
Abstract
Purpose of review
To explain the underlying pathophysiology of cystic fibrosis and to describe current treatment modalities for disease manifestations, including an introduction to novel therapies known as CFTR modulator drugs, which are now available to over 50% of people with cystic fibrosis.
Recent findings
CFTR modulator drugs, aimed at restoring CFTR protein function, have been shown to improve lung function and reduce pulmonary exacerbations, improve BMI and QOL, and are a new added treatment to the traditional therapies for cystic fibrosis.
Summary
With the current modalities of treatment available to patient with cystic fibrosis, the quality of life and the life expectancy continues to improve. Future directions for research include looking at novel anti-inflammatory agents, antimicrobial agents, mucociliary clearance agents, and newer generation CFTR modulator drugs/drugs that restore CFTR function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cystic fibrosis (CF) is a genetic disease caused by an abnormality in the membrane protein, cystic fibrosis transmembrane conductance regulator (CFTR). This abnormality leads to thick secretions throughout the body, and progressive dysfunction of multiple organ systems.
CF is a common disease with nearly 30,000 patients in the USA, 80,000 patients worldwide. One in 29 Caucasians carry a CFTR gene mutation. Currently, median predicted survival is over 43 years, and the majority of the patients (53%) are over 18 years old [1•]. While multiple organ systems are affected, the primary cause of mortality is respiratory.
CF is an autosomal recessive disease, and the gene encoding for the CFTR protein is located on chromosome 7. CFTR is located in the apical cell membrane, and functions to regulate water and ion movement. Specifically, it actively transports chloride out of the cell, and inhibits sodium resorption through inhibition of an associated ion channel, the epithelial sodium channel (ENaC). CFTR dysfunction causes excessive sodium and water resorption into cells, with a resultant dehydration of secretions.
There are over 1800 known mutations that cause CF, with the most common mutation being F508del. Mutations can be divided into 5 classes, based on the error in CFTR protein production. These classes can be used to both provide general prognostic information, and provide specific targets for therapy.
Class I mutations include nonsense mutations, where a single point alteration in DNA creates a stop codon interfering with protein production. The process of translation is thus terminated prematurely, resulting in a CFTR protein that has little or no function. Class II mutations involve mutations that affect protein processing. It results from mutations that cause abnormalities in protein folding, altering its overall shape and function, or abnormalities in protein transport to the cell surface. F508del is the most common mutation in CF, and a class II mutation. Class III mutations involve abnormalities in protein gating, or the ability to remain open to chloride transport. Class IV mutations affect CFTR conduction, or the ability to function once inserted into the cell surface. Class V mutations result in a diminished amount of functioning CFTR at the cell surface. This could result from decreased production or increased protein turnover.
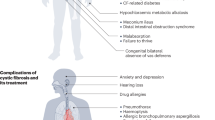
Complications of CF include progressive effects on the respiratory, gastrointestinal, endocrine, and reproductive systems. Within the respiratory system, dehydration of secretions leads to a diminished airway surface liquid layer that is essential to the function of mucociliary clearance. With this diminished surface liquid layer, mucociliary clearance is impaired. Without effective mucus clearance, bacteria chronically colonize and then infect the respiratory system. The most common pathogens in CF include Staphylococcus aureus in children, and Pseudomonas aeruginosa in adolescence and adulthood. Chronic infection leads to chronic inflammation, with release of various factors, and bronchiectasis within the lungs. Inflammation is typically characterized as neutrophilic, with elevated circulating levels of IgG, IgA, IgM, and elevated elastase in the sputum [2]. Sinus disease is also very prevalent in CF [3], involving chronic sinusitis and an increased prevalence of nasal polyps (7–48%) [4, 5]. These polyps are unique in that they are mediated by Th1 neutrophilic inflammation, rather than eosinophilic Th2 inflammation. Chronic inflammation is noted to cause goblet cell hyperplasia, squamous metaplasia, and loss of ciliated cells.
Within the gastrointestinal system, pancreatic and hepatobiliary dysfunction is common. GI disease can present as early as infancy with bowel obstruction and meconium ileus. Pancreatic insufficiency is present in over 85% of CF patients, with resultant effects on nutritional status, including fat soluble vitamin deficiencies. Ten percent of patients will develop pancreatitis [6], and 30% will develop gastroesophageal reflux disease. Unique to CF, distal intestinal obstruction syndrome (DIOS) is obstruction of the ileocecum with fecal matter. Hepatobiliary disease can include duct stenosis, hepatic steatosis, focal biliary cirrhosis, and portal hypertension. Cirrhosis is prevalent in 2 to 15% of patients [7].
Within the endocrine system, the pancreatic dysfunction leads to CF-related diabetes (CFRD) characterized by normal insulin sensitivity and reduced insulin production. This leads to poor nutritional status and increased mortality. The prevalence of CFRD in CF patients is over 20%.
In the reproductive system, 98% of men with CF are infertile due to congenital bilateral absence of the vas deferens. This does not affect women, and men with this condition can utilize assisted reproductive technologies if they wish to have children.
The treatment of CF has advanced with a resultant increase in median survival of less than 15 years old in the 1970s to over 40 years currently. Treatment had been developed to mitigate the effects of each organ dysfunction. However, in 2012, the FDA approved a novel medication, ivacaftor, which targets the underlying cause of CF (CFTR dysfunction). Since then, multiple medications have been approved and are in the drug pipeline as the new class of CFTR modulators.
Chronic treatments of pulmonary manifestations of CF
Airway obstruction from dry and thickened secretions is the central precipitating cause of lung disease in CF. Consequently, airway clearance therapies are an essential part in maintaining lung health.
Dornase alfa
Degeneration of neutrophils, abundant in purulent CF sputum, leads to a significant presence of DNA causing a high viscosity of respiratory secretions. Dornase alfa (recombinant human DNase) is an endonuclease administered by inhalation via nebulizer that selectively cleaves strands of DNA. By cleaving the large amounts of DNA, dornase reduces sputum viscosity and facilitates airway clearance. Studies evaluating the short-term (6–14 days) and long-term (12–96 weeks) efficacy of dornase as compared with placebo showed significant improvement in lung function by measure of FEV1 [8]. A meta-analysis of randomized control trials showed an overall reduction in pulmonary exacerbations in addition to improvement in lung function [8]. Dornase is generally well-tolerated with the most common adverse reaction of voice alteration. The Cystic Fibrosis Foundation (CFF) recommends regular use of DNase for all CF patients 6 years or older regardless of lung disease severity given the significant net benefit and low risk profile [9].
Hypertonic saline
Inhalational hypertonic saline (HS) is thought to hydrate the dried and thickened CF sputum allowing for improved airway clearance. In a multicenter study comparing twice daily treatment with 7% saline compared with normal saline, a 56% reduction in pulmonary exacerbations was seen in the hypertonic saline group [10]. Studies comparing HS with once daily treatment with dornase alfa revealed improvement in FEV1 in both groups but a larger increase was noted in the DNase group [8]. HS is generally well-tolerated but is known to cause bronchospasm and severe cough. Pre-treatment with an inhaled bronchodilator is routinely used to mitigate these symptoms. The CFF recommends regular use of inhaled HS for all CF patients 6 years or older regardless of disease severity given the benefits of reducing pulmonary exacerbation frequency and improving lung function [9].
Chest physiotherapy
Chest physiotherapy is an essential component in the treatment of CF patients and is often used in conjunction with inhalational therapies to facilitate clearance of thick airway secretions. Benefits of chest physiotherapy include improved sputum expectoration, quality of life, and possible improvement in lung function [8]. There are a wide range of techniques and medical devices that may be used for chest physiotherapy in the care of CF patients. Breathing and coughing techniques such as “huffing” or “autogenic drainage” can be taught to patients and be performed independently. Direct percussion of the chest, which can be performed by caregivers, in conjunction with postural drainage, where patients lie and sit in various positions facilitates mobilization of secretions. Devices such as a high-frequency chest wall oscillation device (percussion vest) or a positive expiratory pressure mask can also be used in individuals where cough techniques are inadequate for airway clearance or as an adjunct. Exercise is also recommended to facilitate mobilization of secretions. Adherence is often a challenge as treatments are time consuming.
Bronchodilators
Airflow obstruction in CF manifests due to plugging of airways from thick secretions, bronchial wall thickening, airway destruction, and in a subset of patients, bronchial hyperreactivity. Studies examining short-term treatment with β2 adrenergic receptor agonists have demonstrated improvement in lung function as measured by FEV1 or peak expiratory flow rate especially in patients with known bronchial hyperreactivity [11]. However, the benefits with long-term use were not sustained [9]. While β2 adrenergic receptor agonists are routinely used in conjunction with other inhalational therapies such as HS to mitigate potential for bronchospasm and to facilitate airway clearance, there is insufficient evidence to support chronic use of β2 adrenergic agonists in improving lung function or reducing exacerbations as determined by the CFF [9]. The Cystic Fibrosis Foundation’s Pulmonary Clinical Practice Guidelines Committee was created to help aid practitioners in the use of chronic medications. The committee’s recommendation for the use of chronic inhaled β2 adrenergic agonists remains “low” due to the lack of evidence from RCTs of long-term use β2 adrenergic agonists. We were unable to find any RCTs of use of β2 adrenergic agonists beyond 6 months of time. Additionally, there is insufficient evidence to support routine use of inhaled anticholinergic agents such as ipratropium for chronic pulmonary disease in CF [9].
There is no consensus on the most effective order of therapies for clearance of sputum owing to the paucity of evidence but in practice, chest physiotherapy is typically performed after administration of inhalational therapies starting with bronchodilators followed by HT saline and dornase. Inhaled antibiotics are usually administered following chest physiotherapy.
Anti-inflammatory agents
A pathologic feature of CF is an inappropriate, excessive inflammatory response that leads to airway destruction, bronchiectasis, and ultimately severe obstructive airway disease. Various anti-inflammatory agents including inhaled corticosteroids, oral corticosteroids, non-steroidal anti-inflammatory drugs, and macrolide antibiotics have been evaluated for their potential effects on lung function and pulmonary exacerbations. While inhaled and oral corticosteroids have not demonstrated significant benefit in clinical studies, ibuprofen and azithromycin have demonstrated potential for improvement in lung function [8, 12]. Inhaled corticosteroids are not recommended in the absence of a concurrent diagnosis of asthma and while oral corticosteroids have demonstrated efficacy in CF, they are not recommended due to the greater concern for their side effects with chronic use [9, 12]. The CFF recommends treatment with twice daily ibuprofen in patients ages 6–17 years old with a baseline FEV1 ≥ 60% predicted [9]. A double blind randomized control trial including patients between age of 5 to 39 years with baseline FEV1 of at least 60% predicted on enrollment treated with ibuprofen versus placebo showed a significant decrease in the rate of decline in lung function in the ibuprofen group with the effect most profound in patients less than 13 years old [13]. The study did not find a significant difference in adverse events [13]. Lack of data in patients over the age of 18 years old led the CFF to narrow recommended age range for treatment with ibuprofen [9]. Treatment with ibuprofen requires maintenance of a specific peak plasma concentration in patients [9]. While inflammation in the lungs is present from birth, structural lung disease develops over time as a result of infection and ongoing inflammation. Also, FEV1 declines most rapidly in patients with mild disease (i.e. younger patients). Thus, treatment of inflammation prior to the presence of significant lung disease (in the age group < 13 years old) who are at risk for the largest FEV1 decline would be expected to have the largest treatment effect.
Azithromycin, a macrolide antibiotic, is recommended for chronic treatment in patients with and without persistent Pseudomonas aeruginosa in sputum cultures [9]. Treatment of patients with persistent Pseudomonas has been shown to improve lung function and decrease risk for exacerbations [8]. A systematic review found a significant improvement in FEV1 (3.6–6.2%) in patients treated with azithromycin compared with placebo [14]. Azithromycin is also recommended for patients without chronic colonization with Pseudomonas to reduce risk for pulmonary exacerbations [9, 14]. A large trial evaluating treatment with azithromycin compared with placebo found a 50% reduction in pulmonary exacerbations despite no significant change in lung function [15]. Screening for nontuberculous mycobacteria is routinely performed prior to treatment initiation and azithromycin should not be used if present to avoid induction of macrolide resistance. Macrolides are believed to provide anti-inflammatory and antimicrobial effects. While macrolides do not have bactericidal effects on Pseudomonas, they are believed to inhibit their ability to produce biofilms.
There are other anti-inflammatory agents currently in phase 2 trials, including acebilustat (CTX-4430) and lenabasum (JBT-101). A phase 2 trial of lenabasum completed in 2016 showed that the oral drug reduced the rate of acute pulmonary exacerbations. A phase 2 trial with acebilustat, an oral drug that reduces production of leukotriene B4, is ongoing.
Chronic antibiotic therapy
Chronic infections from multiple organisms lead to intermittent pulmonary exacerbations and a persistent decline in lung function as part of the natural course of CF. Colonization with Pseudomonas aeruginosa is a risk factor for more rapid decline in lung function and decreased survival. In addition to chronic azithromycin therapy as discussed in the previous section, inhaled tobramycin and inhaled aztreonam are recommended by the CFF in patients 6 years or older with mild to severe lung disease and sputum with Pseudomonas aeruginosa [9]. Studies have demonstrated improvement in lung function, decrease in frequency of exacerbations and improvement in quality in life in patients with moderate to severe lung disease who received inhaled tobramycin or aztreonam compared with placebo [8, 9, 16]. In mild lung disease, inhaled tobramycin has been shown to reduce the frequency of pulmonary exacerbations and inhaled aztreonam has been shown to improve lung function, decrease frequency of exacerbations, and improve quality of life [9, 16]. Other inhaled antibiotics including colistin and gentamicin, and oral antibiotics targeting Pseudomonas or Staphylococcus aureus, are not routinely used in the USA as there is insufficient evidence to support their use and the CFF does not recommend their use [9].
Currently, other aerosolized antibiotics including inhaled vancomycin for treatment of MRSA airway infection and inhaled levofloxacin for treatment of chronic lung infections caused by Pseudomonas aeruginosa are in phase 3 trials.
Table 1 reviews the chronic treatments of pulmonary manifestations in cystic fibrosis discussed in this article.
Chronic treatment of extrapulmonary manifestations of CF
Sinusitis
Involvement of nasal and sinus cavities is present in nearly all patients with CF when evaluated radiographically or endoscopically; however, as few as 10% of patients report significant symptoms [17•]. The most common symptoms include chronic nasal congestion, nasal drainage, and headaches. Current treatment is directed towards addressing impairment of mucociliary clearance, management of chronic inflammation, and directed antimicrobial therapy though consensus guidelines for the treatment of CF-related sinusitis do not exist [3, 17•]. Isotonic or hypertonic nasal saline rinses are used to debride crusted secretions and hydrate thick obstructing mucus in the sinuses. Topical corticosteroids have been demonstrated to reduce the size of nasal polyps, which are seen in up to one-third of patients with CF sinusitis, resulting in an improvement in presenting symptoms [3]. Topical tobramycin has been shown to be effective in reducing CF sinusitis symptoms and severity of chronic infections [3]. Chronic macrolide therapy (as discussed in prior section) is also used in management of chronic rhinosinusitis in CF patients based on similar pathophysiology of sinonasal and pulmonary manifestations. In addition to medical management, functional endoscopic sinus surgery may also be required to remove obstructing polyps, clearing obstructive mucus and opening sinus passages to allow drainage of secretions and has been shown to be effective in improving symptoms and may also help with improving lung function [3, 18•].
Nutrition
CF patients with normal weight-for-age, height-for-age, and weight-for-height percentiles have better lung function and overall survival [19]. Consequently, maintaining adequate caloric intake is essential in the care of this patient population. Caloric intake of 110–200% of requirements for a healthy individual of similar age, sex, and size is needed to achieve weight goals. In children and adults with weight deficits, the CFF recommends the use of nutritional supplements to improve weight gain [19].
Pancreatic insufficiency is the most common GI manifestation in CF. Fat malabsorption due to inadequate production of pancreatic enzymes contributes to the risk of malnutrition. Consensus-based guidelines recommend pancreatic enzyme replacement therapy of 500 to 2500 units of lipase per kilogram body weight per meal; however, specific dosing to in relation to growth response is not known [19].
Constipation/DIOS
Constipation from fecal impaction in the colon is experienced by up to 50% of CF patients and is thought to occur due to dehydrated bowel contents, pancreatic insufficiency, and dysmotility [20]. Osmotic laxatives are most commonly used to treat constipation. DIOS is distinguished from constipation as it is characterized by an acute partial or complete obstruction of the ileocecum by intestinal contents. Similar to constipation, abnormal water, and electrolyte composition of intestinal contents, pancreatic insufficiency and bowel dysmotility are all believed to play a role in development of DIOS [21•]. Management with osmotic laxatives, such as GoLytely, and rehydration is essential. In cases of complete obstruction, decompression with a nasogastric tube and treatment with hyperosmolar contrast enemas, such as Gastrografin, are often needed [21•]. Severe cases with peritonitis require surgical intervention.
CFTR modulator drugs
While all the treatment modalities listed above are aimed at treating the manifestations of CF, newer modalities of treatment are aimed at correcting CFTR protein function by targeting specific defects caused by the CFTR gene mutation, and thereby treat the underlying cause for the disease. These exciting new oral drugs have been shown not only to improve lung function (measured as FEV1) but also to reduce the frequency and duration of pulmonary exacerbations, improve nutritional status, and improve quality of life. Since pulmonary exacerbations can often lead to inpatient hospitalizations and permanent loss of lung function, this is a particularly important measurement when evaluating the efficacy of these drugs.
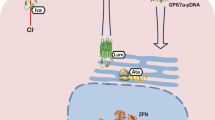
The 2 classes of CFTR modulator drugs currently approved for use are classified as “potentiator” and “corrector” drugs. Potentiator drugs are aimed at increasing the opening of the CFTR channel, while the corrector drugs are aimed at helping the CFTR protein form the right shape so it can be trafficked to the cell surface. These drugs can be used in combination to ensure enough protein makes it to the cell surface and then assist in the channel staying open to allow for ion flow.
Ivacaftor, a CFTR potentiator, was the first CFTR modulator drug to gain approval in 2012. The drug was initially studied in patients with the class III “gating” mutation G551D. Initial data demonstrated a change from baseline through week 24 in the percent predicted FEV1 of 10.6 percentage points in the ivacaftor group (P < 0.001) [22]. These effects were noted on pulmonary function within 2 weeks of initiating the drug, and were sustained through 48 weeks, and even longer in the open label extension study. Patients were also noted to have a reduction in pulmonary exacerbations, to have substantial weight gain, and an improvement in their quality of life measurement. Finally, a notable reduction in sweat chloride measurements also demonstrated the effectiveness on chloride channel function. Since 2012, further research has allowed the approval of the drug to be expanded, and ivacaftor is now available to patients with CF age 6 months and older with “gating” mutations, and several other residual function mutations that produce some CFTR protein on the epithelial cell surface. Of note, in 2017, the FDA allowed for drug testing on cells in the laboratory for label extension to patients with rare mutations who otherwise would not be recruited for larger studies, another exciting step to get effective drugs to patients.
Lumacaftor, a CFTR corrector, in combination with the potentiator ivacaftor was the next CFTR modulator drug to gain approval for CF patients with 2 copies of the F508del mutation in 2015. In vitro, lumacaftor was shown to correct the CFTR misprocessing in the F508del mutation. Monotherapy with ivacaftor or lumacaftor alone was not shown to be efficacious in patients with 2 copies of the F508del mutation. However, when used in combination, these drugs have shown promising clinical outcomes for these patients. Clinical trials of the lumacaftor-ivacaftor drug showed a mean absolute improvement in the percent predicted FEV1 ranging from 2.6 to 4.0 percentage points (P < 0.001), and a pooled analysis of these studies showed that the rate of pulmonary exacerbations was 30 to 39% lower in the lumacaftor-ivacaftor groups than in the placebo group [23•]. Patients were also noted to have weight gain and an improvement in their quality of life measurement score. Some patients, however, experience respiratory side effects with this drug combination, including chest tightness and dyspnea, limiting its ability to be used on all homozygous F508del patients. Currently, the lumacaftor-ivacaftor combination drug is available to people age 2 and older with 2 copies of the F508del mutation.
A newer corrector drug, tezacaftor, in combination with the potentiator ivacaftor, is the most recently approved CFTR modulator drug. This newer generation CFTR modulator drug was approved in 2018, for people with CF age 12 and older with 2 copies of the F508del mutation, and for people with CF who have a single copy of 26 other specified mutations. The tezacaftor–ivacaftor combination drug demonstrated an absolute change from baseline in the percent predicted FEV1 of 4.0 percentage points (P < 0.001). The rate of pulmonary exacerbation was 35% lower in the tezacaftor–ivacaftor group than in the placebo group (P = 0.005) [24••]. This drug also has notably less respiratory side effects than lumacaftor-ivacaftor combination drug.
Finally, recent data published demonstrates that next-generation CFTR correctors (VX-445 and VX-659) in combination with tezacaftor–ivacaftor may lead to the development of promising triple combination CFTR modulator drugs to become available to CF patients. These triple combination drugs have been evaluated in both patients homozygous for the F508del mutation, and in patients with only one copy of F508del mutation and one copy of a minimally functional gene mutation. Phase 2 trials with the triple combination drugs resulted in an increased percentage of predicted FEV1 of up to 13.8 points in the heterozygote F508del patients (P < 0.001), along with a decrease in sweat chloride concentrations and an improvement in the quality of life measurement score [25••, 26••]. Phase 3 trials are ongoing. If one of these promising combinations is approved for use, this would bring CFTR modulator drug availability to approximately 90% of patients with CF.
The CFTR modulator drugs provide a novel approach to disease management in CF. Ivacaftor, the longest available CFTR modulator drug, is currently intended for lifelong use. Review of existing cystic fibrosis registry data indicates that ivacaftor-treated patients have a lower prevalence of CF-related complications, and have lower risk of hospitalizations and pulmonary exacerbations, lower risk of organ transplantation, and lower risk of death, when compared with matched comparators. Thus far long-term registry data for the lumacaftor-ivacaftor combination also demonstrates a rate of progressive decline in lung function that is lower than untreated matched control registry patients [26•]. This data suggests that CFTR modulator drugs are truly disease-modifying agents. Additional CFTR modulator drugs are currently in the research pipeline, such as those listed above, including a new class of CFTR modulator drugs considered CFTR “amplifiers.”
Table 2 provides an overview of the current FDA approved CFTR modulator drugs discussed in this article.
Conclusion
CF is a common inherited genetic disease caused by a defect in the CFTR ion channel. This defect leads to progressive multiorgan dysfunction. Treatments have historically been focused on mitigating the end effects of this dysfunction. This has led to a significant increase in left expectancy. Recently, a new class of medications, the CFTR modulators, has been developed to act directly upon the CFTR protein. This promising class of medications has a large treatment effect and is specific to each mutation. Ongoing development has expanded the treatment population by targeting more CF mutations.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• 2017 Patient Registry Annual Data Report, CF Foundation. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf. This is the most recent CF Foundation registry annual data report.
Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med. 1994;150(2):448–54. https://doi.org/10.1164/ajrccm.150.2.8049828.
Chaaban MR, Kejner A, Rowe SM, Woodworth BA. Cystic fibrosis chronic rhinosinusitis: a comprehensive review. Am J Rhinol Allergy. 2013;27:387–95.
Robertson JM, Friedman EM, Rubin BK. Nasal and sinus disease in cystic fibrosis. Paediatr Respir Rev. 2008;9:213–9.
Nicollas R, Facon F, Sudre-Levillain I, et al. Pediatric paranasal sinus mucoceles: etiologic factors, management and outcome. Int J Pediatr Otorhinolaryngol. 2006;70:905–8.
Walkowiak J, Lisowska A, Blaszczynski M. The changing face of the exocrine pancreas in cystic fibrosis: pancreatic sufficiency, pancreatitis and genotype. Eur J Gastroenterol Hepatol. 2008;20:157–60.
Colombo C, Russo MC, Zazzeron L, et al. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S49–55.
Flume PA, O’Sullivan BP, Robinson KA, et al. Cystic fibrosis guidelines: chronic medications for lung health. Am J Respir Crit Care Med. 2007;176(10):957–69.
Mogayzel PJ, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for lung health. Am J Respir Care Med. 2013;187(7):680–9.
Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–40.
Halfhide C, Evans HJ, Couriel J. Inhaled bronchodilators for cystic fibrosis. Cochrane Database Syst Rev:4, CD003428.
Balfour-Lynn IM, Welch K. Inhaled corticosteroids for cystic fibrosis. Cochrane Database Syst Rev. 2009;(1):CD001915.
Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med. 1995;332:848–54.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev. 2012;11:CD002203
Saiman L, Anstead M, Mayer-Hamblett N, et al. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2010;303:1707–15.
Ryan G, Singh M, Dwan K. Inhaled antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst Rev. 2011;16(3):CD001021.
• Le C, McCray HC, Chang E. Cystic fibrosis sinusitis. Adv Otorhinolaryngol. 2016;79:29–37. This is a nice review of CF-related sinus disease.
• Gallant J, Mitchell MB, Virgin FW. Update on sinus disease in children with cystic fibrosis: advances in treatment modalities, microbiology and health-related quality-of-life instruments. Curr Opin Otolaryngol Head Neck Surg. 2018;26(6):417–20. This is an update on sinus disease and advances in treatment.
Stallings VA, Stark LJ, Robinson KA, et al. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008;108:832–9.
Colombo C, Ellemunter H, Houwen R, et al. Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J Cyst Fibros. 2011;10(Suppl 2):S24–8.
• Ooi CY, Durie PR. Cystic fibrosis from the gastroenterologist’s perspective. Nat Rev Gastroenterol Hepatol. 2016;13(3):175–85. This is a nice review of GI manifestations of CF.
Ramsey BW, Davies J, McElvaney NG, Tullis E, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation: VX08-770-102 Study Group. N Engl J Med. 2011;365(18):1663–72.
• Wainwright CE, Elborn JS, Ramsey BW, Boyle MP, et al. TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31. https://doi.org/10.1056/NEJMoa1409547. This article shows data for the first approved CFTR modulator combination drug.
•• Taylor-Cousar JL, Munck A, McKone EF, Elborn JS, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–23. This is the data for the most recently approved CFTR modulator drug.
•• Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612–20. This shows recent phase 2 data for a triple CFTR modulator drug.
•• Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, et al. VX16-659-101 Study Group. VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1599–611. This shows recent phase 2 data for a triple CFTR modulator drug.
• Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018;73(8):731–40. This article demonstrates the longer term effects of CFTR modulator ivacaftor based on registry data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Timothy Young, Douglas Li, and Patricia Eshaghian have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric Allergy
Rights and permissions
About this article

Cite this article
Young, T.J., Li, D.A. & Eshaghian, P.H. Cystic Fibrosis: an Update on Disease Pathophysiology, Management, and Novel Modalities of Therapy. Curr Treat Options Allergy 6, 226–237 (2019). https://doi.org/10.1007/s40521-019-00211-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40521-019-00211-4