nat Med 26:252–258
Funding
This work was supported by academic grants to Paolo Moghetti from the University of Verona.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to disclose.
Ethical approval
Ethical approval was not required for this review article.
Informed consent
Informed consent was not required for this review article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Moghetti, P., Tosi, F. Insulin resistance and PCOS: chicken or egg?. J Endocrinol Invest 44, 233–244 (2021). https://doi.org/10.1007/s40618-020-01351-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-020-01351-0