
Abstract
Purpose of Review
Atopic dermatitis (AD) is a chronic eczematous skin disease associated with intense pruritus and skin barrier dysfunction. Interleukin-31 (IL-31), which is preferentially produced by T helper 2 (Th2) cells, has attracted attention as a pruritogen in patients with AD. To understand the mechanism of action, production, and roles of IL-31, we reviewed recent advances of the knowledge of IL-31, its receptor as well as results of recent clinical trials targeting IL-31/IL-31R signaling for pruritus in patients with AD.
Recent Findings
Activation of many cytokine pathways has been reported in AD, suggesting potential therapeutic targets with novel biologics. Most notably, the lesional skin of AD exhibits Th2-deviated immune reactions. Recently reported striking efficacy of dupilumab, a monoclonal antibody targeting the IL-4 receptor α subunit reconfirms the importance of Th2-deviated immune reactions in the pathogenesis of AD. A systemic and local administration of IL-31 induces scratching behavior in mice, dogs, and monkeys. In addition, serum levels of IL-31 have been correlated with severity of AD. Finally, an anti-interleukin (IL)-31 receptor α antibody (nemolizumab) has been shown to significantly alleviate pruritus in AD patients.
Summary
The interaction between IL-31 and IL-31R has a role not only in the induction of intense pruritus, but also in the regulation of inflammation and barrier function in AD. In clinical trials, nemolizumab significantly improved pruritus in patients with moderate-to-severe AD. Targeting the IL-31/IL-31R interaction might be a promising therapeutic target to improve the burdensome pruritus experienced by patients with AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atopic dermatitis (AD) is a common inflammatory skin disease associated with eczematous lesions, skin barrier dysfunction, and intense pruritus [1]. Intense pruritus is the major and most burdensome symptom of AD [2,3,4] and induces not only scratch-induced exacerbation of skin inflammation, but also sleep loss and severe disturbances of the quality of life [2, 4]. Sleep disturbance is a major problem that further impacts the ability of patients and their families to function in a school, work, or social environment [5, 6]. Conventional treatments such as emollients for dry skin, topical steroids and tacrolimus for skin inflammation, and oral anti-histamines for pruritus are more or less effective in reducing atopic itch [7]; however, patient satisfaction is generally reported to be low in daily clinics [8]. The development of new anti-pruritic agents has been demanded, but new agents still remain far below patient expectations.
Since discovering the pruritogenic action of interleukin-31 (IL-31) in mice in 2004 [9], IL-31 and its receptor have been studied extensively. Systemic and local administration of IL-31 induces scratching behavior in mice, dogs, and cynomolgus monkeys [9,10,11,12,13, 14•, 15]. IL-31 challenge can also induce delayed itch responses in humans [16]. In addition, serum IL-31 levels are elevated in patients with AD, and activated leukocytes in patients with AD expressed higher levels of IL-31 compared with those in healthy individuals [2]. Therefore, IL-31 has attracted attention as a pruritogen in patients with AD. Indeed, recent clinical trials have demonstrated that injections of an anti-IL-31RA antibody (nemorizumab) significantly improved pruritus in patients with AD [17, 18••, 19•].
In this review, we summarize recent knowledge about roles of the interaction between IL-31 and its receptor in the induction of pruritus. This review also focuses on the roles of IL-31 in the pathogenesis of atopic itch and successful itch control in clinical trials for AD by nemorizumab.
Structure and expression of the IL-31/IL-31R system
IL-31 and cell sources of IL-31
IL-31 was discovered in 2004 as a member of the family of the gp130/IL6 cytokines [9, 20]. Th2 cells from peripheral blood and skin-homing CD45RO+CLA+T cells are the major cellular sources of IL-31 [9, 21, 22] (Fig. 1). Lower levels of IL-31 expression have also been reported in Th1 cells [9]. Other cell sources of IL-31 are CD8+ T cells, monocytes/macrophages, dendritic cells, keratinocytes, eosinophils, basophils, mast cells, and fibroblasts [23,24,25,26] (Fig. 2).

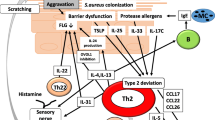
The roles of IL-31/IL-31R system in the pathogenesis of atopic dermatitis. The interaction between IL-31 and IL-31R on neurons induces intense pruritus and nerve fiber elongation. IL-31 also regulates inflammation and barrier function through activation of keratinocytes or immune cells.

Cell sources of IL-31 and IL-31 signaling pathway. IL-31 signals via a heterodimeric receptor complex comprising the IL-31RA and OSMR β. Interaction of the IL-31R complex with IL-31 results in activation of the JAK1/2 and STAT1/3/5 pathways as well as PI3K/AKT and mitogen-activated protein kinase pathways.
Most of these cell types require activation to express IL-31 mRNA and protein [27]. IL-31 production from cell sources seems to be dependent on IL-4 [28, 29]. IL-4 induces gene expression and release of IL-31 protein from human Th2 cells, and IL-33 further potentiates IL-4-induced IL-31 release [28, 29]. Both physical and bacterial stress, such as ultraviolet B irradiation, reactive oxygen species, and staphylococcal enterotoxin B, as well as anti-microbial peptides, also increase IL-31 expression [30].
IL-31R and target cells
IL-31 signals via a heterodimeric receptor complex comprising the IL-31 receptor A (IL-31RA) and oncostatin M receptor β (OSMR β) [20, 31, 32] (Fig. 2). This receptor complex is expressed in macrophages, dendritic cells, basophils, cutaneous neurons, and epithelial cells, including keratinocytes [23,24,25, 33,34,35] (Fig. 1). However, IL-31R expression is not observed in CD3+ T cells [25]. Both human and mouse dorsal root ganglia (DRG) neurons express IL-31RA. Large-diameter DRG neurons do not express IL-31R, whereas approximately half of the small-diameter DRG neurons (including C-fibers, which are important for itch transmission) express IL-31RA [33]. Interferon (IFN)-γ or toll-like receptor 2 (TLR2) ligands significantly induce IL-31RA expression in human keratinocytes, while staphylococcal enterotoxin B or staphylococcal α-toxin induce IL-31RA expression in human macrophages [34, 35]. Human dendritic cells significantly upregulate IL-31RA expression upon stimulation with IFN-γ, and IL-31–stimulated dendritic cells release several proinflammatory cytokines [36] (Fig. 1).
IL-31 binds predominantly to IL-31RA, but not to OSMR β, in the IL-31R complex. However, on coupling, OSMR β converts IL-31R into the high-affinity receptor and increases IL-31 binding [31]. Interaction of the IL-31R complex with IL-31 results in activation of the Janus kinase (JAK)1/JAK2 and signal transducer and activator of transcription (STAT)3 (also to less extent STAT1 and STAT5) pathways as well as phosphatidylinositol-3 kinase (PI3K)/AKT and mitogen-activated protein kinase pathways [20, 28, 34, 35, 37, 38] (Fig. 2).
Several new drugs such as dupilumab (anti-IL-4 receptor α antibody), phosphodiesterase 4 inhibitors, and JAK inhibitors have been evaluated or are currently being evaluated in clinical trials [39,40,41,42,43,44]. The anti-pruritic effects of these agents might be partly due to the inhibition of IL-31 signaling.
Itch and the IL-31/IL-31R system
IL-31 induced itch in mice, dogs, cynomolgus monkeys, and humans
The ability of IL-31 to cause an itch response has been implicated in various species such as mice, dogs, and cynomolgus monkeys [9,10,11,12,13,14,15]. Severe pruritus and AD-like skin lesions developed in IL-31 transgenic mice that overexpress IL-31 [9]. Wild-type mice treated with IL-31 locally or systemically also developed scratching behavior and AD-like skin lesions [9, 10, 33, 45, 46•]. Excessive scratching behavior induces similar histopathological changes in the skin lesions as those in AD [4, 45, 46•]. The scratching behavior in an AD-like murine model is inhibited by treatment with an anti-mouse IL-31 antibody [47]. The scratching behavior induced by an intravenous injection of IL-31 is inhibited by pretreatment with an anti-IL-31RA-neutralizing antibody [10]. In contrast, it is not inhibited significantly by anti-histamine agent, dexamethasone, tacrolimus, or the μ-opioid receptor antagonist [10]. Just as in the case of mice, systemically or locally injected canine IL-31 also evokes scratching behavior in dogs [11, 12, 14•, 15] and the anti-canine IL-31 antibody (lokivetmab) significantly inhibits scratching in canine AD [48, 49]. Besides, administration of cynomolgus IL-31 can elicit a scratching response in cynomolgus monkeys, which is inhibited by humanized anti-human IL-31 monoclonal antibody (mAb) administration [14•, 15]. In a recent study, IL-31 did not induce an immediate itch response but did induce late-onset pruritus in humans in an IL-31 prick test, indicating that IL-31 exerts its pruritic action via an indirect mechanism that may involve keratinocytes [16].
The expression of IL-31RA is identified in cutaneous neurons and DRG in mice, cynomolgus monkeys, and humans [15, 25, 33]. The human and mouse IL-31RA+ DRG neurons co-express transient receptor potential cation channel vanilloid subtype 1 (TRPV1) [33] (Fig. 1). IL-31-mediated itch is significantly reduced in TRPV1 KO mice or TRPA1 KO mice suggesting that TRP channels are critical to IL-31-mediated itch [33]. IL-31 stimulation of cultured murine DRG neurons induce ERK1/2 phosphorylation, and blockage of ERK1/2 with a specific inhibitor reduces scratching behavior in vivo [33], suggesting that ERK1/2 is required for IL-31-induced itch transmission.
Atopic itch and IL-31
IL-31 has been reported to play a role in the mechanism underlying pruritus in patients with AD. IL-31 is expressed more in lesional AD skin than non-lesional skin [50]. Activated leukocytes in patients with AD expressed higher levels of IL-31 compared with those in healthy individuals [21, 51, 52]. Human eosinophils can release IL-31 protein and the IL-31 release by eosinophils is higher in AD patients than in normal volunteers [26]. In addition, serum IL-31 levels are elevated in patients with AD, and there is a significant correlation with disease severity [53, 54]. Exposure to staphylococcal superantigen, which is frequently found in the skin of patients with AD, rapidly induces IL-31 expression in patients with AD [50]. IL-31RA is expressed by keratinocytes from both patients with AD and healthy control subjects. However, the levels of IL-31RA expressed on keratinocytes from AD skin are consistently higher than the levels observed in skin biopsy specimens from healthy control subjects [21]. These data strongly indicate that IL-31 can be functionally related to pruritus in AD.
Epidermal hyperinnervation or elongation of sensory nerve fibers in AD may cause itchy and sensitive skin [2, 33]. A recent study revealed that elongation and branching of cutaneous nerve fibers were seen in IL-31-transgenic and IL-31 pump equipped mice in addition to AD-like skin lesions and scratching [46•]. The same phenomena are seen in murine small-diameter DRG neurons treated with IL-31 in vitro, and these are independent of Trpv1 but dependent on STAT3 signaling [46•]. Interestingly, STAT3-dependent reactive astrogliosis in the spinal dorsal horn mediates chronic itch in mice [55]. These findings suggested that IL-31 might promote sensitivity to minimal stimuli and self-aggravates skin hypersensitivity in human AD.
It has been reported that IL-31RA and OSMR β are expressed on keratinocytes [56, 57]. With regard to pruritus, keratinocytes release various pruritogenic mediators, which directly interfere with sensory neurons [2]. Therefore, in addition to the direct effects of IL31 on neurons, an additional indirect role of IL-31 in pruritus through producing of pruritogenic mediators from keratinocytes also can be considered [2].
The potential proinflammatory and barrier-disrupting role of the IL-31/IL-31R system in vitro
In addition to induction of itch, the effects of IL-31 on release of proinflammatory cytokines and barrier disruption of keratinocytes might be involved in the pathogenesis of AD.
Several chemokines such as GRO1α (CXCL1), TARC (CCL17), MIP3β (CCL19), MDC (CCL22), MIP-3 (CCL23), MIP-1β (CCL4) and I-309 are induced in normal human epidermal keratinocytes stimulated with human IL-31 [9] (Fig. 1). These chemokines upregulated by IL-31 in keratinocytes are mostly elevated in AD as well [9, 58]. IL-31 also significantly induces the release of proinflammatory cytokines (IL-1β, IL-6) and AD-related chemokines (CXCL1, CXCL8, CCL2, and CCL18) from eosinophils, via functional cell surface IL-31R (Fig. 1). Such induction was further enhanced upon the co-culture of eosinophils and keratinocytes [59]. These findings suggest that a crucial role of IL-31 in AD through activation of keratinocytes or the eosinophils-keratinocytes system, and IL-31 might play a role in the recruitment of inflammatory cells into the lesional skin of AD [9, 58].
In three-dimensional skin models, IL-31 induces disturbed keratinocyte differentiation caused by reduced expression of physical skin barrier-related molecules, such as desmoglein, desmocollin, corneodesmosin and filaggrin, resulting in impaired skin barrier function [60] (Fig. 1). As FLG mutations or downregulation of filaggrin are strongly associated with AD [61,62,63], IL-31-induced downregulation of filaggrin must be involved in the development of AD from the aspect of skin barrier dysfunction. Conversely, low doses of IL-31 upregulate the expression of anti-microbial peptides, including S100A7, S100A8, S100A9, β-defensin 2, and β-defensin 3, and promote the anti-microbial skin barrier [60] (Fig. 1). These findings demonstrate that IL-31 control the physical and anti-microbial skin barrier in multiple ways.
A new potential therapeutic target for pruritus in human AD
Nemolizumab (CIM331), a humanized anti-IL-31RA mAb, binds to IL-31RA and inhibits IL-31 signaling.
As the first human study, a randomized, double-blind, placebo-controlled phase I/Ib clinical trial of nemolizumab was conducted in healthy volunteers and in AD patients to assess tolerability, safety, and preliminary efficacy [17]. In the study, nemolizumab was administered in a single subcutaneous injection at different doses (0.003, 0.01, 0.03, 0.1, 0.3, 1.0, or 3 mg/kg). No serious adverse events (AEs) or dose-dependent increases in the incidence of AEs were reported in the study. A single administration of nemolizumab significantly reduced pruritus visual analogue scale (VAS) scores at week 1 (− 24%, − 24%, and − 33% in the 0.3, 1.0, and 3.0 mg/kg nemolizumab groups, respectively, versus − 9% in the placebo group). At week 4, nemolizumab further reduced the pruritus VAS scale score (− 50%, − 44%, and − 48% in the 0.3, 1.0, and 3.0 mg/kg nemolizumab groups, respectively, versus − 20% in the placebo group). In addition to the decrease in pruritus, a decrease in the use of topical steroids and an improvement of sleep efficiency were observed [17].
The phase II trial (NCT01986933) was performed in two parts (part A and part B) [18••, 19•] to evaluate the optimal dose, safety, and efficacy of nemolizumab. In part A, adult patients with moderate-to-severe AD were randomly assigned into four groups (nemolizumab 0.1, 0.5, or 2.0 mg/kg every 4 weeks [Q4W] or 2.0 mg/kg every 8 weeks [Q8W]) and a placebo group (Q4W). All groups received treatment or placebo subcutaneously for 12 weeks [18••]. At week 12, there was a significant, dose-dependent reduction in the percentage change from baseline in pruritus VAS scores as compared with placebo. The percentage reductions were − 43.7%, − 59.8%, and − 63.1% in the 0.1, 0.5, and 2.0 mg/kg nemolizumab groups, respectively, as compared with − 20.9% in the placebo group (all P < 0.01) [18••]. The percentage changes in the Eczema Area and Severity Index (EASI) were − 23.0%, − 42.3%, and − 40.9% in the nemolizumab groups versus − 26.6% in the placebo group; however, there was no significant difference [18••]. Several parameters assessing sleep quality and the Dermatology Quality of Life Index improved in the nemolizumab group compared with the placebo group. Regarding AEs, exacerbation of AD, nasopharyngitis, upper respiratory tract infections, peripheral edema, and increased creatine phosphokinase levels occurred more frequently in patients receiving nemolizumab than in those receiving placebo [18••]. No serious side effects were observed in association with nemolizumab.
To assess long-term efficacy and safety of nemolizumab, an extension study (phase 2) was then conducted (part B) [19•]. During part B, patients continued the previous nemolizumab dose (0.1, 0.5, or 2.0 mg/kg Q4W or 2.0 mg/kg Q8W). The improvement from the baseline in pruritus VAS score observed in part A was maintained or increased from week 12 to week 64 in patients randomized to receive nemolizumab throughout the 64-week study period [19•]. The greatest improvement throughout the study was observed in the 0.5-mg/kg nemolizumab group. The percentage change from baseline in the EASI score and sleep disturbance VAS score were also maintained or increased from week 12 to week 64 in patients who had received nemolizumab in part A. No new safety concerns were identified after long-term use of nemolizumab [19•].
These findings in clinical trials provide evidence that nemolizumab might become a useful and potent therapy for severe pruritus of AD.
Conclusions
Although intense pruritus is the major and most burdensome subjective symptom of AD [1,2,3, 5], the satisfaction of most AD patients with conventional treatments for pruritus remains very limited. Since IL-31 was reported to induce itch, numerous studies investigating the biology of IL-31 have been reported. Current knowledge shows that the interaction between IL-31 and IL-31R has a role not only in the induction of intense pruritus, but also in the regulation of inflammation and barrier function in AD. Targeting the IL-31/IL-31R interaction is a promising therapeutic target to improve the burdensome pruritus experienced by patients with AD. However, further studies are warranted to understand the exact mechanism involved in IL-31 regulation of itch and any unfavorable events associated with inhibition of the IL-31 pathway.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387:1109–22.
Furue M, Yamamura K, Kido-Nakahara M, Nakahara T, Fukui Y. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy. 2018;73:29–36.
Saeki H, Nakahara T, Tanaka A, Kabashima K, Sugaya M, Murota H, et al. Clinical practice guidelines for the management of atopic dermatitis 2016. J Dermatol. 2016;43:1117–45.
Takeuchi S, Yasukawa F, Furue M, Katz SI. Collared mice: a model to assess the effects of scratching. J Dermatol Sci. 2010;57:44–50.
Furue M, Kadono T. New therapies for controlling atopic itch. J Dermatol. 2015;42:847–50.
Chamlin SL, Mattson CL, Frieden IJ, Williams ML, Mancini AJ, Cella D, et al. The price of pruritus: sleep disturbance and cosleeping in atopic dermatitis. Arch Pediatr Adolesc Med. 2005;159:745–50.
Sher LG, Chang J, Patel IB, Balkrishnan R, Fleischer AB Jr. Relieving the pruritus of atopic dermatitis: a meta-analysis. Acta Derm Venereol. 2012;92:455–61.
Murota H, Takeuchi S, Sugaya M, Tanioka M, Onozuka D, Hagihara A, et al. Characterization of socioeconomic status of Japanese patients with atopic dermatitis showing poor medical adherence and reasons for drug discontinuation. J Dermatol Sci. 2015;79:279–87.
Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–60.
Kasutani K, Fujii E, Ohyama S, Adachi H, Hasegawa M, Kitamura H, et al. Anti-IL-31 receptor antibody is shown to be a potential therapeutic option for treating itch and dermatitis in mice. Br J Pharmacol. 2014;171:5049–58.
Gonzales AJ, Humphrey WR, Messamore JE, Fleck TJ, Fici GJ, Shelly JA, et al. Interleukin-31: its role in canine pruritus and naturally occurring canine atopic dermatitis. Vet Dermatol. 2013;24:48–53.e11–2.
Gonzales AJ, Bowman JW, Fici GJ, Zhang M, Mann DW, Mitton-Fry M. Oclacitinib (APOQUEL(®)) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J Vet Pharmacol Ther. 2014;37:317–24.
Gonzales AJ, Fleck TJ, Humphrey WR, Galvan BA, Aleo MM, Mahabir SP, et al. IL-31-induced pruritus in dogs: a novel experimental model to evaluate anti-pruritic effects of canine therapeutics. Vet Dermatol. 2016;27:34–e10.
• Lewis KE, Holdren MS, Maurer MF, Underwood S, Meengs B, Julien SH, et al. Interleukin (IL) 31 induces in cynomolgus monkeys a rapid and intense itch response that can be inhibited by an IL-31 neutralizing antibody. J Eur Acad Dermatol Venereol. 2017;31:142–50 This study demonstrated that in cynomolgus monkeys, IL-31 (cIL-31) administration elicited itch response, and treatment with the IL-31 mAb inhibited the cIL-31-mediated scratching response.
Oyama S, Kitamura H, Kuramochi T, Higuchi Y, Matsushita H, Suzuki T, et al. Cynomolgus monkey model of interleukin-31-induced scratching depicts blockade of human interleukin-31 receptor A by a humanized monoclonal antibody. Exp Dermatol. 2018;27:14–21.
Hawro T, Saluja R, Weller K, Altrichter S, Metz M, Maurer M. Interleukin-31 does not induce immediate itch in atopic dermatitis patients and healthy controls after skin challenge. Allergy. 2014;69:113–7.
Nemoto O, Furue M, Nakagawa H, Shiramoto M, Hanada R, Matsuki S, et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor A antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br J Dermatol. 2016;174:296–304.
•• Ruzicka T, Hanifin JM, Furue M, Pulka G, Mlynarczyk I, Wollenberg A, et al. Anti-interleukin-31 receptor A antibody for atopic dermatitis. N Engl J Med. 2017;376:826–35 This study demonstrated the promising therapeutic potential of nemolizumab for the treatment of itch in AD.
• Kabashima K, Furue M, Hanifin JM, Pulka G, Wollenberg A, Galus R, et al. Nemolizumab in patients with moderate-to-severe atopic dermatitis: Randomized, phase II, long-term extension study. J Allergy Clin Immunol. 2018. https://doi.org/10.1016/j.jaci.2018.03.018 This study showed the long-term efficacy and safety of nemolizumab.
Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008;19:347–56.
Bilsborough J, Leung DY, Maurer M, Howell M, Boguniewicz M, Yao L, et al. IL-31 is associated with cutaneous lymphocyte antigen-positive skin homing T cells in patients with atopic dermatitis. J Allergy Clin Immunol. 2006;117:418–25.
McCandless EE, Rugg CA, Fici GJ, Messamore JE, Aleo MM, Gonzales AJ. Allergen-induced production of IL-31 by canine Th2 cells and identification of immune, skin, and neuronal target cells. Vet Immunol Immunopathol. 2014;157:42–8.
Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor b, and TNF-a: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2016;138:984–1010.
Raap U, Gehring M, Kleiner S, Rüdrich U, Eiz-Vesper B, Haas H, et al. Human basophils are a source of - and are differentially activated by IL-31. Clin Exp Allergy. 2017;47:499–508.
Kato A, Fujii E, Watanabe T, Takashima Y, Matsushita H, Furuhashi T, et al. Distribution of IL-31 and its receptor expressing cells in skin of atopic dermatitis. J Dermatol Sci. 2014;74:229–35.
Kunsleben N, Rüdrich U, Gehring M, Novak N, Kapp A, Raap U. IL-31 induces chemotaxis, calcium mobilization, release of reactive oxygen species, and CCL26 in eosinophils, which are capable to release IL-31. J Invest Dermatol. 2015;135:1908–11.
Ferretti E, Corcione A, Pistoia V. The IL-31/IL-31 receptor axis: general features and role in tumor microenvironment. J Leukoc Biol. 2017;102:711–7.
Maier E, Werner D, Duschl A, Bohle B, Horejs-Hoeck J. Human Th2 but not Th9 cells release IL-31 in a STAT6/NF-κB-dependent way. J Immunol. 2014;193:645–54.
Stott B, Lavender P, Lehmann S, Pennino D, Durham S, Schmidt-Weber CB. Human IL-31 is induced by IL-4 and promotes TH2-driven inflammation. J Allergy Clin Immunol. 2013;132:446–54.
Cornelissen C, Brans R, Czaja K, Skazik C, Marquardt Y, Zwadlo-Klarwasser G, et al. Ultraviolet B radiation and reactive oxygen species modulate interleukin-31 expression in T lymphocytes, monocytes and dendritic cells. Br J Dermatol. 2011;165:966–75.
Diveu C, Lak-Hal AH, Froger J, Ravon E, Grimaud L, Barbier F, et al. Predominant expression of the long isoform of GP130-like (GPL) receptor is required for interleukin-31 signaling. Eur Cytokine Netw. 2004;15:291–302.
Dreuw A, Radtke S, Pflanz S, Lippok BE, Heinrich PC, Hermanns HM. Characterization of the signaling capacities of the novel gp130-like cytokine receptor. J Biol Chem. 2004;279:36112–20.
Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: involvement of TRPV1 and TRPA1. J Allergy Clin Immunol. 2014;133:448–60.
Kasraie S, Niebuhr M, Baumert K, Werfel T. Functional effects of interleukin 31 in human primary keratinocytes. Allergy. 2011;66:845–52.
Kasraie S, Niebuhr M, Werfel T. Interleukin (IL)-31 activates signal transducer and activator of transcription (STAT)-1, STAT-5 and extracellular signal-regulated kinase 1/2 and down-regulates IL-12p40 production in activated human macrophages. Allergy. 2013;68:739–47.
Horejs-Hoeck J, Schwarz H, Lamprecht S, Maier E, Hainzl S, Schmittner M, et al. Dendritic cells activated by IFN-γ/STAT1 express IL-31 receptor and release proinflammatory mediators upon IL-31 treatment. J Immunol. 2012;188:5319–26.
Edukulla R, Singh B, Jegga AG, Sontake V, Dillon SR, Madala SK. Th2 cytokines augment IL-31/IL-31RA interactions via STAT6-dependent IL-31RA expression. J Biol Chem. 2015;290:13510–20.
Perrigoue JG, Li J, Zaph C, Goldschmidt M, Scott P, de Sauvage FJ, et al. IL-31-IL-31R interactions negatively regulate type 2 inflammation in the lung. J Exp Med. 2007;204:481–7.
Heratizadeh A, Werfel T. Anti-inflammatory therapies in atopic dermatitis. Allergy. 2016;71:1666–75.
Furue M, Chiba T, Tsuji G, Ulzii D, Kido-Nakahara M, Nakahara T, et al. Atopic dermatitis: immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol Int. 2017. https://doi.org/10.1016/j.alit.2016.12.002.
Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335–48.
Nemoto O, Hayashi N, Kitahara Y, Furue M, Hojo S, Nomoto M, et al. Effect of topical phosphodiesterase 4 inhibitor E6005 on Japanese children with atopic dermatitis: results from a randomized, vehicle-controlled exploratory trial. J Dermatol. 2016;43:881–7.
Samrao A, Berry TM, Goreshi R, Simpson EL. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch Dermatol. 2012;148:890–7.
Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. 2016;175:902–11.
Singh B, Jegga AG, Shanmukhappa KS, Edukulla R, Khurana GH, Medvedovic M, et al. IL-31-Driven skin remodeling involves epidermal cell proliferation and thickening that lead to impaired skin-barrier function. PLoS One. 2016;11:e0161877.
• Feld M, Garcia R, Buddenkotte J, Katayama S, Lewis K, Muirhead G, et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J Allergy Clin Immunol. 2016;138:500–8 This study was the first to demonstrate that IL-31 can promote nerve fiber elongation and branching of murine small-diameter DRG neurons.
Grimstad O, Sawanobori Y, Vestergaard C, Bilsborough J, Olsen UB, Grønhøj-Larsen C, et al. Anti-interleukin-31-antibodies ameliorate scratching behaviour in NC/Nga mice: a model of atopic dermatitis. Exp Dermatol. 2009;18:35–43.
Michels GM, Ramsey DS, Walsh KF, Martinon OM, Mahabir SP, Hoevers JD, et al. A blinded, randomized, placebo-controlled, dose determination trial of lokivetmab (ZTS-00103289), a caninized, anti-canine IL-31 monoclonal antibody in client owned dogs with atopic dermatitis. Vet Dermatol. 2016;27:478–e129.
Michels GM, Walsh KF, Kryda KA, Mahabir SP, Walters RR, Hoevers JD, et al. A blinded, randomized, placebo-controlled trial of the safety of lokivetmab (ZTS-00103289), a caninized anti-canine IL-31 monoclonal antibody in client-owned dogs with atopic dermatitis. Vet Dermatol. 2016;27:505–e136.
Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117:411–7.
Szegedi K, Kremer AE, Kezic S, Teunissen MB, Bos JD, Luiten RM, et al. Increased frequencies of IL-31-producing T cells are found in chronic atopic dermatitis skin. Exp Dermatol. 2012;21:431–6.
Yamamura K, Uruno T, Shiraishi A, Tanaka Y, Ushijima M, Nakahara T, et al. The transcription factor EPAS1 links DOCK8 deficiency to atopic skin inflammation via IL-31 induction. Nat Commun. 2017;8:13946.
Ezzat MH, Hasan ZE, Shaheen KY. Serum measurement of interleukin-31 (IL-31) in paediatric atopic dermatitis: elevated levels correlate with severity scoring. J Eur Acad Dermatol Venereol. 2011;25:334–9.
Raap U, Wichmann K, Bruder M, Ständer S, Wedi B, Kapp A, et al. Correlation of IL-31 serum levels with severity of atopic dermatitis. J Allergy Clin Immunol. 2008;122:421–3.
Shiratori-Hayashi M, Koga K, Tozaki-Saitoh H, Kohro Y, Toyonaga H, Yamaguchi C, et al. STAT3-dependent reactive astrogliosis in the spinal dorsal horn underlies chronic itch. Nat Med. 2015;21:927–31.
Nobbe S, Dziunycz P, Mühleisen B, Bilsborough J, Dillon SR, French LE, et al. IL-31 expression by inflammatory cells is preferentially elevated in atopic dermatitis. Acta Derm Venereol. 2012;92:24–8.
Nattkemper LA, Martinez-Escala ME, Gelman AB, Singer EM, Rook AH, Guitart J, et al. Cutaneous T-cell lymphoma and pruritus: the expression of IL-31 and its receptors in the skin. Acta Derm Venereol. 2016;96:894–8.
Esaki H, Takeuchi S, Furusyo N, Yamamura K, Hayashida S, Tsuji G, et al. Levels of immunoglobulin E specific to the major food allergen and chemokine (C-C motif) ligand (CCL)17/thymus and activation regulated chemokine and CCL22/macrophage-derived chemokine in infantile atopic dermatitis on Ishigaki Island. J Dermatol. 2016;43:1278–82.
Cheung PF, Wong CK, Ho AW, Hu S, Chen DP, Lam CW. Activation of human eosinophils and epidermal keratinocytes by Th2 cytokine IL-31: implication for the immunopathogenesis of atopic dermatitis. Int Immunol. 2010;22:453–67.
Hänel KH, Pfaff CM, Cornelissen C, Amann PM, Marquardt Y, Czaja K, et al. Control of the physical and antimicrobial skin barrier by an IL-31-IL-1 signaling network. J Immunol. 2016;196:3233–44.
Weidinger S, Rodríguez E, Stahl C, Wagenpfeil S, Klopp N, Illig T, et al. Filaggrin mutations strongly predispose to early-onset and extrinsic atopic dermatitis. J Invest Dermatol. 2007;127:724–6.
Park J, Jekarl DW, Kim Y, Kim J, Kim M, Park YM. Novel FLG null mutations in Korean patients with atopic dermatitis and comparison of the mutational spectra in Asian populations. J Dermatol. 2015;42:867–73.
Takei K, Mitoma C, Hashimoto-Hachiya A, Uchi H, Takahara M, Tsuji G, et al. Antioxidant soybean tar Glyteer rescues T-helper-mediated downregulation of filaggrin expression via aryl hydrocarbon receptor. J Dermatol. 2015;42:171–80.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Takeshi Nakahara declares that he has no conflict of interest. Masutaka Furue declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Urticaria and Atopic Dermatitis
Rights and permissions
About this article

Cite this article
Nakahara, T., Furue, M. Nemolizumab and Atopic Dermatitis: the Interaction Between Interleukin-31 and Interleukin-31 Receptor as a Potential Therapeutic Target for Pruritus in Patients With Atopic Dermatitis. Curr Treat Options Allergy 5, 405–414 (2018). https://doi.org/10.1007/s40521-018-0191-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40521-018-0191-3