
Abstract
Purpose of Review
Colonization of the gastrointestinal (GI) tract with Candida albicans (CA), the most common human fungal pathogen, is the first step towards the development of invasive infection. Yet, the fungal virulence factors and host factors that modulate CA GI colonization are still poorly understood. In this review, we will review emerging evidence of the importance of select CA genetic determinants and CA’s interaction with the host that contribute to its successful adaptation as a pathobiont in the human GI tract.
Recent Findings
Recent data reveal the importance of (1) CA genetic determinants, (2) host factors, and (3) environmental factors in modulating CA GI colonization in humans.
Summary
As evidence continues to grow supporting the notion that the GI tract and its resident microbiota are an integral part of the host immune system, it will be critical for studies to interrogate the interaction of CA with the host (including both the host innate and adaptive immune system as well as the endogenous gut microbiota) in order to dissect the mechanisms of CA pathogenesis and thus lay the foundation for novel therapeutic approaches to prevent and/or treat invasive fungal infections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Candida albicans (CA), the most common human fungal pathogen, manifests as a number of distinct infectious disease phenotypes including a mucosal infection (oral candidiasis), localized organ infections (dermatitis or vaginitis), chronic or persistent infection (chronic mucocutaneous candidiasis), and acute invasive/disseminated infections, which will be the focus of this review.
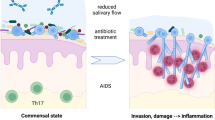
As with many invasive infections, the first or antecedent step of CA invasive infection requires colonization of a host mucosal surface. In fact, 95% percent of all infectious agents enter through mucosal surfaces, most notably the linings of the respiratory, gastrointestinal (GI), and genitourinary tracts [1]. Interestingly, humans are considered a natural reservoir for CA, with the genitourinary tract and GI tract being the main repository of CA. CA GI colonization alone does not induce a pathophysiologic state in either humans or other mammals (e.g., mice)—despite reports in the lay press and internet attributing symptoms of fatigue and malaise to “Candida overgrowth.” CA is often classified as a commensal organism, in that it does not provide any known direct benefit to the mammalian host but itself likely benefits itself from possible nutrient access and a host niche or reservoir. CA is better defined as a pathobiont, a potentially pathological organism which, under normal circumstances, lives as a commensal or symbiont [2].
The major concern for pathobionts residing in the GI tract is that these microbes will translocate to extraintestinal organs, notably the liver and spleen, and ultimately in the bloodstream. In cancer and stem cell transplant patients, CA colonizes the GI tract with subsequent translocation into extraintestinal organs in the setting of chemotherapy-induced neutropenia and GI mucosal damage [3]. In these patients, the role of the gut as a source for disseminated candidiasis was first suggested by older autopsy studies [4] and recently substantiated by molecular methods [5•]. Interestingly, Candida parapsilosis bloodstream isolates do not correspond to rectal isolates [5•], confirming prior studies suggesting that C. parapsilosis infections do not originate from the GI tract [4] and also highlighting the important of Candida species-specific differences.
Three factors are critical for preventing both bacterial and fungal GI translocation in humans and mice: (1) a balanced gut microbiome, with low abundance of pathobiont bacteria and/or fungi; (2) robust intestinal barrier function, and (3) intact cellular immunity, particularly neutrophils [6,7,8,9]. It is becoming evident that all three factors can be modulated by (1) the host (innate and adaptive immune responses, mucosal and cellular immune responses); (2) the pathobiont itself, specifically genetic determinants that promote either GI colonization or dissemination; and finally, (3) the gut microbiome and its effect on both the host and the pathobiont.
Therefore, understanding the conditions both in CA and in the host that promote the change from commensal to pathogen could lead to significant insights into CA invasive infection pathophysiology. Hence, this review will focus on recent insights regarding (1) CA genetic determinants; (2) host factors, including the gut microbiome; and (3) environmental factors that modulate CA’s ability to colonize the mammalian GI tract.
CA Genetic Determinants of GI Colonization in the Mammalian Host
Morphogenesis
CA is a dimorphic fungus that can exist in the yeast (round/oval) and the filamentous/hyphal form. The ability to transition between the yeast and hyphal form (morphogenesis) has been intimately tied to CA virulence (e.g., CA mutants unable to filament are less virulent) [10]. Our group has shown that filament-locked CA is actually less virulent in a murine model of CA GI colonization and dissemination after immunosuppression [11••]. The decreased virulence in our preclinical model, however, was most likely secondary to a deficiency in GI colonization of the mutant (2–3 log fold lower than the wildtype CA strain), as both bacterial and fungal GI translocation is directly proportional to microbial gut burden [11••, 12••, 13]. These data suggest that the hyphal form of CA may be less suited for GI colonization.
Indeed, a recent study showed that CA almost uniformly adopts the yeast form in germ-free mice [14••]. Interestingly, CA mutants lacking the transcription regulator genes ZCF8, ZFU2, and TRY4 had reduced fitness in the GI tract of germ-free mice, which was attributable to a predilection for the filamentous form. Finally, ZCF8, ZFU2, and TRY4 promote CA adherence in the gut in a mucin-dependent fashion. As for a teleological explanation as to why the yeast form would be preferable in the mammalian GI tract, the authors postulate that the hyphal or filamentous form may be more immunogenic and thus less beneficial for the survival of CA in the gut—supported by the observation that a host immune effector (granulocyte colony stimulating factor) was significantly more abundant in intestinal tissue in mice colonized with filamentous CA compared to counterparts colonized with the yeast form [14••].
GUT Morphology
In terms of other CA-specific adaptive responses which would promote GI colonization, an unusual morphology, termed the gastrointestinally induced transition (GUT) phenotype [15••, 16], has been described as a specialized form of CA adapted to the GI tract. Noble and colleagues noted that the introduction of CA into the mammalian GI tract triggers a developmental switch, driven by the Wor1 transcription factor, to this commensal cell type. CA Wor1 deletion mutants showed a significant GI fitness disadvantage compared to wildtype CA. Overexpression of Wor1 resulted in a gain of function phenotype and a competitive advantage over the wildtype strain. Of note, Wor1 had previously been shown to be important for controlling CA white-opaque switch in mating [17, 18], and only rare cell types had been shown to be competent for WOR1 expression in vitro. Overexpression of Wor1 has also been shown to increase susceptibility to bile salts, which may explain a transient defect in the initial stages of GI colonization observed by different groups [16, 19] and also enhanced adhesion to the murine GI mucosa [19], perhaps explaining the competitive fitness advantage exhibited by the mutant in later stages of GI colonization. These data suggest that the GI tract environment may induce CA phenotype changes, such as the GUT phenotype, as an adaptive response to mammalian GI tract environmental signals and cues.
Candidalysin
GI epithelial damage is a critical factor required for CA GI translocation [11••, 12••]. Recently, the first fungal cytolytic peptide toxin (candidalysin) in CA was identified [20••]. Candidalysin is a short 31 amino acid long peptide generated from the hyphae-associated cell elongation 1 gene (ECE1). Interestingly, CA strains lacking candidalysin do not activate or damage epithelial cells and have a colonization defect in an animal model of oral candidiasis [20••]. These same mutants, however, do not exhibit a colonization defect in a mouse model of CA vulvovaginitis [21]. With regard to the lower GI tract, the importance of candidalysin in both colonization and dissemination is unclear. Candidalysin does, however, appear to be critical for translocation through intestinal epithelial cells in vitro and appears to have a direct effect on gut microbiota (personal communication, Bernard Hube). Of note, the three host factors that are critical for preventing both bacterial and fungal GI translocation in humans and mice include (1) balanced gut microbiome, with low abundance of potentially pathogenic bacteria and/or fungi; (2) intact intestinal barrier function, and (3) intact cellular immunity, particularly neutrophils [6,7,8,9]. Our group has shown that pathogenic bacteria (e.g., Pseudomonas aeruginosa) unable to induce gut intestinal damage (e.g., mutants unable to produce type III secretion exotoxins) are unable to translocate from the mouse GI tract despite being highly abundant in the gut and the host being severely neutropenic [22]. Thus, candidalysin may be a critical factor in determining CA’s ability to translocate from the mammalian GI tract.
Host Factors Promoting CA Colonization Resistance
Gut Microbiome
The concept of colonization resistance, the notion that the gut microbiota promotes colonization resistance to pathogens, was first noted over 50 years when mice treated with antibiotics developed Salmonella infection with an inoculum 100,000-fold less than required for untreated mice [23]. The relevance of colonization resistance to fungal infections has been well-established in the medical literature that treatment with anti-bacterial antibiotics can lead to the development of “yeast, infections,” specifically vulvovaginal candidiasis [24]. While the importance of the gut microbiota for providing protection against these infections was strongly suggested by these data, the underlying mechanisms of colonization resistance have only recently been elucidated.
One important mechanism by which commensal bacteria promote colonization resistance is via induction of host GI epithelial immune effectors. Our group leveraged the observation that mice are naturally resistant to CA GI colonization but can be colonized after administration of antibiotics. We used different clinically relevant antibiotics to induce variable CA GI colonization phenotypes [12••]. Interestingly, antibiotics most effective in depleting commensal anaerobes resulted in the highest CA GI colonization levels, comparable to levels seen in germ-free mice. Of note, the data supporting that mice are resistant to CA GI colonization, including our own, was generated using the popular laboratory CA strain SC5314. Various clinical CA isolates, particularly those recovered from the human gut, can colonize the murine GI tract without the use of antibiotics to various degrees—highlighting the importance of CA strain variability (A.Y Koh, unpublished observation).
Using gut microbiome profiling techniques, we were then able to identify specific commensal anaerobic bacteria which promoted reduction of CA GI colonization in germ-free mice [12••, 13]. Ultimately, we identified a mechanism by which specific commensal anaerobic bacteria induce the transcription factor HIF-1α, a key regulator of mammalian innate immunity [25], which then increases expression of the antimicrobial peptide LL-37/cathelin-related antimicrobial peptide (CRAMP) in intestinal epithelial cells. LL-37, which has activity against CA [26] and can also inhibit CA adhesion to epithelial surfaces [27]. By inducing HIF-1α, via the pharmacologic HIF-1α agonist mimosine, we were able to increase LL-37/CRAMP expression, reduce CA GI colonization, and decrease CA dissemination in mice, whereas these effects were nullified in mice lacking HIF-1α in their intestinal tissue [12••]. Not surprisingly, the protective effect of commensal anaerobic bacteria was abrogated in mice lacking HIF-1α or CRAMP.
What was most striking about these findings was that a 1–2 log-fold reduction in CA GI colonization was sufficient to significantly decrease CA dissemination or mortality. Similar findings have been reported with regard to bacterial dissemination from the gut [28]. These data suggest that complete eradication or absence of pathobiont GI colonization is not needed to achieve a significant decrease in dissemination. As to whether these findings are relevant to humans, an expansion of GI Enterococcus spp. or Enterobacteriaceae (along with a concomitant depletion of commensal anaerobic microbiota) in adult stem cell transplant patients is associated with a significantly increased risk of developing bloodstream infection with the same bacterial species [29, 30]. While there are data showing that CA bloodstream isolates recovered from patients are genetically similar to CA GI isolates from the same patient [5•], there are no data confirming that CA GI burden is directly proportional to the risk of invasive CA infection in patients.
Lactobacillus spp. probiotic therapies have been used with some success in both animal models and human patients to reduce CA GI colonization. For example, L. acidophilus can reduce the size of Candida-induced gastric ulcers and decrease Candida GI colonization in animals [31]. L. rhamnosus oral therapy induces significant reductions in Candida GI colonization in both premature babies [32] and elderly adults [33]. While these results are intriguing, one caveat that must be considered is that some Lactobacillus probiotic therapies have no effect on host gut microbiota composition or levels [34, 35]. Further studies using probiotics, particularly preparations including commensal anaerobic gut microbiota, will need to be conducted in order to determine whether precision probiotic therapy can modulate CA GI colonization in humans.
As to whether gut microbiota induce a direct effect on CA growth or colonization, we have shown that specific commensal anaerobic gut microbiota do not directly inhibit CA growth in vitro and vice versa [12••]. Furthermore, co-colonization with other pathobiont bacteria, such as P. aeruginosa or Escherichia coli, has no effect on CA GI colonization in germ-free mice [13]. Gut microbiota, however, also produce metabolites (e.g., short-chain fatty acids, SCFAs) that could potentially have a direct effect on CA. SCFAs inhibit CA yeast to hyphal transition [36] and inhibits its growth in vitro [37]. To further confound issues, however, SCFAs can also induce GI epithelial cells to produce immune effectors, such as antimicrobial peptides (e.g., LL-37) [38]. Ultimately, the maintenance of CA colonization resistance in the mammalian host is most likely dependent on both gut microbiota and gut microbiota-derived metabolite effects.
Host Immune System
While lymphocyte deficiency (e.g., patients with HIV and AIDS) results in oral and esophageal candidiasis [39, 40], lymphocytes do not appear to be important for modulating CA GI colonization, as noted by studies using athymic mice [41] and recombinase-activating gene-deficient mice [11••]. In contrast to the critical importance of neutrophils for controlling disseminated fungal disease [11••, 42,43,44], their role in modulating CA GI colonization appears to be negligible [11••]. Similarly, neither macrophages [11••] nor NK cells [45] appear to affect CA GI colonization.
The mammalian innate immune system utilizes pattern recognition receptors (PRRs) to recognize fungi. Dectin-1, a C-type lectin receptor PRR, has been shown to be critical for the control of fungal infections, including CA, in both mice and humans [46, 47]. Interestingly, dectin-1 is essential for the control of GI invasion or translocation during systemic infection in mice, manifested as impairment in fungal clearance and dysregulated cytokine production [48•]. Surprisingly, dectin-1, however, is not required for the control of mucosal colonization of the GI tract, in terms of either fungal burdens or cytokine response [48•]. In light of the importance of commensal gut microbiota in maintaining CA colonization resistance, there is both older [49] and more recent data [50, 51] to suggest that Toll-like receptors (TLR), specifically TLR2 and TLR4, are important for promoting CA colonization resistance. These data are consistent with studies showing that TLR4 and TLR5 are essential to for maintaining colonization resistance to pathobiont bacteria [28, 52]. In total, these data suggest that the mechanisms of maintaining colonization resistant to bacterial and fungal pathobionts may share common pathways: commensal gut microbiota signal through PRRs (e.g., TLRs) to induce gut epithelial immune effectors, most notably antimicrobial peptides (i.e., LL-37/CRAMP [12••, 26], alpha-defensins [53, 54], beta-defensins [55, 56]) that have activity against a variety of pathogens, including CA.
Environmental Factors
A major disparity in studies focused on CA GI colonization rests with the observation that mice are resistant to CA colonization, whereas 40–80% of humans are colonized with CA [57]. The human CA colonization data, however, is based on culture-based data from humans living in Western societies [58]. More recent studies of humans living in remote and traditional societies, however, exhibit widespread Candida GI carriage (e.g., C. krusei), but CA GI carriage rate of less than 10% [59••, 60]. Thus, CA might not be a “normal” commensal of the human gut, but a more recently acquired commensal resulting from medical advances (particularly antibiotics) and adoption of Western diets.
Antibiotics and Chemotherapy
The impact of antibiotics on CA GI colonization resistance cannot be overstated. As noted before, almost all murine models of CA GI colonization utilize antibiotics to establish sustained CA GI colonization, but the CA GI colonization levels achieved can vary widely depending on the antibiotics used [11••, 61,62,63,64,65,66]. Antibiotics that are most effective in depleting anaerobic bacteria, which are the majority of commensal gut microbiota, are the most effective in promoting high CA GI colonization levels. In fact, penicillin (and not clindamycin or metronidazole) has been shown to be most effective in depleting endogenous murine anaerobic gut microbiota and promoting overgrowth and translocation of Enterobacteriaceae [67]. Of note, mice treated with penicillin achieve CA GI colonization levels comparable to those seen in germ-free mice [12••]. These data further suggest that commensal anaerobic gut microbiota are essential for GI colonization resistance to both bacterial and fungal pathobionts and that the host mechanisms for maintaining pathobiont GI colonization resistance may utilize redundant functional pathways and/or strategies.
A recent study screened more than 1000 marketed drugs against 40 representative gut microbiota (all bacteria) and found that a large number of non-antibiotic medications inhibited the growth of gut microbiota, including commensal anaerobes [68]. One of the major medication classes that had a negative effect on gut microbiota were cancer chemotherapy agents. Interestingly, there are prior reports suggesting that cancer chemotherapy can lead to reduced overall numbers of gut microbiota and also lead to changes in gut microbiota taxonomic composition in both preclinical models [69, 70] and human patients [71, 72]. The two classes of medications most frequently given to cancer and stem cell transplant patients are chemotherapeutic agents and antibiotics. This begs the question as to whether these medications contribute to the fact that these patients are at such high risk of developing CA invasive infections originating from the gut [3]. Unfortunately, there are no studies in either animals or humans that have examined the effect of cancer chemotherapeutic agents on the gut mycobiome; so, further studies are merited.
Diet
One obvious environmental factor that differs between mice and humans is diet. Thus, if a mouse were to adopt a human “Western society” diet, could the mouse be colonized with CA without the use of antibiotics? In fact, when mice are a fed “purified” diets consisting of significant amounts of cornstarch, sucrose, and soybean oil [73, 74••], they can be colonized with CA in the absence of antibiotics, albeit at levels 1–2 log fold lower than seen in antibiotic-treated or germ-free mice. There is some data to suggest that the use of this specific diet leads to gut microbiota taxonomic changes that results in decreased amounts of gut microbiota-derived organic acid, which as noted previously may have a direct inhibitory effect on CA [74••]. Yet, it is still unclear whether the effects of the diet on CA GI colonization resistance are more a result of gut microbiota taxonomic changes (and thus changes in host immune effectors) or whether this may be due to direct effects on CA metabolism secondary to the increased availability of refined carbohydrates and fat. Further studies are needed to dissect the mechanisms by which dietary changes can modulate CA GI colonization resistance in the mammalian host.
Conclusion
In sum, the regulation of CA GI colonization and dissemination is a dynamic and complex process that involves CA genetic determinants, host factors (including the gut microbiome), and environmental elements, such as exposure to medication and dietary choices (Table 1). Commensal microbiota outnumber mammalian host cells by a factor of 10:1, with the microbial genetic repertoire 100-fold more abundant than the host. The gut microbiome contributes to the metabolic, nutritional, and immunological status of the mammalian host. As such, the field of host-pathogen interactions must now include host-pathogen-commensal interactions, which inherently increases the complexity of exploring these pathophysiologic processes. But given the advances in molecular biology approaches and multi-omic analyses, we can now pursue more mechanistic insight into how and why the commensal CA transforms into the pathogenic CA and thus provide the platform for innovative diagnostic and therapeutic approaches to preventing harmful CA infections in the future.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Mostov K, Su T, ter Beest M. Polarized epithelial membrane traffic: conservation and plasticity. Nat Cell Biol. 2003;5(4):287–93. https://doi.org/10.1038/ncb0403-287.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23. https://doi.org/10.1038/nri2515.
Koh AY. The microbiome in hematopoietic stem cell transplant recipients and cancer patients: opportunities for clinical advances that reduce infection. PLoS Pathog. 2017;13(6):e1006342. https://doi.org/10.1371/journal.ppat.1006342.
Nucci M, Anaissie E. Revisiting the source of candidemia: skin or gut? Clin Infect Dis. 2001;33(12):1959–67. https://doi.org/10.1086/323759.
• Miranda LN, van der Heijden IM, Costa SF, Sousa AP, Sienra RA, Gobara S, et al. Candida colonisation as a source for candidaemia. J Hosp Infect. 2009;72(1):9–16. https://doi.org/10.1016/j.jhin.2009.02.009. Provides molecular evidence that the gut can be a source for CA bloodstream infections.
Berg RD. Bacterial translocation from the gastrointestinal tract. Adv Exp Med Biol. 1999;473:11–30.
Pasqualotto AC, Nedel WL, Machado TS, Severo LC. Risk factors and outcome for nosocomial breakthrough candidaemia. J Infect. 2006;52(3):216–22. https://doi.org/10.1016/j.jinf.2005.04.020.
Rosen GP, Nielsen K, Glenn S, Abelson J, Deville J, Moore TB. Invasive fungal infections in pediatric oncology patients: 11-year experience at a single institution. J Pediatr Hematol Oncol. 2005;27(3):135–40.
Shoham S, Levitz SM. The immune response to fungal infections. Br J Haematol. 2005;129(5):569–82. https://doi.org/10.1111/j.1365-2141.2005.05397.x.
Sudbery PE. Growth of Candida albicans hyphae. Nat Rev Microbiol. 2011;9(10):737–48. https://doi.org/10.1038/nrmicro2636.
•• Koh AY, Kohler JR, Coggshall KT, Van Rooijen N, Pier GB. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 2008;4(2):e35. https://doi.org/10.1371/journal.ppat.0040035. This study demonstrates that gut microbiota dysbiosis (high CA levels), neutropenia and GI mucosal damage are all required for CA dissemination from the gut.
•• Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat Med. 2015;21(7):808–14. https://doi.org/10.1038/nm.3871. Provides a specific mechanism by which commensal gut microbiota inhibit CA GI colonization and demonstrates that manipulation of this specific pathway, in the absence of gut microbiota, reduces CA GI colonization and dissemination in mice.
Lopez-Medina E, Fan D, Coughlin LA, Ho EX, Lamont IL, Reimmann C, et al. Candida albicans inhibits Pseudomonas aeruginosa virulence through suppression of pyochelin and pyoverdine biosynthesis. PLoS Pathog. 2015;11(8):e1005129. https://doi.org/10.1371/journal.ppat.1005129.
•• Bohm L, Torsin S, Tint SH, Eckstein MT, Ludwig T, Perez JC. The yeast form of the fungus Candida albicans promotes persistence in the gut of gnotobiotic mice. PLoS Pathog. 2017;13(10):e1006699. https://doi.org/10.1371/journal.ppat.1006699. Demonstrates that the CA yeast form is preferred for GI colonization in germ free mice and is dependent on specific CA transcriptional regulators.
•• Noble SM, Gianetti BA, Witchley JN. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol. 2017;15(2):96–108. https://doi.org/10.1038/nrmicro.2016.157. First study to describe the gastrointestinally induced transition (GUT) phenotype and why this morphology is better suited for the mammalian GI tract.
Pande K, Chen C, Noble SM. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet. 2013;45(9):1088–91. https://doi.org/10.1038/ng.2710.
Huang G, Wang H, Chou S, Nie X, Chen J, Liu H. Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc Natl Acad Sci U S A. 2006;103(34):12813–8. https://doi.org/10.1073/pnas.0605270103.
Zordan RE, Galgoczy DJ, Johnson AD. Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc Natl Acad Sci U S A. 2006;103(34):12807–12. https://doi.org/10.1073/pnas.0605138103.
Prieto D, Roman E, Alonso-Monge R, Pla J. Overexpression of the transcriptional regulator WOR1 increases susceptibility to bile salts and adhesion to the mouse gut mucosa in Candida albicans. Front Cell Infect Microbiol. 2017;7:389. https://doi.org/10.3389/fcimb.2017.00389.
•• Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532(7597):64–8. https://doi.org/10.1038/nature17625. Discovery of the first reported CA cytolytic peptide toxin and its importance in CA pathogenesis.
Richardson JP, Willems HME, Moyes DL, Shoaie S, Barker KS, Tan SL, et al. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun. 2017;86:e00645–17. https://doi.org/10.1128/IAI.00645-17.
Koh AY, Priebe GP, Pier GB. Virulence of Pseudomonas aeruginosa in a murine model of gastrointestinal colonization and dissemination in neutropenia. Infect Immun. 2005;73(4):2262–72. https://doi.org/10.1128/IAI.73.4.2262-2272.2005.
Miller CP, Bohnhoff M, Rifkind D. The effect of an antibiotic on the susceptibility of the mouse’s intestinal tract to Salmonella infection. Trans Am Clin Climatol Assoc. 1957;68:51–8.
Xu J, Sobel JD. Antibiotic-associated vulvovaginal candidiasis. Curr Infect Dis Rep. 2003;5(6):481–7.
Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9(9):609–17. https://doi.org/10.1038/nri2607.
Lopez-Garcia B, Lee PH, Yamasaki K, Gallo RL. Anti-fungal activity of cathelicidins and their potential role in Candida albicans skin infection. J Investig Dermatol. 2005;125(1):108–15. https://doi.org/10.1111/j.0022-202X.2005.23713.x.
Tsai PW, Yang CY, Chang HT, Lan CY. Human antimicrobial peptide LL-37 inhibits adhesion of Candida albicans by interacting with yeast cell-wall carbohydrates. PLoS One. 2011;6(3):e17755. https://doi.org/10.1371/journal.pone.0017755.
Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455(7214):804–7. https://doi.org/10.1038/nature07250.
Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55(7):905–14. https://doi.org/10.1093/cid/cis580.
Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest. 2010;120(12):4332–41. https://doi.org/10.1172/JCI43918.
Zwolinska-Wcislo M, Brzozowski T, Mach T, Budak A, Trojanowska D, Konturek PC, et al. Are probiotics effective in the treatment of fungal colonization of the gastrointestinal tract? Experimental and clinical studies. J Physiol Pharmacol. 2006;57(Suppl 9):35–49.
Manzoni P, Mostert M, Leonessa ML, Priolo C, Farina D, Monetti C, et al. Oral supplementation with Lactobacillus casei subspecies rhamnosus prevents enteric colonization by Candida species in preterm neonates: a randomized study. Clin Infect Dis. 2006;42(12):1735–42. https://doi.org/10.1086/504324.
Hatakka K, Ahola AJ, Yli-Knuuttila H, Richardson M, Poussa T, Meurman JH, et al. Probiotics reduce the prevalence of oral candida in the elderly—a randomized controlled trial. J Dent Res. 2007;86(2):125–30.
Crovesy L, Ostrowski M, Ferreira D, Rosado EL, Soares-Mota M. Effect of Lactobacillus on body weight and body fat in overweight subjects: a systematic review of randomized controlled clinical trials. Int J Obes. 2017;41:1607–14. https://doi.org/10.1038/ijo.2017.161.
Uyeno Y, Sekiguchi Y, Kamagata Y. Impact of consumption of probiotic lactobacilli-containing yogurt on microbial composition in human feces. Int J Food Microbiol. 2008;122(1–2):16–22. https://doi.org/10.1016/j.ijfoodmicro.2007.11.042.
Noverr MC, Huffnagle GB. Regulation of Candida albicans morphogenesis by fatty acid metabolites. Infect Immun. 2004;72(11):6206–10. https://doi.org/10.1128/IAI.72.11.6206-6210.2004.
Cottier F, Tan AS, Chen J, Lum J, Zolezzi F, Poidinger M, et al. The transcriptional stress response of Candida albicans to weak organic acids. G3 (Bethesda). 2015;5(4):497–505. https://doi.org/10.1534/g3.114.015941.
Schauber J, Svanholm C, Termen S, Iffland K, Menzel T, Scheppach W, et al. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut. 2003;52(5):735–41.
Farah CS, Elahi S, Pang G, Gotjamanos T, Seymour GJ, Clancy RL, et al. T cells augment monocyte and neutrophil function in host resistance against oropharyngeal candidiasis. Infect Immun. 2001;69(10):6110–8. https://doi.org/10.1128/IAI.69.10.6110-6118.2001.
Jones-Carson J, Vazquez-Torres A, Warner T, Balish E. Disparate requirement for T cells in resistance to mucosal and acute systemic candidiasis. Infect Immun. 2000;68(4):2363–5.
Helstrom PB, Balish E. Effect of oral tetracycline, the microbial flora, and the athymic state on gastrointestinal colonization and infection of BALB/c mice with Candida albicans. Infect Immun. 1979;23(3):764–74.
Boyne R, Arthur JR. The response of selenium-deficient mice to Candida albicans infection. J Nutr. 1986;116(5):816–22.
Jensen J, Warner T, Balish E. Resistance of SCID mice to Candida albicans administered intravenously or colonizing the gut: role of polymorphonuclear leukocytes and macrophages. J Infect Dis. 1993;167(4):912–9.
Mullick A, Elias M, Harakidas P, Marcil A, Whiteway M, Ge B, et al. Gene expression in HL60 granulocytoids and human polymorphonuclear leukocytes exposed to Candida albicans. Infect Immun. 2004;72(1):414–29.
Greenfield RA, Abrams VL, Crawford DL, Kuhls TL. Effect of abrogation of natural killer cell activity on the course of candidiasis induced by intraperitoneal administration and gastrointestinal candidiasis in mice with severe combined immunodeficiency. Infect Immun. 1993;61(6):2520–5.
Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8(1):39–46. https://doi.org/10.1038/ni1425.
Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8(1):31–8. https://doi.org/10.1038/ni1408.
• Vautier S, Drummond RA, Redelinghuys P, Murray GI, MacCallum DM, Brown GD. Dectin-1 is not required for controlling Candida albicans colonization of the gastrointestinal tract. Infect Immun. 2012;80(12):4216–22. https://doi.org/10.1128/IAI.00559-12. This study shows that dectin-1 is required for modulating invasive CA infection but is not required for CA GI colonization resistance.
Brown GD. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol. 2011;29:1–21. https://doi.org/10.1146/annurev-immunol-030409-101229.
Choteau L, Vancraeyneste H, Le Roy D, Dubuquoy L, Romani L, Jouault T, et al. Role of TLR1, TLR2 and TLR6 in the modulation of intestinal inflammation and Candida albicans elimination. Gut Pathog. 2017;9:9. https://doi.org/10.1186/s13099-017-0158-0.
Prieto D, Carpena N, Maneu V, Gil ML, Pla J, Gozalbo D. TLR2 modulates gut colonization and dissemination of Candida albicans in a murine model. Microbes Infect. 2016;18(10):656–60. https://doi.org/10.1016/j.micinf.2016.05.005.
Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313(5790):1126–30. https://doi.org/10.1126/science.1127119.
Chairatana P, Chiang IL, Nolan EM. Human alpha-defensin 6 self-assembly prevents adhesion and suppresses virulence traits of Candida albicans. Biochemistry. 2017;56(8):1033–41. https://doi.org/10.1021/acs.biochem.6b01111.
Porter EM, van Dam E, Valore EV, Ganz T. Broad-spectrum antimicrobial activity of human intestinal defensin 5. Infect Immun. 1997;65(6):2396–401.
Vylkova S, Li XS, Berner JC, Edgerton M. Distinct antifungal mechanisms: beta-defensins require Candida albicans Ssa1 protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob Agents Chemother. 2006;50(1):324–31. https://doi.org/10.1128/AAC.50.1.324-331.2006.
Vylkova S, Nayyar N, Li W, Edgerton M. Human beta-defensins kill Candida albicans in an energy-dependent and salt-sensitive manner without causing membrane disruption. Antimicrob Agents Chemother. 2007;51(1):154–61. https://doi.org/10.1128/AAC.00478-06.
Odds FC. Molecular phylogenetics and epidemiology of Candida albicans. Future Microbiol. 2010;5(1):67–79. https://doi.org/10.2217/fmb.09.113.
Bougnoux ME, Diogo D, Francois N, Sendid B, Veirmeire S, Colombel JF, et al. Multilocus sequence typing reveals intrafamilial transmission and microevolutions of Candida albicans isolates from the human digestive tract. J Clin Microbiol. 2006;44(5):1810–20. https://doi.org/10.1128/JCM.44.5.1810-1820.2006.
•• Angebault C, Djossou F, Abelanet S, Permal E, Ben Soltana M, Diancourt L, et al. Candida albicans is not always the preferential yeast colonizing humans: a study in Wayampi Amerindians. J Infect Dis. 2013;208(10):1705–16. https://doi.org/10.1093/infdis/jit389. This work uses next-generation sequencing approaches to demonstrate that indigenous humans living in the Amazon forest are GI colonized with Candida spp. but have low (< 10%) CA carriage rates.
Xu J, Mitchell TG. Geographical differences in human oral yeast flora. Clin Infect Dis. 2003;36(2):221–4. https://doi.org/10.1086/345672.
Clemons KV, Gonzalez GM, Singh G, Imai J, Espiritu M, Parmar R, et al. Development of an orogastrointestinal mucosal model of candidiasis with dissemination to visceral organs. Antimicrob Agents Chemother. 2006;50(8):2650–7. https://doi.org/10.1128/AAC.00530-06.
Cole GT, Lynn KT, Seshan KR, Pope LM. Gastrointestinal and systemic candidosis in immunocompromised mice. J Med Vet Mycol. 1989;27(6):363–80.
Samonis G, Anaissie EJ, Rosenbaum B, Bodey GP. A model of sustained gastrointestinal colonization by Candida albicans in healthy adult mice. Infect Immun. 1990;58(6):1514–7.
Shankar J, Solis NV, Mounaud S, Szpakowski S, Liu H, Losada L, et al. Using Bayesian modelling to investigate factors governing antibiotic-induced Candida albicans colonization of the GI tract. Sci Rep. 2015;5:8131. https://doi.org/10.1038/srep08131.
White SJ, Rosenbach A, Lephart P, Nguyen D, Benjamin A, Tzipori S, et al. Self-regulation of Candida albicans population size during GI colonization. PLoS Pathog. 2007;3(12):e184. https://doi.org/10.1371/journal.ppat.0030184.
Wiesner SM, Jechorek RP, Garni RM, Bendel CM, Wells CL. Gastrointestinal colonization by Candida albicans mutant strains in antibiotic-treated mice. Clin Diagn Lab Immunol. 2001;8(1):192–5. https://doi.org/10.1128/CDLI.8.1.192-195.2001.
Berg RD. Promotion of the translocation of enteric bacteria from the gastrointestinal tracts of mice by oral treatment with penicillin, clindamycin, or metronidazole. Infect Immun. 1981;33(3):854–61.
Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555(7698):623–8. https://doi.org/10.1038/nature25979.
Lin XB, Dieleman LA, Ketabi A, Bibova I, Sawyer MB, Xue H, et al. Irinotecan (CPT-11) chemotherapy alters intestinal microbiota in tumour bearing rats. PLoS One. 2012;7(7):e39764. https://doi.org/10.1371/journal.pone.0039764.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–6. https://doi.org/10.1126/science.1240537.
van Vliet MJ, Harmsen HJ, de Bont ES, Tissing WJ. The role of intestinal microbiota in the development and severity of chemotherapy-induced mucositis. PLoS Pathog. 2010;6(5):e1000879. https://doi.org/10.1371/journal.ppat.1000879.
Zwielehner J, Lassl C, Hippe B, Pointner A, Switzeny OJ, Remely M, et al. Changes in human fecal microbiota due to chemotherapy analyzed by TaqMan-PCR, 454 sequencing and PCR-DGGE fingerprinting. PLoS One. 2011;6(12):e28654. https://doi.org/10.1371/journal.pone.0028654.
Kadosh D, Najvar LK, Bocanegra R, Olivo M, Kirkpatrick WR, Wiederhold NP, et al. Effect of antifungal treatment in a diet-based murine model of disseminated candidiasis acquired via the gastrointestinal tract. Antimicrob Agents Chemother. 2016;60(11):6703–8. https://doi.org/10.1128/AAC.01144-16.
•• Yamaguchi N, Sonoyama K, Kikuchi H, Nagura T, Aritsuka T, Kawabata J. Gastric colonization of Candida albicans differs in mice fed commercial and purified diets. J Nutr. 2005;135(1):109–15. https://doi.org/10.1093/jn/135.1.109 . Demonstrates that diet modification can promote CA colonization in mice, in the absence of antibiotic therapy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Koh reports grants from NIH/NIAID and grants from Centers for Disease Control, during the conduct of the study; others from Merck Research Laboratories, outside the submitted work.
Dr. Animesh A. Mishra has no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Mycology
Rights and permissions
About this article

Cite this article
Mishra, A.A., Koh, A.Y. Adaptation of Candida albicans During Gastrointestinal Tract Colonization. Curr Clin Micro Rpt 5, 165–172 (2018). https://doi.org/10.1007/s40588-018-0096-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40588-018-0096-8