
Abstract
Background
Deficiency of 17β-hydroxysteroid dehydrogenase type 3 (17β-HSD3) is a rare autosomal recessive 46,XY disorder of sex development (DSD). It is due to pathogenetic variants in the HSD17B3 gene. Mutated genes encode an abnormal enzyme with absent or reduced ability to convert Δ4-androstenedione (Δ4-A) to testosterone (T) in the fetal testis. Affected individuals are usually raised as females and diagnosis is made at puberty, when they show virilization.
Methods
A girl with a presumptive diagnosis of complete androgen insensitivity syndrome underwent endocrine and genetic assessment. Long-term follow-up was reported.
Results
The diagnosis of 17β-HSD3 deficiency was made (stimulated T/Δ4-A ratio: 0.15; HSD17B3 gene analysis: c.277+4A>T in intron 3/c.640_645del (p.Glu214_Glu215del) in exon 9. After extensive information, parents decided to maintain female sex. Gonadal removal was performed and histological evaluation demonstrated deep fibrosis of testicular tissue. Follow-up till 8.5 years of age showed somatic and neuro-psychological development fitting with the female sex.
Conclusions
Management of a child with the rare 17β-HSD3 deficiency remains challenging. Any decision must be carefully evaluated with parents. Long-term follow-up must be warranted by a multidisciplinary DSD team to evaluate the adequacy of the choices made on quality of life in later life.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Disorders (or differences) of sex development (DSD) are congenital conditions, whereby the sex chromosome, gonads and/or genital phenotype are atypical in respect to the usual embryonic developmental pathways. The term 46,XY DSD refers to the group with 46,XY karyotype and impaired synthesis or function of androgens [1]. Among them, 17β-hydroxysteroid dehydrogenase type 3 (17β-HSD3) deficiency, also known as 17-keto-reductase deficiency (OMIM #264300), is a very rare autosomal recessive DSD at least in Western countries, whereas it is relatively frequent in some Arab populations, as that of Gaza strip [2, 3].
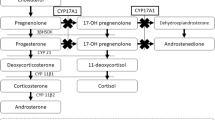
17β-HSD3 deficiency consists of a defect in the last step of testicular steroidogenesis, in which Δ4-androstenedione (Δ4-A) is converted into testosterone (T); it is due to impaired activity of 17β-HSD3 isoenzyme, which is highly expressed in the developing testes [2, 4]. Pathogenetic loss of function variants in the HSD17B3 gene are the cause of 17β-HSD3 deficiency [4, 5]. The impairment of T synthesis during foetal development results in undervirilization of genitalia in 46,XY foetuses [2, 3, 6]. Thus, newborns with 17β-HSD3 deficiency may have a phenotypic spectrum ranging from normal-appearing female external genitalia with a blind vaginal pouch to micro-phallus with hypospadias or variable degrees of genital ambiguity; the testes are often found in the inguinal canal or in a bifid scrotum/labia majora [2, 3, 6]. The Wolffian structures maybe present, almost rudimentary [2, 3, 6]. Most affected children are raised as females at least in Italy [7, 8] as in other countries, albeit some more virilised subjects are raised as males [5, 6, 9,10,11,12]. Virilization occurs at puberty: the adolescents may experience enlargement of the phallic structures, deepening of the voice, male body appearance and male secondary hair growth [2, 3, 6]. Virilization may depend on the higher levels of Δ4-A itself from puberty onward; in fact, Δ4-A is a weak androgen which binds androgen receptor and may induce androgenic effects. Indeed, T synthesis may occur through alternative 17β-HSD isoenzyme pathways or by the residual activity of mutated 17β-HSD3 enzyme [2, 4, 13].
Hormonal profile is characterized by low serum levels of T associated with increased concentrations of Δ4-A [2]. Thus, T:Δ4-A ratio less than 0.8 is considered the diagnostic hallmark of 17-βHSD3 deficiency [9], when other defects of testosterone synthesis including Leydig cell hypoplasia and testicular dysgenesis have been ruled out [14].
Here, an additional girl with proven 17-βHSD3 deficiency is reported. She was early diagnosed and follow-up till 8 years of age described.
Patients and methods
Clinical report
The girl was born at term after a first uncomplicated pregnancy and an uneventful delivery from non-consanguineous Italian healthy parents; family history was unremarkable. At birth, she showed female phenotype, but bilateral inguinal hernia. Sonography showed the presence of bilateral testes and karyotyping demonstrated a 46,XY karyotype. Diagnosis of complete androgen insensitivity syndrome (CAIS) was made. She was referred to us at the age of 2 to verify the diagnosis.
Methods
Height and weight data were expressed as SD according to WHO standards (https://www.who.int/childgrowth/software/en/). Mid-parental height was calculated using measured parental heights adjusted for female sex: father’s height + mother’s height/2 − 6.5 cm. Genital phenotype was scored using external masculinization score (EMS) [15].
Baseline blood samples for anti-müllerian hormone (AMH), LH, and FSH measurements were obtained in the fasting state between 8:00 and 9:00 a.m.; normative levels were derived from values reported in a recent review [16]. Basal and stimulated levels of Δ4-A, T, and DHT were measured by commercially available kits; hCG test was performed using recombinant hCG (Ovitrelle®, Merck Europe B.V. 250 µg) as previously described [17], adopting the normative values proposed by Oliveira et al. [18].
Genomic DNA was extracted from peripheral blood sample according to standard procedures and sequencing of exons and the intron–exon boundary of androgen receptor (AR), SRD5A2, and HSD17B3 genes was done using previously described procedures [7].
Results
At the first visit, the girl presented with normal female height and weight, fitting with her mid-parental height (Table 1). EMS was 3. Ultrasound scanning did not demonstrate any Müllerian structures. The endocrine evaluation showed slightly increased gonadotropin levels; basal steroid gonadal hormone measurements were uninformative; low values of AMH were detected (Table 2). hCG test demonstrated an increase of Δ4-A, while T increase was blunted determining a stimulated T:Δ4-A ratio below 0.8 (Table 2). Abnormal T/DHT ratio was also detected (Table 2).
Genetic analyses demonstrated the presence of two genetic variants in the HSD17B3 gene: c.277+4A>T in intron 3 and c.640_645del in exon 9, the latter causing the deletion of two amminoacids in the protein (p.Glu214_Glu215del). The former variant was inherited from the father and it is reported as causative of 17β-HSD3 deficiency [5, 9, 12, 19], while the latter was inherited from the mother and has not been identified before, to the best of our knowledge (Human Gene Mutation Database: https://www.hgmd.cf.ac.uk/ac/all.php; LOVD3 database: https://databases.lovd.nl/shared/variants/HSD17B3/unique). No variants were found in AR or in SRD5A2 genes. After ample discussion regarding the diagnosis of 17-βHSD3 deficiency, including reported evolution at puberty and fertility issues as well as management options, the parents chose to maintain female sex after accurate neuro-psychological evaluation of the girl; the decision to remove gonads were also done. Histological evaluation demonstrated immature male gonads with deep fibrosis of tunica vaginalis and interstitium. Ectopic adrenal rest is present near to left epididymis. Findings of neoplastic degeneration were not found. The girl continued to grow according to her female percentile; clinical data at last evaluation are in Table 1; during follow-up, she showed age and female sex appropriate neuropsychological development, choose girl's clothes and played with typical girl’s toys.
Discussion
Here, an additional Italian girl with 17-βHSD3 deficiency is reported. A wrong diagnosis of CAIS was firstly performed, as it is not unusual for this disease [7, 9,10,11,12, 19]. The endocrine evaluation may also suggest a 5α-reductase-2 deficiency, that is the other main differential diagnosis [3, 6, 7, 9,10,11,12]. Thus, molecular analyses are often mandatory to reach a definite diagnosis.
17-βHSD3 deficiency is considered the commonest DSD due to a biosynthetic testosterone defect (3, 6, 7), but it is very rare at least in European countries. A frequency of 1:147.000 was estimated in a Dutch study [9]. In Italy, only 11 patients were collected in a multicentric study of the Italian DSD Study Group, which is an association connecting the main Italian centres involved in the care of DSD [8]. The large dsd-LIFE study collecting data from 14 centres in six European countries (France, Germany, the Netherlands, Poland, Sweden, United Kingdom) also individuated 11 patients with 17-βHSD3 deficiency (~ 0.4% of all 46,XY DSD group) [20]. In our previous paper, five girls were reported and two of them were from the same area of the present one [7], suggesting a higher frequency of this 46,XY DSD in some regions of South Italy. Anyway, no founder effect seems to operative because different genetic variants were found in this girl and the other ones coming from the same region (Table 3). As far as we know, there are 67 mutations identified in this gene at this time (https://www.hgmd.cf.ac.uk/ac/gene.php?gene=HSD17B3). These mutations include intronic splice sites, exonic deletions, missense and non-sense mutations, four of which are small deletions. The present study describes a girl that proved to be a compound heterozygote for the gene responsible for 17βHSD3 production. The genetic variants were inherited from the parents. The c.277+4A>T in intron 3 has been previously reported in several patients of various countries [9,10,11,12, 17] and it may be spread worldwide by North European common founders [9]. The c.277+4A>T variant was demonstrated to be pathogenetic by disrupting normal splicing [5, 9]. The c.640_645del (rs747176985) is a new in frame variant in exon 9 of gene HSD17B3 (NM_000197.1), it is not located in a repeat region and determines the loss of two Glu residues (p.Glu214_Glu215del), its GnomAD exomes allele frequency is 0.00000812, smaller than the threshold for the recessive HSD17B3 gene. Although mutations throughout the gene have been described, a mutation cluster region in exon nine with complete elimination of 17β-HSD3 activity was identified in many populations. Our variant is not reported in ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar) and in other Locus Specific database (HGMD and LOVD3) and in silico predictions indicated its pathogenic role (ACMG classifications: Pathogenic computational verdict because of one pathogenic prediction from GERP vs no benign predictions). Functional studies will be needed to confirm the pathogenetic role of the unreported variant in exon 9 of HSD17B3 gene.
Decision on the sex of rearing is challenging in several 46,XY DSDs. The Chicago Consensus indicates that diagnosis, genital appearance, surgical options, need for life-long replacement therapy, the potential for fertility, views of the family and circumstances relating to cultural practices must be considered [1]. The Global DSD Update Consortium recommended male sex assignment [21]. Indeed, such conclusion was likely based on data of the Arab population of Gaza strip [2]. Gaza people with 17-βHSD3 deficiency are usually reared as girl before puberty, when they undergo to virilization and the majority adopt a male gender identity [2]. This scenario may be related to local social and cultural influences [2, 6]. In several countries, most children with 17-βHSD3 deficiency and almost female genitalia are raised as girls and underwent gonadal surgery [5, 7, 9,10,11,12, 22], as in the present girl, who presented female somatic and behavioural growth pattern during follow-up as also reported in other cases [10, 23]. The long term outcome of these patients remains largely unknown, but a recent paper reported that girls with early gonadal removal retained female gender identity with follow-up extending to 26 years of age [10]. These authors concluded that there is insufficient data to support male gender reassignment in girls with very early diagnosis and that early gonadal removal may be an option in view of retention of female sex [10]. This conclusion is in line with our own experience with the girls of our previous paper [7], who now reached young adulthood without any signs of gender dysforia (unreported data). The same Global DSD Update Consortium stated that there is evidence of satisfactory sexual function both among people with 17-βHSD3 deficiency raised as males and those raised as females [21]. Sex difference in brain development and postnatal behaviour are largely determined by in utero gonadal secretion; T exposure largely contributes for guiding one’s sexual orientation and to a lesser extend gender identity [24]. The present girl likely received very low androgen exposure during intrauterine life and in the postnatal minipuberty, because the phenotype was minimum masculinized and testosterone production assessed in infancy was severely impaired. Anyway, data remain scarce and longer follow-up in larger homogeneous samples selected on the basis of objective clinical score, as EMS or structural variation of the brain by MR scanning, are needed. Fertility issue is an additional point, that must be taken into account. While spontaneous or assisted paternity has been documented in 5α-reductase 2 deficiency [25]—that is due to impaired conversion of T to DHT—fertility is poorly addressed in 17-βHSD3 deficiency [10]. To the best of our knowledge, only an Arab male patient self-reported fatherhood [26]. Indeed, testicular histology is often not consistent with reproductive possibility and spermatogenesis is absent in adulthood [2, 10, 11]; in the present girl, hormonal parameter predictive of reproductive function [27] were very poor and severe fibrosis of testicular tissue was detected. Histological data did not show evidence of malignancy at surgery, but immunostaining for the presence of precursor malignant lesions by recent biomarkers were not performed [10, 11, 22]. The risk of germ cell tumours has been estimated as 28% in this DSD [28]. Indeed, the overall risk of gonadal neoplasia in patients with 17-βHSD3 deficiency is currently largely unknown, due to the paucity data on gonadal histology. Two recent reviews reported seven patients with malignancy: patients showed findings of carcinoma in situ and two patients (21 and 61 years old) presented with Leydig cell tumour [29, 30]. The youngest reported case was 13 years old and it has been suggested that cell abnormalities can be present from the second decade of life [29]. Thus, they may be undetectable at gonadal surgery in the present girl. Anyway, patients who maintained the gonads may require surgical, medical or radiological therapies adjunctively impairing endocrine function and fertility in adolescence or adulthood.
Conclusions
Jürgensen et al. [23] pointed out that there are no generally established “right” or “wrong” decision in the difficult field of sex assignment in some rare 46,XY DSD as 17β-HSD deficiency, but every family has to find its own path when dealing with the specific nature of their child [29]. At any rate, the decision about sex of rearing should be made in light of the best possible prediction of future sexual function, virilization, and satisfaction with gender identity. At this regard, it has been suggested that early gonadal removal likely results in retention of female gender identity at adolescence in very poor virilised children [10]. A more conservative approach regarding gonads may be done today, managing puberty with GnRH analogs to permit the involvement of affected persons in the decision-making process, allowing them to better explore their gender identity [31].
References
Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group, ESPE Consensus Group (2006) Consensus statement on management of intersex disorders. Arch Dis Child 91:554–563. https://doi.org/10.1136/adc.2006.098319
Rosler A (2006) 17β-Hydroxysteroid dehydrogenase 3 deficiency in the Mediterranean population. Pediatr Endocrinol Rev 3(Suppl 3):455–461
Faienza MF, Giordani L, Delvecchio M, Cavallo L (2008) Clinical, endocrine, and molecular findings in 17β-hydroxysteroid dehydrogenase type 3 deficiency. J Endocrinol Investig 31:85–91. https://doi.org/10.1007/bf03345572
Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, Elliston KO, Wilson JD, Russell DW, Andersson S (1994) Male pseudo-hermaphroditism caused by mutations of testicular. Nat Genet 1994(7):34–39. https://doi.org/10.1038/ng0594-34
Andersson S, Geissler WM, Wu L, Davis DL, Grumbach MM, New MI, Schwarz HP, Blethen SL, Mendonca BB, Bloise W, Witchel SF, Cutler GB Jr, Griffin JE, Wilson JD, Russel DW (1996) Molecular genetics and pathophysiology of 17β-hydroxysteroid dehydrogenase 3 deficiency. J Clin Endocrinol Metab 81:130–136. https://doi.org/10.1210/jcem.81.1.8550739
George MM, New MI, Ten S, Sultan C, Bhangoo A (2010) The clinical and molecular heterogeneity of 17βHSD-3 enzyme deficiency. Horm Res Paediatr 74:229–240. https://doi.org/10.1159/000318004
Bertelloni S, Balsamo A, Giordani L, Fischetto R, Russo G, Del Vecchio M, Gennari M, Nicoletti A, Maggio MC, Concolino D, Cavallo L, Cicognani A, Chiumello G, Hiort O, Baroncelli GI, Faienza MF (2009) 17β-Hydroxysteroid dehydrogenase-3 deficiency: from pregnancy to adolescence. J Endocrinol Investig 32:666–670. https://doi.org/10.1007/bf03345738
Meroni SLC, Bertelloni S, Di Mase R, Balsamo A, Baldazzi L, Baldinotti F, Salerno M, Marchese P, Stancampiano MR, Ungaro C, Russo G (2017) Deficit di 17β-idrossisteroido-deidrogenasi tipo 3 (17βHSD3): report italiano multicentrico del “It-Dsd Study Group”. In: Procceding XXX Congresso Nazionale SIEDP, Padova, September 27–29, abs 141
Boehmer ALM, Brinkmann AO, Sandkuijl LA, Halley DJJ, Niermeijer MF, Andersson S, de Jong FH, Kayserili H, de Vroede MA, Otten BJ, Rouwe CW, Mendonca BB, Rodrigues C, Bode HH, de Ruiter PE, Delemarre-van de Waal HA, Drop SLS (1999) 17β-Hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J Clin Endocrinol Metab 84:4713–4721. https://doi.org/10.1210/jcem.84.12.6174
Chuang J, Vallerie A, Breech L, Saal HM, Alam S, Crawford P, Rutter MM (2013) Complexities of gender assignment in 17β-hydroxysteroid dehydrogenase type 3 deficiency: is there a role for early orchiectomy? Int J Pediatr 2013(1):15. https://doi.org/10.1186/1687-9856-2013-15
Lee YS, Kirk JM, Stanhope RG, Johnston DI, Harland S, Auchus RJ, Andersson S, Hughes IA (2007) Phenotypic variability in 17β-hydroxysteroid dehydrogenase-3 deficiency and diagnostic pitfalls. Clin Endocrinol 67:20–28. https://doi.org/10.1111/j.1365-2265.2007.02829.x
Castro CC, Guaragna-Filho G, Calais FL, Coeli FB, Leal IR, Cavalcante-Junior EF, Monlleó IL, Pereira SR, Silva RB, Gabiatti JR, Marques-de-Faria AP, Maciel-Guerra AT, Mello MP, Guerra-Junior G (2012) Clinical and molecular spectrum of patients with 17β-hydroxysteroid dehydrogenase type 3 (17-β-HSD3) deficiency. Arq Bras Endocrinol Metabol 56:533–539. https://doi.org/10.1590/s0004-27302012000800012
Werner R, Kulle A, Sommerfeld I, Riepe FG, Wudy S, Hartmann MF, Merz H, Döhnert U, Bertelloni S, Holterhus PM, Hiort O (2012) Testosterone synthesis in patients with 17β-hydroxysteroid dehydrogenase 3 deficiency. Sex Dev 6:161–168. https://doi.org/10.1159/000336605
Faisal Ahmed S, Iqbal A, Hughes IA (2000) The testosterone:androstenedione ratio in male undermasculinization. Clin Endocrinol 53:697–702
Ahmed SF, Khwaja O, Hughes IA (2000) The role of a clinical score in the assessment of ambiguous genitalia. BJU Int 85:120–124. https://doi.org/10.1046/j.1365-2265.2000.01166.x
Fanelli F, Baronio F, Ortolano R, Mezzullo M, Cassio A, Pagotto U, Balsamo A (2018) Normative basal values of hormones and proteins of gonadal and adrenal functions from birth to adulthood. Sex Dev 12:50–94. https://doi.org/10.1159/000486840
Bertelloni S, Russo G, Baroncelli GI (2018) Human chorionic gonadotropin test: old uncertainties, new perspectives, and value in 46, XY disorders of sex development. Sex Dev 12:41–49. https://doi.org/10.1159/000481552
Oliveira LR, Homma TK, Woloszynek RR, Brito VN, Longui CA (2016) Gonadal response after a single-dose stimulation test with recombinant human chorionic gonadotropin (rhCG) in patients with isolated prepubertal cryptorchidism. Basic Clin Androl 26:13. https://doi.org/10.1186/s12610-016-0039-2(eCollection 2016)
Twesten W, Holterhus P, Sippell WG, Morlot M, Schumacher H, Schenk B, Hiort O (2000) Clinical, endocrine, and molecular genetic findings in patients with 17β-hydroxysteroid dehydrogenase deficiency. Horm Res 53:26–31. https://doi.org/10.1159/000023509
Röhle R, Gehrmann K, Szarras-Czapnik M, Claahsen-van der Grinten H, Pienkowski C, Bouvattier C, Cohen-Kettenis P, Nordenström A, Thyen U, Köhler B, dsd-LIFE group (2017) Participation of adults with disorders/differences of sex development (DSD) in the clinical study dsd-LIFE: design, methodology, recruitment, data quality and study population. BMC Endocr Disord 17:52. https://doi.org/10.1186/s12902-017-0198-y
Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S, Global DSD Update Consortium (2016) Global disorders of sex development update since 2006: perceptions, approach and care. Horm Res Paediatr 85:158–180. https://doi.org/10.1159/000442975
Alikaşifoğlu A, Vurallı D, Hiort O, Gönç N, Özön A, Kandemir N (2015) Severe undervirilisation in a 46, XY case due to a novel mutation in HSD17B3 gene. J Clin Res Pediatr Endocrinol 7:249–252. https://doi.org/10.4274/jcrpe.2069
Jürgensen M, Hampel E, Hiort O, Thyen U (2006) “Any decision is better than none” decision-making about sex of rearing for siblings with 17β-hydroxysteroid-dehydrogenase-3 deficiency. Arch Sex Behav 35:359–371. https://doi.org/10.1007/s10508-006-9034-6
Bramble MS, Lipson A, Vashist N, Vilain E (2017) Effects of chromosomal sex and hormonal influences on shaping sex differences in brain and behavior: lessons from cases of disorders of sex development. J Neurosci Res 95:65–74. https://doi.org/10.1002/jnr.23832
Bertelloni S, Baldinotti F, Baroncelli GI, Caligo MA, Peroni D (2019) Paternity in 5α-reductase-2 deficiency: report of two brothers with spontaneous or assisted fertility and literature review. Sex Dev 13:55–59. https://doi.org/10.1159/000497400
Farkas A, Rosler A (1993) Ten years experience with masculinizing genitoplasty in male pseudohermaphroditism due to 17β-hydroxysteroid dehydrogenase deficiency. Eur J Pediatr 152(Suppl 2):S88–90. https://doi.org/10.1007/bf02125448
Freire AV, Grinspon RP, Rey RA (2018) Importance of serum testicular protein hormone measurement in the assessment of disorders of sex development. Sex Dev 12:30–40. https://doi.org/10.1159/000479572
Cools M, Looijenga LH, Wolffenbuttel KP, Drop SL (2009) Disorders of sex development: update on the genetic background, terminology and risk for the development of germ cell tumors. World J Pediatr 5:93–102. https://doi.org/10.1007/s12519-009-0020-7
Folsom LJ, Hjaige M, Liu J, Eugster EA, Auchus RJ (2019) Germ cell neoplasia in situ complicating 17β-hydroxysteroid dehydrogenase type 3 deficiency. Mol Cell Endocrinol 489:3–8. https://doi.org/10.1016/j.mce.2018.11.014
Yang Z, Ye L, Wang W, Zhao Y, Wang W, Jia H, Dong Z, Chen Y, Wang W, Ning G, Sun S (2017) 17β-Hydroxysteroid dehydrogenase 3 deficiency: three case reports and a systematic review. J Steroid Biochem Mol Biol 174:141–145. https://doi.org/10.1016/j.jsbmb.2017.08.012(Epub ahead of print)
Cocchetti C, Ristori J, Mazzoli F, Prunas A, Bertelloni S, Magini A, Vignozzi L, Maggi M, Fisher AD (2020) 5α-Reductase-2 deficiency: is gender assignment recommended in infancy? Two case-reports and review of the literature. J Endocrinol Investig. https://doi.org/10.1007/s40618-020-01193-w
Funding
This study did not receive any specific fund or grant.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
For this type of study, clinical standard procedures for the investigation of people with DSD in our Departments were used, so that formal consent is not required. The clinical investigations were in accordance with the ethical standards of the Helsinki Declaration.
Informed consent
Written informed consent was obtained from the parents before any clinical procedures. No further measures were taken beyond those of routine clinical practice.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Faienza, M.F., Baldinotti, F., Marrocco, G. et al. 17β-hydroxysteroid dehydrogenase type 3 deficiency: female sex assignment and follow-up. J Endocrinol Invest 43, 1711–1716 (2020). https://doi.org/10.1007/s40618-020-01248-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-020-01248-y