
Abstract
Purpose of the review
Pediatric sinonasal and skull base lesions are rare and complex disorders. The safe and comprehensive management of this diverse group of pathologies requires the expertise of an experienced multidisciplinary skull base team.
Recent findings
With significant evolution and improved outcomes achieved in adult endoscopic skull base surgery (ESBS), similar principles have been applied to the pediatric population. Pediatric endoscopic surgery for sinonasal and skull base tumors has therefore also evolved due to a variety of technologic and surgical advances, multidisciplinary team approaches, and continued innovation.
Summary
Pediatric sinonasal and skull base tumors require complex multidisciplinary management for optimal outcome. Detailed understanding of the sinonasal anatomy will improve surgical outcomes. The surgical approaches and techniques are dependent on the location of the lesion, the pathology, extent of disease, and surgical goals. Reconstruction following tumor extirpation follows the same principles as adult reconstruction, with the ultimate goal of achieving a watertight seal that separates the nasal cavity from the brain. Complications can be catastrophic and rely on immediate recognition and management to prevent long-term sequelae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pediatric sinonasal and skull base lesions are rare and complex disorders with a highly variable presentation (Table 1). Patients may be asymptomatic, with a lesion identified incidentally on imaging, or may present with a range of symptoms including nasal obstruction, epistaxis, rhinorrhea, endocrinopathies, headaches, vision changes, other cranial neuropathies, or other neurologic changes. A comprehensive history and physical examination should be performed, with particular attention to neurologic, ophthalmologic, and rhinologic evaluations, including nasal endoscopy when possible. Imaging of the sinuses, orbits, and skull base, usually including both computed tomography (CT) and magnetic resonance imaging (MRI), is recommended.
A multidisciplinary team approach to the management of these patients is critical. The comprehensive management of these complex lesions may include Otolaryngology, Neurosurgery, Ophthalmology, Neurology, Hematology-Oncology, Radiation Oncology, Endocrinology, Intensivists, and others. Many centers now have a Pediatric- or Skull Base–specific multidisciplinary tumor board as well to facilitate optimal treatment planning. The collaborative partnership between Otolaryngology and Neurosurgery has improved surgical success and outcomes [1]. Surgeon experience and meticulous surgical techniques result in optimal tumor extirpation and reconstruction while preserving sinonasal and neurologic structure and function [2,3,4,5,6].
Post-operative hospitalization, with an available pediatric intensive care unit, allows for appropriate recovery following surgery, prompt recognition of possible post-operative complications, and expert management of potential neurologic, metabolic, cardiopulmonary, or endocrinological issues. Long-term post-operative management is essential for optimal outcomes to ensure proper wound healing, perform necessary wound debridements, manage adjuvant treatment as indicated, and monitor for recurrence.
Congenital skull base lesions
Dermoid cysts
Embryologic developmental abnormalities may result in skull base lesions. The fonticulus frontalis and the prenasal space are two transient spaces that may allow the dural diverticulum to extend from the anterior cranial fossa to the skin. The fonticulus frontalis temporarily separates the frontal and nasal bones, while the prenasal space is between the nasal bones and developing nasal cartilage. As the frontal and nasal bones grow, the dural diverticulum and prenasal spaces involute and regress. At the same time, the cribriform plate develops due to the fusion of the fonticulus frontalis and foramen cecum. Failure of these steps results in incomplete separation of dura from the overlying skin and can result in nasal dermoid, glioma, or encephalocele formation [7].
Nasal dermoids occur due to an incomplete separation and involution of the dural diverticulum through the foramen cecum from the overlying skin [8, 9]. Incomplete closure of the path of the diverticulum results in a persistent attachment of the dura to the dermis, resulting in trapped epithelium along the diverticulum path. As time passes, the trapped epithelium produces a dermoid with hair and glandular structures [10]. A telltale sign is a midline pit in the nasal skin, usually at the rhinion. CT and MRI are obtained to assist with diagnosis as well as determine if there is intracranial extension [11]. Dermoids with intracranial extension are at an increased risk for infection [12]. Surgery is recommended, though the approach and scope of dissection is dependent on the location, overlying skin involvement, and extension and size of the tumor [13]. The external approach includes an ellipse around the pit combined with an external rhinoplasty [14]. Larger tumors with intracranial extension require neurosurgical involvement and can include a bicoronal approach and craniotomy. More recently, endoscopic endonasal or endoscopic-assisted techniques have been applied to these tumors [12].
Encephaloceles
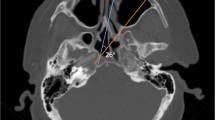
Congenital defects of the skull base can lead to the development of meningoencephaloceles, or the herniation of brain, cerebrospinal fluid (CSF), and dural structures into the sinonasal cavity. Though nasal dermoid cysts are the most common midline nasal mass, encephaloceles and gliomas are important diagnoses to consider. Patients may present with an array of complaints. Occasionally, these may be identified incidentally on imaging, or patients may present with nasal obstruction, respiratory distress, failure to thrive, infectious symptoms including meningitis, or clear rhinorrhea (Fig. 1a). In unclear clinical scenarios, the clinician can consider collecting clear rhinorrhea to evaluate the sample for Beta-2 transferrin, which has a high specificity and sensitivity [15]. Trauma may also result in the acquired development of an encephalocele. A full history and physical, including nasal endoscopy if the patient is cooperative, is necessary. Pre-operative imaging includes both CT and MRI [9]. Ma et al. reviewed 23 children with a mean age of 7.0 years undergoing endoscopic transnasal surgery for repair of CSF leak with or without meningocele or encephalocele. Defect sites were found in the ethmoid roof in ten patients, cribriform plate in five patients, lateral to the foramen cecum in three patients, in the sphenoid sinus in two, and at the posterior wall of the frontal sinus in one. The authors reported favorable clinical outcomes without recurrence in all patients with an average follow-up of 61.1 months [16]. Congenital defects require surgery to achieve a watertight seal and prevent future complications including CSF rhinorrhea and meningitis and can be safely and successfully treated with endoscopic surgery [17]. This minimally invasive, single-stage surgery can decrease hospital stay, cost of treatment, and result in better cosmesis [18]. With the continued improvement in surgical technology, optics, imaging, and experience, this disease process can be treated in even some of the youngest children with favorable outcomes [19].

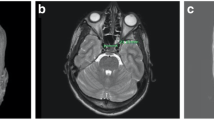
a Computed tomography of a 6-month-old male with a patent craniopharyngeal canal and meningocele. b Computed tomography of an 11-year-old male with juvenile nasopharyngeal angiofibroma. c Magnetic resonance imaging of a 10-year-old male with rhabdomyosarcoma. d Magnetic resonance imaging of an 11-year-old female with chordoma. (Images are property of Columbia University Division of Rhinology).
Gliomas
Gliomas, which contain mature neuroglial tissue, are the least common congenital midline mass. Gliomas share a similar embryologic origin to encephaloceles, though they differ in that gliomas lack communication to the subarachnoid space. Frequently, gliomas present as a pale mass in the nasal cavity and may have extension from the nostril. These masses are non-compressible, smooth, and do not exhibit pulsation or expansion with straining or crying. Surgery is recommended early in the patient’s life to prevent complications such as infection and cosmetic deformity. Up to 25% of nasal gliomas have a fibrous stalk that extend towards the skull base [20, 21]
Neoplasms
Juvenile nasopharyngeal angiofibroma
Juvenile nasopharyngeal angiofibromas (JNAs) are benign, highly vascular tumors, thought to arise from the pterygoid canal and may extend laterally to the pterygopalatine fossa (PPF) and/or infratemporal fossa (ITF), medially through the sphenopalatine foramen into the nasal cavity [22], superiorly through the middle cranial fossa, and inferiorly into the buccal, masseteric, and other deep spaces of the neck. Patients are generally adolescent males who present with severe, recurrent epistaxis and nasal obstruction.
JNAs tend to be locally aggressive, invading the nasal cavity, nasopharynx, PPF, ITF, clivus, orbit, or other adjacent structures, and extend through bony foramina (Fig. 1b). Biopsy in the clinic is not recommended due to the highly vascular nature of these tumors. Pre-operative CT and MRI can be pathognomonic and can guide counseling and treatment. Anterior displacement of the posterior wall of the maxillary sinus seen on CT scan, also called the Holman-Miller sign, is diagnostic for JNA. Several staging systems have been created and are based on the extent of disease including imaging and clinical examination [23, 24]. Authors from the University of Pittsburgh Medical Center developed an endoscopic staging system which considered residual vascularity from the internal carotid artery following embolization of external carotid tributaries. Snyderman et al. found that compared to previous staging systems, tumor size and extent of sinus disease were less important in predicting complete tumor removal during endoscopic resection. The authors identified that the route of skull base extension and residual vascularity provided better prediction of immediate morbidity, strongly correlating with blood loss, requirement for multiple procedures, and residual or recurrent tumor.
Hormonal and genetic factors are thought to explain why JNAs are found almost exclusively in adolescent males, though the associations remain incompletely defined. These tumors express estrogen, progesterone, and androgen receptors, and exogenous testosterone has been found to cause tumor growth [25, 26]. Beta-catenin, a coactivator of androgen receptors, and vascular endothelial growth factor (VEGFR-2) are overexpressed in JNAs [27]. Though hormonal therapy is not routinely recommended for the treatment of JNA, androgen receptor antagonists have been used to achieve partial regression of disease [28]. The effectiveness of Bevacizumab, a monoclonal antibody that inhibits vascular endothelial growth factor A, is under current investigation [29].
Surgery is the mainstay for treating JNAs. Open and endoscopic approaches have been described, yielding similar results. Prior to surgery, pre-operative embolization should be sought to decrease intra-operative blood loss. Embolization should occur 24 to 48 h before surgery. Delaying surgery beyond this time period may allow for revascularization of tumor-feeding vessels. Residual disease can be treated with either revision surgery or radiation. Rowan et al. found that of the 34 patients in their series, 33 underwent an exclusively endoscopic surgical approach, with six (18%) requiring planned staged operations. Ten patients (29%) had residual disease, and three (9%) underwent further surgical resection [30•] Table 2. Surveillance with nasal endoscopy and MRI can identify disease recurrence.
Rhabdomyosarcoma
Sinonasal rhabdomyosarcoma is a rare malignancy with poor outcomes. The nasal cavity is the most common primary site (37.1%). These patients often present with advanced disease and rapid onset of symptoms including cranial neuropathies [31] (Fig. 1c). Five-, 10-, and 20-year disease-specific survival rates are 60.2%, 46.1%, and 20.6%, respectively [32]. Initial surgical intervention, after appropriate imaging, includes a biopsy of the lesion and workup of metastatic disease. In a recent retrospective review of 16 patients with sinonasal rhabdomyosarcoma by Thompson et al. [33], the authors found that age less than 18 years and the embryonal or botryoid subtypes were positive prognostic factors, while the alveolar subtype was a poor prognostic factor. These patients tend to present more often with regional and distant metastases, and have increased recurrence rates, and decreased survival. Multimodality treatment, which often includes surgery, radiation, and chemotherapy, is associated with the best survival outcomes [34]. Optimal surgery includes primary resection with negative margins, which can limit radiation therapy, though in instances of intraorbital or intracranial extension, this may not be achievable [35]. Second-look surgery after completion of treatment may identify recurrent or persistent disease, though additional studies are required [31].
Pituitary adenomas
Pituitary adenomas are rare in children, accounting for 3% of all intracranial neoplasms in children and 5% of all pituitary adenomas [36]. Pituitary adenomas either can be asymptomatic and detected incidentally on imaging or may present with endocrine abnormalities or visual loss. Functional pituitary adenomas are more common than non-functional adenomas in children, with prolactinomas and adrenocorticotrophic hormone-secreting tumors being most commonly encountered, followed by growth hormone–secreting and non-secreting tumors [36]. Macroadenomas are classified as pituitary adenomas greater than 1 cm in diameter; microadenomas are less than 1 cm. Symptoms are often the result of compression to nearby structures. Patients may complain of visual loss or headache. Surgery is routinely recommended for these pathologies to restore neurologic and endocrine function. Hanba et al. [37] demonstrated a significantly decreased length of stay and hospital costs in children with pituitary adenomas undergoing endoscopic transsphenoidal approaches compared to the transfrontal approach. In a literature review of 37 publications regarding pediatric pituitary adenomas examining 1,284 patients, surgical cure was achieved in 65% of patients. Complications included pituitary insufficiency (23%), permanent visual dysfunction (6%), chronic diabetes insipidus (3%), and post-operative cerebrospinal fluid (CSF) leak (4%) [36]. Patients younger than 10 years of age are more likely to have complications including post-operative hydrocephalus, panhypopituitarism, and diabetes insipidus [37].
Rathke’s cleft cyst
Rathke’s cleft cyst is a benign cystic lesion located in close proximity to the pituitary gland. These cysts are ectopic remnants of Rathke’s pouch, with the minority of these tumors diagnosed in childhood [38]. Frequently, these are found incidentally but may also be identified in patients with recurrent headaches, visual changes, or endocrine pathology. On MRI, a Rathke’s cleft cyst is non-enhancing and often has an intracystic nodule with high signal intensity on T1-weighted images [39]. The majority of pediatric Rathke’s cleft cysts are followed with observation, especially if discovered incidentally [40]. Surgery is indicated when there are significant or progressive symptoms related to the cyst, increasing cyst size, or diagnostic uncertainty [41]. Surgery involves fenestration and aspiration of the cyst with partial or complete removal of the cyst wall. Kuan et al. [42] found that in 52 consecutive adult patients undergoing fenestration, intra-operative CSF leak rate occurred in 27% of cases and was associated with larger Rathke’s cleft cyst size. Post-operative diabetes insipidus can occur in approximately 20% of patients [43, 38].
Craniopharyngiomas
Craniopharyngiomas are rare tumors with a reported incidence of 300–400 new cases per year in the USA [44], despite being the most commonly diagnosed benign tumor in the sellar and suprasellar region in children. These tumors arise from epithelial remnants of Rathke’s pouch and are benign; however, craniopharyngiomas pose distinct surgical dilemmas and can result in serious morbidity [45]. Craniopharyngiomas frequently have both a solid and cystic component with a propensity to recur years after resection. Patients with craniopharyngiomas typically present with endocrine deficits, visual changes, and occasionally mental status changes due to mass effect secondary to their anatomic location or hydrocephalus. Pre-operative MRI is necessary to determine the extent of the disease.
Gross total resection (GTR) is desired though not necessarily practical in all instances, particularly in larger tumors or tumors with significant supradiaphragmatic extension. In a recent case series of 45 consecutive pediatric patients undergoing endoscopic transsphenoidal resection, 20 patients required repeat surgeries. The authors found GTR was more likely in patients undergoing primary surgery (98%) compared to the revision surgery group (75%). Among the patients in which GTR was achieved, 12% experienced tumor recurrence with a mean follow-up of 7.8 years. In the primary surgery cohort, 80% of patients had worsening of pituitary function and 83% developed diabetes insipidus. In the repeat surgery cohort, 100% of patients developed these complications [46•]. The authors concluded that GTR should be the goal for the first surgical attempt. Patel and coauthors similarly found post-operative pituitary dysfunction and diabetes insipidus in 63.6% and 46.7% of a cohort of 16 pediatric patients undergoing endoscopic craniopharyngioma resection [47]. Fear of hypothalamic injury may cause surgeons to consider debulking or subtotal resection with post-operative radiation as major complication including post-operative CSF leak and death rates can occur in 19% and 12.5% of cases, respectively (Patel, 2017). Alalade et al. [48] reported on their experience and noted that GTR was achieved in 45% of patients, while subtotal resection, near-total resection, or biopsy was performed in the remaining patients to avoid hypothalamic injury. Due to the known severe complications possible after attempted GTR in these patients (including hypothalamic injury, morbid obesity, and visual loss), many centers have advocated for limited surgery with post-operative radiation therapy [49]. They have demonstrated equivalent long-term tumor control rates comparing GTR and limited surgery with radiation therapy, but with reduced post-operative surgical and endocrine complications. Regardless of the surgical plan, longitudinal tumor surveillance is necessary with serial MRI scans due to the high incidence of recurrence and complications in a benign disease that places children at risk of severe morbidity [50].
Chordoma
Chordomas, occurring most commonly in the clivus, are malignant lesions that exhibit locally destructive behavior with a low metastatic potential [51]. However, metastases to the lung, bone, soft tissue, lymph nodes, liver, and skin have been reported in up to 65% of patients, especially those with advanced disease [52]. These tumors arise from an embryologic remnant of the notochord. The most common presenting symptom is abducens nerve palsy [53]. Both CT and MRI are usually obtained as they provide complementary information on the extent of disease (Fig. 1d). Chordomas tend to be midline, a factor that can sometimes differentiate chordoma from chondrosarcoma, which tend to be lateral to the midline. On pathology, chordomas demonstrate physaliferous cells with a “soap bubble” appearance. Optimal treatment strategies include surgical resection followed by radiotherapy. Chemotherapy may be an option in select cases [54]. Surgical goals include total resection of the involved bone, though in some instances, this is not feasible due to involvement of vital neurovascular structures including the internal carotid artery or brainstem. Radiation or radiation with chemotherapy may be considered in recurrent or persistent disease [55]. In children with chordomas less than 5 years of age, a worse prognosis is observed. Clival chordomas appear to be at a particular high risk for post-operative CSF leak in a recent publication by Stapleton et al. [56]. In a recent retrospective review, Rassi et al. [53] studied 31 patients with skull base chordomas and found that long-term overall and progression-free survival was achievable with gross total resection and proton-beam therapy.
Fibro-osseous lesions
Fibrous dysplasia
An important skull base pathology that deserves mention is fibrous dysplasia of the skull base bones [57]. Fibrous dysplasia is a benign fibro-osseous disorder that can involve any areas of the skull base in the pediatric population and are diagnosed on radiographic imaging. CT imaging demonstrates a ground-glass appearance of the bone, pathognomonic for fibro-osseous lesions. It is thought that these lesions are slow-growing and can be observed. However, when causing symptoms such as visual disturbance, nasal obstruction, or obstructive sinusitis, surgery may be indicated. In a recent retrospective review from the University of Pittsburg, 14 patients aged 2–18 were identified. Five patients (36%) had proptosis, while four (29%) had diplopia [58]. Two patients (14%) had cranial nerve VI palsy. Gross total resection was achieved with single-stage surgery in 10 patients (71%). Two additional patients (14%) required a second surgery and achieved gross total resection. The authors concluded that for benign fibro-osseous tumors of the skull base, endoscopic surgery is safe and effective to excise these lesions in the pediatric population.
Osteoma
Osteomas are benign bony tumors that occur in the paranasal sinuses. In children, these growths are very rare, as the pediatric literature involves case reports and small case series. Most paranasal sinus osteomas are slow-growing, found incidentally, and often asymptomatic. As the osteoma grows, the patient can become symptomatic. Symptoms vary depending on the location and can include proptosis, diplopia, congestion, facial pain, sinusitis episodes, and cosmetic changes [59]. Surgical excision also depends on tumor location and can be through open, endoscopic, or combined approaches, though endoscopic resection is often possible.
Juvenile ossifying fibroma
Juvenile ossifying fibromas are rare benign fibro-osseous lesions that can involve the paranasal sinuses and skull base [60]. These tumors tend to occur in the first or second decade of life and grow rapidly. Complete removal of this tumor, often achieved with extensive surgery, is necessary due to its aggressive and locally destructive course [61]. In a small case series of 11 patients undergoing endoscopic transnasal resection, ten patients were cured following surgery after mean follow-up of 25.8 months [62].
Skull base reconstruction
Skull base reconstruction is a critical component of successful skull base surgery. The goal of skull base reconstruction is to achieve a watertight seal in order to completely separate the sinonasal cavity from the brain, thereby preventing post-operative cerebrospinal fluid (CSF) leak, pneumocephalus, and infection. Post-operative CSF leak may manifest as rhinorrhea, meningitis, abscess, or death. Identification of a CSF leak warrants immediate repair. The principles applied to pediatric skull base reconstruction are mostly extrapolated from the adult skull base experience. Still, attention must be paid to balance pediatric skull base reconstruction to the cumulative impact of multimodality treatment on craniofacial growth, donor-site morbidity, and the potential for serious psychosocial issues [63].
Both onlay and inlay grafts may be used for skull base reconstruction. Onlay grafts are placed within the paranasal sinuses, abutting the skull base defect. These grafts are usually free mucosal grafts, which, when used alone, should be limited to small, low-flow CSF leak defects as elevated intracranial pressure can displace the reconstruction [64]. Often, onlay grafts are part of a multi-layered closure. Inlay grafts, also called underlay grafts, are placed intracranially between the dura and bone. These grafts are usually autologous fascia, biologic dural substitute, or rigid materials such as bone or porous polyethylene (Medpor, Stryker Inc, Kalamazoo MI). The presence of a circumferential ledge of bony is required to secure an inlay graft.
Skull base reconstruction is dependent on the size of the defect and the rate in which CSF is leaking. Small post-operative defects without an obvious CSF leak or only a low-flow CSF leak can be reconstructed with free grafts. Free grafts range from fascia (typically taken from the lateral thigh, the tensor fascia lata, or the temporalis), muscle, fat, cartilage, mucosa, bone, or a combination of these materials [65]. Abdominal fat also plays a critical role in the obliteration of dead space after tumor extirpation, conforming to the skull base defect in three dimensions, and is frequently used in conjunction with other reconstructive options to achieve a watertight seal [66, 67]. Tensor fascia lata or temporalis fascia provide pliable tissue that functionally replaces absent dura. Both fat and fascia have the disadvantage of a second surgical site and a finite tissue availability.
Larger skull base defects or defects in which there is a high-flow CSF leak warrant a multi-layered closure. Vascularized flaps are best suited for these defects as the robust blood supply and larger surface area ensure proper healing, which decreases post-operative complications. The nasoseptal flap (NSF), supplied by the posterior septal branch of the sphenopalatine artery, is a robust and reliable flap for endoscopic skull base repair [68]. The NSF can be easily harvested at the start of the case and has the distinct advantages of providing a large mucosal surface area with a wide arc of rotation. Bilateral NSF may be harvested in cases in which large defects are expected [69]. The superior incision for the NSF should extend anteriorly from the sphenoid ostia to avoid injury to the pedicle, while remaining 1 cm below the cribriform to avoid olfactory nerve injury [70]. The inferior incision is made parallel to the superior incision and may be at the inferior portion of the septum along the junction of the floor of the nose and septum or carried out along the nasal floor if additional tissue is needed.
Placement of the flap requires the removal of mucosa around the defect to prevent post-operative mucocele formation [71]. The NSF has been shown to reliably provide adequate flap coverage in children with sellar [72] and suprasellar defects [73••]. Ben-Ari et al. demonstrated the utility of the NSF in a case series of 12 pediatric patients undergoing endoscopic repair of anterior skull base lesions for benign and malignant diseases. The authors found craniofacial growth to be unimpaired and that the NSF had a high success rate and low complication rate [74•].
Other vascularized flaps may be utilized in certain surgical scenarios. The middle turbinate flap is pedicled posteriorly on the middle turbinate branch of the sphenopalatine artery [75]. The external pericranial flap provides a large, vascularized flap based upon the supraorbital and supratrochlear arteries. This flap can reconstruct the entire skull base if necessary but requires the creation of a bony window at the nasion to pass the flap into the nasal cavity [76]. The external temporoparietal fascia flap, based on the superficial temporal artery, is another option that provides large size, pliability, and bulk [77].
The gasket-seal closure is a reconstructive technique that has been recently described with excellent post-operative outcomes [78]. In a gasket-seal closure, a piece of tensor fascia lata (or acellular dermis) that is about 30% larger than the skull base defect is countersunk into the subdural space with a porous polyethylene plate. The gasket-seal closure is tailored to the defect, and then covered with a NSF to complete the multi-layered closure and is associated with a low rate of post-operative CSF leak. Reconstruction efforts are held in place with tissue sealants such as Duraseal, Adherus, or Tisseel and absorbable nasal packing. Another well-established reconstructive technique is the bilayered button, in which two pieces of fascia, one piece 25% larger than the skull base defect and the second the size of the defect, are sewn together [79]. The larger piece serves as the intracranial inlay graft, while the smaller piece functions as the onlay graft.
Conservative measures may play a role in assisting with skull base reconstruction techniques. Elevating the head of the bed 30°, stool softeners, avoiding strenuous activity and Valsalva maneuvers, and CSF diversion techniques such as lumbar drain or ventriculoperitoneal shunting can aid with removing intracranial pressure placed upon the reconstruction site. Lumbar drainage and other diversion methods are often times surgeon- or institution-specific [80].
Complications
Despite advances in technology, surgical technique, optics, and multidisciplinary team approaches, devastating complications may occur in endoscopic sinus and skull base surgery (Table 3). Prompt identification and proper management of these complications require an experienced and prepared surgical team.
Endocrine complications, most notably diabetes insipidus (DI), are frequently encountered in endoscopic transsphenoidal surgery. DI is the result of a decrease in antidiuretic hormone (ADH), resulting in a systemic water imbalance [81]. Symptoms include polyuria and polydipsia. If untreated, electrolyte and metabolic complications can result. Hallmarks of DI include large volumes of dilute urine (> 250 mL/hr for at least 2–3 consecutive hours), urine osmolality less than 200 mOsm/kg, increasing serum sodium levels (> 140–145 mEq/L), and urine specific gravity of 1.005 or less. Post-operative DI can be either transient or permanent and can be prevented by meticulous dissection [82]. Preservation of the pituitary stalk and superior hypophyseal arteries is critical to minimize the risk of DI. Elevated serum sodium can be treated with boluses of ½ normal saline. High urine output is treated with DDAVP [83]. Neuro-endocrinology consultant is often warranted in the pediatric intensive care setting for post-operative patients with DI. Other endocrine complications include hypocortisolemia or panhypopituitarism. Low cortisol levels may require maintenance oral steroids. Lifelong medication requirements or fertility issues should be discussed with patients and their families prior to surgery. An important complication related to craniopharyngioma surgery is post-operative obesity and hyperphagia [84, 85]. Alalade et al. [86] demonstrated that body mass index increased by 9% or more in 18% of patients undergoing endoscopic endonasal resection.
Meticulous hemostasis during surgery is essential for both patient safety and optimal intra-operative visualization. Bleeding can occur from discrete arterial supply and also be the result of dissection within the tumor bed, the cavernous sinus, intracranial arterial perforators, or catastrophic hemorrhage from the internal carotid artery [87, 88]. Control of bleeding can be achieved in a variety of different techniques. Topical vasoconstrictive agents, mono- or bi-polar electrocautery, or application of hemostatic agents, such as Floseal (Baxter Health Corp., Deerfield, Il) or Gelfoam (Pfizer Inc., New York, NY), all aid in the intra-operative control of bleeding.
Injury to the internal carotid artery (ICA), fortunately a rare complication, can be devastating. A thorough review of the course of the ICA on pre-operative imaging and intra-operative utilization of image guidance and Doppler ultrasound can avoid vascular injury [89]. If ICA injury occurs, direct pressure is necessary to temporize the bleeding. Transfer under general anesthesia to the Interventional Neuroradiology suite for angiography and possible occlusion should be performed.
Inadequate reconstruction of the skull base defect can result in post-operative complications. CSF leak, meningitis, pneumocephalus, and additional surgeries can lead to significant morbidity and mortality [90]. Lumbar drains are sometimes placed to assist with the reconstructive effort. By diverting CSF, pressure on the reconstruction site is reduced. However, lumbar drain placement is associated with complications including headache, retained foreign body, pneumocephalus, and prolonged immobility leading to deep vein thrombosis and/or pulmonary embolism [91].
Infectious complications, though uncommon, can have devastating outcomes [92]. Meningitis or intracranial abscess should be diagnosed promptly, with targeted management and treatment started immediately. Acute rhinosinusitis occurs rarely after ESBS but should be diagnosed and treated with cultured-directed antibiotics and nasal saline irrigations [93]. Management of chronic rhinosinusitis also requires thorough post-operative care.
Rhinologic complications can occur following ESBS. In the adult population, sinonasal quality of life may be reduced in the first 3–6 months after surgery, though generally quality of life scores return to baseline [94]. Sinonasal quality of life data is limited in the pediatric population though it may be extrapolated. Nasal crusting, synechiae, and sinusitis should be treated with debridement, saline rinses, and topical or oral therapies if indicated. A decreased sense of smell is noted following surgery in the adult population, though it tends to return to baseline within a year. Understanding pediatric sinonasal quality of life requires additional study.
Conclusion
Sinonasal and skull base lesions in children encompasses a variety of pathology that is best addressed with a multidisciplinary team of experts who are familiar and comfortable with treating these complex diseases. Intimate knowledge of the surgical anatomy will improve surgical and patient outcomes. Surgical approaches, techniques, objectives, and reconstruction are dependent on the location, pathology, extent of disease, and surgical goals. Reconstruction should follow an algorithmic ladder to achieve a watertight seal, separating the nasal cavity from the brain. Complications can be catastrophic and rely on prompt recognition and treatment to prevent long-term morbidity and mortality.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Chivukula S, Koutourousiou M, Snyderman CH, Fernandez-Miranda JC, Gardner PA, Tyler-Kabara EC. Endosocpic endonasal skull base surgery in the pediatric population. J Neurosurg Pediatric. 2013;11(3):227–41.
Ottenhausen M, Banu MA, Placantonakis DG, Tsiouris AJ, Khan OH, Anand VK, et al. Endoscopic endonasal resection of suprasellar meningiomas: the importance of case selection and experience in determing extent of resection, visual improvement, and complications. World Neurosurg. 2014;82(3-4):442–9.
Turri-Zanoni M, Zocchi J, Lambertoni A, Giovannardi M, Karligkiotis A, Battaglia P, et al. Endosocpic endonasal reconstruction of anterior skull base defects: what factors really affect the outcomes? World Neurosurg. 2018. https://doi.org/10.1016/j.wneu.2018.04.225.
LoPresti MA, Sellin JN, Demonte F. Developmental considerations in pediatric skull base surgery. J Neurol Surg B Skull Base. 2018;79(1):3–12.
Alalade AF, Oganda-Rivas E, Boatey J, Souweidane MM, Anand VK, Greenfield JP, et al. Suprasellar and recurrent pediatric craniopharyngiomas: expanding indications for the extended endoscopic transsphenoidal approach. J Neurosurg Pediatr. 2018;21(1):72–80.
Banu MA, Guerrero-Maldonado A, McCrea HJ, Garcia-Navarro V, Souweidane MM, Anand VK, et al. Impact of skull base development on endonasal endoscopic surgical corridors. J Neurosurg Pediatr. 2014;13(2):155–69.
Van Wyhe RD, Chamata ES, Hollier LH. Midline craniofacial masses in children. Semin Plast Surg. 2016;30(4):176–80.
Zapata S, Kearns DB. Nasal dermoids. Curr Opin Otolaryngol Head Neck Surg. 2006;14:406–11.
Hedlund G. Congenital frontonasal masses: developmental anatomy, malformations, and MR imaging. Pediatr Radiol. 2006;36:647–62 [quiz: 726–7].
Szeremeta W, Parikh TD, Widelitz JS. Congenital nasal malformations. Otolaryngol Clin N Am. 2007;40:97–112.
Fornadley JA, Tami TA. The use of magnetic resonance imaging in the diagnosis of the nasal dermoid sinus-cyst. Otolaryngol Head Neck Surg. 1989;101:397–8.
Pinheiro-Neto CD, Snyderman CH, Fernandez-Miranda J, et al. Endoscopic endonasal surgery for nasal dermoids. Otolaryngol Clin N Am. 2011;44:981–7ix.
Rahbar R, Shah P, Mulliken JB. The presentation and management of nasal dermoid: a 30-year experience. Arch Otolaryngol Head Neck Surg. 2003;129:464–71.
Rohrich RJ, Lowe JM, Schwartz MR. The role of open rhinoplasty in the management of nasal dermoid cysts. Plast Reconstr Surg. 1999;104:1459–66.
Skedros DG, Cass SP, Hirsch BE, Kelly RH. Beta-2 transferrin assay in clinical management of cerebral spinal fluid and perilymphatic fluid leaks. J Otolaryngol. 1993;22(5):341–4.
Ma J, Huang Q, Li X, Huang D, Xian J, Cui S, et al. Endoscopic transnasal repair of cerebrospinal fluid leaks with and without an encephalocele in pediatric patients: from infants to children. Childs Nerv Syst. 2015;31(9):1493–8.
Zeinalizadeh M, Sadrehosseini SM, Habibi Z, Nejat F, Silva HB, Singh H. Endonasal management of pediatric congenital transsphenoidal encephaloceles: nuances of a modified reconstruction technique. Technical note and report of 3 cases. J Neurosurg Pediatr. 2017;19(3):312–8.
Keshri AK, Shah SR, Patadia SD, Sahu RN, Behari S. Transnasal endoscopic repair of pediatric meningoencephalocele. J Pediatr Neurosci. 2016;11(1):42–5.
Gump WC. Endoscopic endonasal repair of congenital defects of the anterior skull base: Developmental considerations and surgical outcomes. J Neurol Surg B Skull Base. 2015;76(4):291–5.
Rahbar R, Resto VA, Robson CD. Nasal glioma and encephalocele: diagnosis and management. Laryngoscope. 2003;113(12):2069–77.
Patterson K, Kapur S, Chandra RS. Nasal gliomas and related brain heteropias: a pathologist’s perspective. Pediatr Pathol. 1986;5(3-4):353–62.
Liu Z, Wang D, Sun X, Wang J, Li H, Dai P. The site of origin and expansive routes of juvenile nasopharyngeal angiofibroma. Int J Pediatr Otorhinolaryngol. 2011;75(9):1088–92.
Snyderman CH, Pant H, Carrau RL, et al. A new endoscopic staging system for angiofibromas. Arch Otolaryngol Head Neck Surg. 2010;136:588–94.
Radkowski D, McGill T, Healy GB, et al. Angiofibroma. Changes in staging and treatment. Arch Otolaryngol Head Neck Surg. 1996;122:122–9.
Lee DA, Rao RB, Meyer JS, Prioleau PG, Bauer WC. Hormonal receptor determination in juvenile nasopharyngeal angiofibromas. Cancer. 1980;46:547–51.
Riggs S, Orlandi RR. Juvenile nasopharyngeal angiofibroma recurrence associated with exogenous testosterone therapy. Head Neck. 2010;32:812–5.
Ponti G, Losi L, Pellacani G, et al. Wnt pathway, angiogenetic and hormonal markers in sporadic and familial adenomatous polyposis-associated juvenile nasopharyngeal angiofibromas (JNA). Appl Immunohistochem Mol Morphol. 2008;16:173–8.
Thakar A, Gupta G, Bhalla AS, et al. Adjuvant therapy with flutamide for presurgical volume reduction in juvenile nasopharyngeal angiofibroma. Head Neck. 2011;33:1747–53.
Saylam G, Yücel OT, Sungur A, Onerci M. Proliferation, angiogenesis and hormonal markers in juvenile nasopharyngeal angiofibroma. Int J Pediatr Otorhinolaryngol. 2006;70:227–34.
• Rowan NR, Zwagerman NT, Heft-Neal ME, Gardner PA, Snyderman CH. Juvenile nasopharyngeal angiofibromas: a comparison of modern staging systems in an endoscopic era. J Neurol Surg B Skull Base. 2017;78(1):63–7 Updated staging system for JNAs that examines important pre-operative characteristics leading to residual disease and/or planned staged operations.
Reilly BK, Kim A, Pena MT, Dong TA, Rossi C, Murnick JG, et al. Rhabdomyosarcoma of the head and neck in children: review and update. Int J Pediatr Otorhinolaryngol. 2015;79(9):1477–83.
Chung SY, Unsal AA, Kilic S, Baredes S, Liu JK, Eloy JA. Pediatric sinonasal malignancies: a population-based analysis. Int J Pediatr Otorhinolaryngol. 2017;98:97–102.
Thompson CF, Kim BJ, Lai C, Grogan T, Elashoff D, St John MA, et al. Sinonasal rhabdomyosarcoma: prognostic factors and treatment outcomes. Inf Forum Allergy Rhinol. 2013;3(8):678–83.
Haubler SM, Stromberger C, Olze H, Seifert G, Knopke S, Bottcher A. Head and neck rhabdomyosarcoma in children: a 20-year retrospective study at a tertiary referral center. J Cancer Res Clin Oncol. 2018;144(2):371–479.
Yang A, Wickremesekera A, Parker A, Davis C. Surgical management of craniofacial and skull base rhabdomyosarcomas. J Craniofac Surg. 2009;20(5):1388–93.
Perry A, Graffeo CS, Marcellino C, Pollock BE, Wetjen NM, Meyer FB. Pediatric pituitary adenoma: case series, review of the literature, and a skull base treatment paradigm. J Neurol Surg B Skull Base. 2018;79(1):91–114.
Hanba C, Svider PF, Shkoukani MA, Sheyn A, Jacob JT, Eloy JA, et al. Pediatric pituitary resection: characterizing surgical approaches and complications. Int Forum Allergy Rhinol. 2017;7(1):72–9.
Zada G, Ditty B, McNatt SA, McComb JG, Krieger MD. Surgical treatment of Rathke cleft cysts in children. Neurosurgery. 2009;64(6):1132–7author reply 1137-1138.
Byun WM, Kim OL, Kim D. MR Imaging findings of rathke’s cleft cysts: significance of intracystic nodules. Am J Neuroradiol. 2000;21(3):485–8.
Lim HH, Yang SW. Risk factor for pituitary dysfunction in children and adolescents with Rathke’s cleft cysts. Korean J Pediatr. 2010;53(7):759–65.
Kim E. Symptomatic Rathke cleft cyst: clinical features and surgical outcomes. World Neurosurg. 2012;78(5):527–34.
Kuan EC, Yoo F, Chyu J, Bergseider M, Wang MB. Treatment outcomes of Rathke’s Cleft Cysts managed with marsupialization. J Neurol Surg B Skull Base. 2017;78(2):112–5.
Jahangiri A, Molinaro AM, Tarapore PE, et al. Rathke cleft cysts in pediatric patients: presentation, surgical management, and postoperative outcomes. Neurosurg Focus. 2011;31(1):E3.
Bunin GR, Surawicz TS, Witman PA, Preston-Martin S, Davis F, Bruner JM. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89:547–51.
Fernandez-Miranda JC, Gardner PA, Snyderman CH, Devaney KO, Strojan P, Suarez C, et al. Craniopharyngioma: a pathologic, clinical and surgical review. Head Neck. 2012Jul;34(7):1036–44.
Yamada S, Fukuhara N, Yamaguchi-Okada M, Nishioka H, Takeshita A, Takeuchi Y, et al. Therapeutic outcomes of transsphenoidal surgery in pediatric patients with craniopharyngiomas: a single-center study. J Neurosurg Pediatr. 2018;30:1–14 Gross total resection is more likely in primary surgery with lower likelihood for recurrence. Postoperative CSF leak is more likely in repeat surgery patients.
Patel VS, Thamboo A, Quon J, Nayak JV, Hwang PH, Edwards M, et al. Outcomes after endoscopic endonasal resection of craniopharyngiomas in the pediatric population. World Neurosurgery. 2017;108:6–14.
Alalade AF, Oganda-Rivas E, Boatey J, Souweidane MM, Anand VK, Greenfield JP, et al. Suprasellar and recurrent pediatric craniopharyngiomas: expanding indications for the extended endoscopic transsphenoidal approach. J Neurosurg Pediatr. 2018;21(1):72–80.
Merchant TE, Kiehna EN, Sanford RA, Mulhern RK, Thompson SJ, Wilson MW, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int J Radiat Oncol Biol Phys. 2002;53(3):533–42.
Graffeo CS, Perry A, Link MJ, Daniels DJ. Pediatric craniopharyngiomas: a primer for the skull base surgeon. J Neurol Surg B Skull Base. 2018;79(1):65–80.
Mohyeldin A, Prevedello DM, Jamshidi AO, Ditzel Filho LF, Carrau RL. Nuances in the treatment of malignant tumors of the clival and retroclival region. Int Arch Otorhinolaryngol. 2014;18(Suppl 2):S157–72.
Campbell RG, Prevedello DM, Ditzel Filho L, Otto BA, Carrau RL. Contemporary management of clival chordomas. Curr Opin Otolaryngol Head Neck Surg. 2015;23:153–61.
Rassi MS, Hulou MM, Almefty K, Bi WL, Pravdenkova S, Dunn IF, et al. Pediatric clival chordoma: a curable disease that conforms to Collins’ Law. Neurosurgery. 2018;82(5):652–60.
Bilginer B, Turk CC, Narin F, Hanalioglu S, Oguz KK, Ozgen B, et al. Enigmatic entity in childhood: clival chordoma from a tertiary center’s perspective. Acta Neurochir. 2015;157(9):1587–93.
Tsitouras V, Wang S, Dirks P, Drake J, Bouffet E, Hawkins C, et al. Management and outcomes of chordomas in the pediatric populations: the Hospital for Sick Children experience and review of the literature. J Clin Neurosci. 2016;34:169–76.
Stapleton AL, Tyler-Kabara EC, Gardner PA, Snyderman CH, Wang EW. Risk factors for cerebrospinal fluid leak in pediatric patients undergoing endoscopic endonasal skull base surgery. Int J Pediatr Otorhinolaryngol. 2017;93:163–6.
Wilson M, Snyderman C. Fibro-Osseous lesions of the skull base in pediatric population. J Neurol Surg B Skull Base. 2018;79(1):31–6.
Stapleton AL, Tyler-Kabara EC, Gardner PA, Snyderman CH. Endoscopic endonasal surgery for benign fibro-osseous lesions of the pediatric skull base. Laryngoscope. 2015;125(9):2199–203.
Ishii T, Sakamoto Y, Miwa T, Yoshida K, Kishi K. A giant osteoma of the ethmoid sinus. J Craniofac Surg. 2018;29(3):661–2.
Carter J, Winters R, Yang C, St Hilaire H, Rodriguez K. Juvenile ossifying fibroma of the middle turbinate. J La State Med Soc. 2014;166(3):100–2.
Kim DY, Lee OH, Choi GC, Cho JH. A case of juvenile psammomatoid ossifying fibroma on skull base. J Craniofac Surg. 2018;29(5):e497–9.
Wang M, Zhou B, Cui S, Li Y. Juvenile psammomatoid ossifying fibroma in paranasal sinus and skull base. Acta Otolaryngol. 2017;137(7):743–9.
Duek I, Pener-Tessler A, Yanko-Arzi R, Zaretski A, Abergel A, Safadi A, et al. Skull base reconstruction in the pediatric patient. J Neurol Surg B Skull Base. 2018;79(1):81–90.
Marks SC. Middle turbinate graft for repair of cerebral spinal fluid leaks. Am J Rhinol. 1998;12(6):417–9.
Ting JY, Metson R. Free graft techniques in skull base reconstruction. Adv Otorhinolaryngol. 2013;74:33–41.
Ziu M, Jimenez DF. The history of autologous fat graft use for prevention of cerebrospinal fluid rhinorrhea after transsphenoidal approaches. World Neurosurg. 2013;80(5):554–62.
Wormald PJ, McDonogh M. The bath-plug closure of anterior skull base cerebrospinal fluid leaks. Am J Rhinol. 2003;17(5):299–30.
Hadad G, Bassagasteguy L, Carrau RL, et al. A novel reconstructive technique after endoscopic expanded endonasal approaches: vascular pedicle nasoseptal flap. Laryngoscope. 2006;116(10):1882–6.
Nyquist GG, Anand VK, Singh A, Schwartz TH. Janus flap: bilateral nasoseptal flaps for anterior skull base reconstruction. Otolaryngol Head Neck Surg. 2010;142(3):327–31.
Upadhyay S, Buohliqah L, Dolci RLL, Otto BA, Prevedello DM, Carrau RL. Periodic olfactory assessment in patients undergoing skull base surgery with preservation of the olfactory strip. Laryngoscope. 2017;127(9):1970–5.
Verillaud B, Genty E, Leboulanger N, Zerah M, Garabedian EN, Roger G. Mucocele after transnasal endoscopic repair of traumatic anterior skull base fistula in children. Int J Pediatr Otorhinolaryngol. 2011;75(9):1137–42.
Purcell PL, Shinn JR, Otto RK, Davis GE, Parikh SR. Nasoseptal flap reconstruction of pediatric sellar defects: a radiographic feasibility study and case series. Otolaryngol Head Neck Surg. 2015;152(4):746–51.
Ghosh A, Hatten K, Learned KO, Rizzi MD, Lee JY, Storm PB, et al. Pediatric nasoseptal flap reconstruction for suprasellar approaches. Laryngoscope. 2015;125(11):2451–6 Nasoseptal flap is a viable option for pediatric skull base reconstruction, providing adequate length and width.
Ben-Ari O, Wengier A, Ringel B, Carmel Neiderman NN, Ram Z, Margalit N, et al. Nasoseptal flap for skull base reconstruction in children. J Neurol Surg B Skull Base. 2018;79(1):37–41 Nasoseptal flap can be utilized for skull base reconstruction and does not appear to alter facial growth.
Prevedello DM, Barges-Coll J, Fernandez-Miranda JC, et al. Middle turbinate flap for skull base reconstruction: cadaveric feasibility study. Laryngoscope. 2009;119(11):2094–8.
Patel MR, Shah RN, Snyderman CH, et al. Pericranial flap for endoscopic anterior skull-base reconstruction: clinical outcomes and radioanatomic analysis of preoperative planning. Neurosurgery. 2010;66(3):506–12discussion 512.
Fortes FS, Carrau RL, Snyderman CH, et al. Transpterygoid transposition of a temporoparietal fascia flap: a new method for skull base reconstruction after endoscopic expanded endonasal approaches. Laryngoscope. 2007;117(6):970–6.
Garcia-Navarro V, Anand VK, Schwartz TH. Gasket seal closure for extended endonasal endoscopic skull base surgery: efficacy in a large case series. World Neurosurg. 2013;80(5):563–8.
Luginbuhl AJ, Campbell PG, Evans J, Rosen M. Endoscopic repair of high-flow cranial base defects using a bilayer button. Laryngoscope. 2010;120(5):876–80.
Greenfield JP, Anand VK, Kacker A, et al. Endoscopic endonasal transethmoidal transcribriform transfovea ethmoidalis approach to the anterior cranial fossa and skull base. Neurosurgery. 2010;66(5):883–92discussion 892.
Tabaee A, Anand VK, Barron Y, Hiltzik DH, Brown SM, Kacker A, et al. Schwartz TH: predictors of short-term outcomes following endoscopic pituitary surgery. Clin Neurol Neurosurg. 2009;111:119–22.
Nemergut EC, Zuo Z, Jane J, Laws ER. Predictors of diabetes insipidus after transsphenoidal surgery: a review of 881 patients. J Neurosurg. 2005;103:448–54.
Kiran Z, Sheikh A, Momin SN, Majeed I, Awan S, Rashid O, et al. Sodium and water imbalance after sellar, suprasellar, and parasellar surgery. Endocr Pract. 2017;23(3):309–17.
Sorva R. Children with craniopharygioma: early growth failure and rapid postoperative weight gain. Acta Paediatr Scand. 1988;77(4):587–92.
Curtis J, Daneman D, Hoffman HJ, Erlich RM. The endocrine outcome after surgical removal of craniopharyngiomas. Pediatr Neurosurg. 1994;21(suppl 1):24–7.
Alalade AF, Oganda-Rivas E, Boatey J, Souweidane MM, Anand VK, Greenfield JP, et al. Suprasellar and recurrent pediatric craniopharyngiomas: expanding indications for the extended endoscopic transsphenoidal approach. J Neurosurg Pediatr. 2018;21(1):72–80.
Vaz-Guimaraes F, Su SY, Fernandez-Miranda JC, Wang E, Synderman CH, Gardner P. Hemostasis in endoscopic endonasal skull base surgery. J Neurol Surg B. 2015;76:296–302.
Gardner PA, Tormenti MJ, Pant H, Fernandez-Miranda JC, Snyderman CH, Horowitz MB. Carotid artery injury during endoscopic endonasal skull base surgery: incidence and outcomes. Neurosurgery. 2013;72:261–9.
Romero DCB, Gangadharan JL, Gobin YP, Anand VK, Schwartz TH. Managing arterial injury in endoscopic skull base surgery. Case series and review of the literature. Operative. Neurosurgery. 2017;13(1):138–49.
Banu MA, Szentirmai O, Mascarenhas L, Al Amin S, Anand VK, Schwartz TH. Pneumocephalus patterns following endonasal endoscopic skull base surgery as predictors of post-operative CSF leak. J Neurosurg. 2014;4:1–15.
Naunheim MR, Sedaghat AR, Lin DT, Bleier BS, Holbrook EH, Curry WT, et al. Immediate and delayed complications following endoscopic skull base surgery. J Neurol Surg B Skull Base. 2015Sep;76(5):390–6.
Brown SM, Anand VK, Tabaee A, Schwartz TH. Role of perioperative antibiotics in endoscopic skull base surgery. Laryngoscope. 2007Sep;117(9):1528–32.
Nyquist GG, Friedel ME, Singhal S, Beahm DD, Farrell CJ, Evans JJ, et al. Surgical management of rhinosinusitis in endoscopic-endonasal skull-base surgery. Int Forum Allergy Rhinol. 2015;5(4):339–43.
Bedrosian JC, McCoul ED, Raithatha R, Akselrod OA, Anand VK, Schwartz TH. A prospective study of postoperative symptoms in sinonasal quality of life following endoscopic skull-base surgery: dissociations based on specific symptoms. Int Forum Allergy Rhinol. 2013;3:664–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Charles A. Riley declares that he has no conflict of interest. Christian P. Soneru declares that he has no conflict of interest. Marc L. Otten declares that he has no conflict of interest. David A. Gudis declares that he has no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric Allergy
Rights and permissions
About this article

Cite this article
Riley, C.A., Soneru, C.P., Otten, M.L. et al. Management of Pediatric Sinonasal and Skull Base Lesions. Curr Treat Options Allergy 6, 253–271 (2019). https://doi.org/10.1007/s40521-019-00216-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40521-019-00216-z