
Abstract
Background and aim
Mortality is a highly complex trait influenced by a wide array of genetic factors.
Methods
We examined a population of 1200 mice that were F2 generation offspring of a 4-way reciprocal cross between C57BL6/J and DBA2/J strains. Animals were sacrificed at age 200, 500, or 800 days and genotyped at 96 markers. The 800 days old cohort, which were the survivors of a much larger breeding group, were examined for enriched frequency of alleles that benefit survival and depletion of alleles that reduce survival.
Results
Loci on Chr 13 in males and on Chr X in females were significantly distorted from Mendelian expectations, even after conservative correction for multiple testing. DBA2/J alleles between 35 and 80 Mb on Chr 13 were underrepresented in the age 800 male animals. D2 genotypes in this region were also associated with premature death during behavioral testing. Furthermore, confirmatory analysis showed BXD recombinant inbred strains carrying the D2 alleles in this region had shorter median survival. Exploration of available pathology data indicated that a syndrome involving dental malocclusions, pancreatic islet hypertrophy, and kidney lipidosis may have mediated the effects of DBA alleles on mortality specifically in male mice. The heterozygote advantage locus on the X Chr was not found to be associated with any pathology.
Conclusions
These results suggest a novel locus influencing survival in the B6/D2 genetic background, perhaps via a metabolic disorder that emerges by 200 days of age in male animals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Longevity, the quintessential complex trait, likely reflects all aspects of an organism’s life history. In humans, the estimated heritability of age at death is estimated at 25–33 % [1]. Genetic contributions to mortality rates are thus of great interest and may aid in the understanding of disease etiology and the process of aging itself [2]. Genetic linkage studies of long-lived human families identified a longevity locus while candidate gene approaches have been used to identify and confirm the association between specific variants in the FOXO3A gene and human longevity [3–7]. Genome-wide association studies have also been used to identify the association of APOE with life span and have yielded insights into potential biological pathways and processes related to aging. Despite these successes, several problems are inherent in human longevity studies including potentially high degrees of environmental heterogeneity, genetic diversity, and lack of birth matched controls, among others [8].
Inbred mouse strains represent a powerful alternative for identifying genes underlying complex trait genes such as longevity [9]. Initial mapping approaches include quantitative trait locus (QTL) analysis that represents an unbiased association analysis to map genomic regions linked to variation of a phenotype. Previously we conducted QTL analyses of recombinant inbred mice derived from C57BL/6J (B6) and DBA/2J (D2) mice that replicated several previously identified longevity associated loci and identified 5 new QTLs [10], which built upon earlier work in these strains [11].
Specific variants of interest can be detected by analyzing deviations from expected gene frequency ratios in aged populations. For example, when comparing youth to centenarians, certain genetic markers may become more common if they aid survival or become less prevalent if they cause earlier onset of death. This methodology was used to find and confirm the association between specific variants in the FOXO3A gene and human longevity [3–7]. Genetic studies on the diseases and biomarkers of aging in animal models also contain data ready for analysis of these age-related allelic distortions.
Here, we have extended this analysis to search for genotypes related to survival to the age of 800 days in a population of a reciprocal F2 cross between (B6) and (D2) mice. Since QTL for longevity in mice have shown strong sex specificity [10, 12], we conducted sex-specific analyses. In addition, we also determined whether there were any change in pathology changes associated with the loci that showed frequency distortions with aging. To confirm the associations of the loci of interest with longevity and pathology, we performed replication analyses on a panel of BXD recombinant inbred strains.
Methods
Animals
F2 population. Reciprocal hybrid crosses of C57BL/6J and DBA/2J mice were used to produce 1659 F2 offspring (equal numbers from each of the four combination of F1 parents).
BXD RI strains. Twenty-three BXD RI strains were represented: 2, 6, 8, 11, 12, 13, 14, 16, 19, 22, 24, 27, 28, 29, 30, 31, 32, 33, 34, 38, 39, 40, and 42, along with the two parental inbred strains, B6 and D2, for a total of 2138 inbred mice (~80/strain distributed across the 3 age groups).
Housing. Siblings were housed at weaning in same sex groupings (four animals per cage with micro-isolators) at age 25 days, except for the males of several RI strains (8, 11, and 24) that were housed individually to avoid injury from fighting. All mice were produced and tested in a specific pathogen-free barrier facility and were fed sterilized Purina mouse chow 5010 (autoclavable; approximately equivalent to Purina 5001) and provided sterilized water ad libitum. Temperature within the barrier facility was maintained at 72 ± 2 °F with a 12/12 light/dark cycle (7am–7pm).
Phenotyping. Mice began behavioral and serological phenotyping at approximately 150, 450, or 750 days of age, and then were sacrificed at approximately 200, 500, or 800 days of age. To provide about 400 animals for testing at each age group, about 400 extra mice had to be bred for the oldest age group to account for the predicted 50 % mortality rate prior to phenotyping (hence ~1600 mice bred to get our sample size of 1200 mice in the F2 animals; even more additional breeding was needed to provide a sufficient sample of RI mice at the older ages). Mice that died before phenotyping were not genotyped, but were submitted for preliminary necropsy reports by a veterinarian before detailed histological analyses by veterinary pathologists.
General phenotyping schedule
Different cohorts of mice were phenotyped at different ages. The youngest testing began testing at approximately 117 days of age and completed testing at 203 days (in this paper, we refer to them as the 200-day group). The 500-day group began testing at approximately 423 days and finished at approximately 511 days. The 800-day group was tested from ~725 to ~810 days of age.
The testing regime included urine collection under basal and 24 h water deprived conditions, brief exposures to a novel environment [13, 14], testing for balance on a rotorod, a test of grip strength, repeated warming and restraint for heart rate/blood pressure monitoring [15], and blood collection procedures [16] as described previously. For the panel of serum chemistry outcomes studied in the current project, data are only available for the 450-day age group.
The series of tests described above was conducted in a fixed order across 1 week, then repeated twice more at 1 month intervals (yielding a total of three measurement points for each mouse for every trait). Mice that were identified as moribund during the three months of testing were marked for early sacrifice and included in analyses. Approximately 1 month after the third measurement, surviving mice were sacrificed for a variety of analyses on blood, brain, muscle, bone, and pathology [13–17] phenotypes. After specific tissues (brain regions and hind limbs) were dissected immediately upon sacrifice, carcasses were preserved in buffered formalin until comprehensive pathological examinations were completed.
Genotyping and genetic analyses
Mice that began the phenotyping procedure in each of the 3 age groups were genotyped at 96 microsatellite markers [13–17]. Distortions in the frequency of alleles with aging were detected by Chi-squared tests conducted for each locus. The expected number of mice of each genotype was assumed to be 25 % of the surviving population for each homozygote genotype and 50 % for heterozygotes (except for males on the X chromosome, where mice were hemizygotic, 50 % carrying B6 and 50 % carrying D2 alleles). The significance threshold for the Chi-squared tests was adjusted for multiple testing at the 96 markers with Bonferroni’s correction (separately for each sex). Thus, the null hypothesis was rejected when allelic distortions were significant at p < 0.00052 (0.05/96, or 1/1920). Since this assumption of 96 independent tests does not account for the covariance between adjacent markers on the same chromosome (there are actually fewer than 96 independent tests), this correction is quite conservative.
Other statistical analyses
Our subsequent search for pathologies was focused on their relationship to a single genomic region (proximal Chr 13), which showed age-dependent distortion in males in the above analyses. For the binary (present vs. not present) pathology outcomes, logistic regression was used to test for associations with sex, genotype, and age (SAS v. 9.2, Proc Logistic). For the quantitative serum chemistry outcomes, linear regression models were used to test for associations with sex and genotype, as only one age group was available (SAS v. 9.2, Proc Mixed). Since these tests, like all statistical tests presented in this article, are exploratory in nature, level of significance is indicated at each juncture. When p < 0.05 in analyses with large sample sizes, the results are considered only suggestive; p < 0.001 or p < 0.0001 are somewhat more compelling.
Results
Survival in the F2 mice
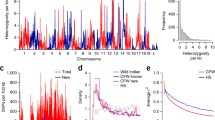
In the 200 days age group, 3/395 mice (1 %) died before the initiation of testing and an additional 12 (3 %) of the 392 that began testing died during the 3 months long testing period. In the 500 days age group, 37/462 (8 %) died before testing and 22/425 (5 %) died during the testing period. In the 800 days age group, 337/782 (43 %) died before testing began and an additional 122/445 mice (27 %) died during the testing procedure. The mortality rate during testing increased with age in the expected accelerating fashion, with median lifespan of 740 days (see Fig. 1). No significant differences in mortality rate were seen between males and females, or between the reciprocal crosses to produce the F2 mice. Thus, about as many mice died prior to genotyping as survived to testing in the 800 days cohort.

Survival curve for mice in the DBA/2J × C57BL/6J F2 breeding colony for this study
Allelic frequency distortions consistent with age-related mortality in the F2 population
Based on the hypothesis that major causes of mortality would be linked to specific loci in this population, we did a genome-wide search for allele frequency distortions in mice surviving to inclusion in the 800 days age group. To visualize these data, we plotted the inverse of the p value of the χ 2 test at each allele across the 19 autosomes and the X chromosome (Figs. 1, 2).

Genome-wide significant allelic distortions in 800 days old mice. The line at 1/1920 indicates our significance threshold
This genome-wide scan identifies two sex-specific QTL one D13Mit125 (80.86 Mb, additive B6 survival advantage in males) and the other at DXMit135 (161.22 Mb, heterozygote advantage in females) in the 800 days age group. Genotype frequencies at the proximal adjacent loci at D13Mit198 (35.06 Mb) and DXMit79 (127.40 Mb) were suggestive, in the same direction. These loci showed no evidence of allelic distortions in the 200 and 500 days age groups (Table 1), suggesting that the reduced prevalence of certain loci was driven by mortality after 500 days of age.
As a follow-up analysis, we evaluated whether the increased mortality in these under-represented genotypic groups continued during the 3 months long phenotyping procedure. The expected trend was observed in the surviving male homozygotes for the D2 allele at D13Mit125. In these animals, 7/32 (22 %) became moribund during the phenotyping period compared to 13/90 (14 %) of the heterozygotes and 16/73 (11 %) of the B6 homozygotes. However, due to the small sample size, this finding only reached a suggestive level of significance (p = 0.087). The female mice with heterozygosity at DXMit135 showed no trend toward a change in risk of moribundity compared to the other genotypes during this period.
Independent confirmation of a survival effect in this region of Chr 13 QTL in recombinant inbred strains
We hypothesized that a genotype that increased mortality in the F2 mice would be associated with shorter median lifespan in the BXD recombinant inbred panel. We used WebQTL [18] to map loci associated with median lifespan in an RI panel on Chr 13 [10]. We excluded data for strains that showed very early mortality, as defined by the first quartile of age at death within strain earlier than 500 days of age (e.g. BXD13 which dies early from seizures).
Lang et al. [10] had previously searched for a genome-wide significant peak in this region and found none. However, we predicted that there would be a small peak favoring longevity in the B6 strain exclusively between 35 and 80 megabases on Chr 13 (i.e. between the significant and adjacent suggestive markers for allelic distortion in the F2 population). We also predicted that this peak would be present for males but not females. The predicted small, sex-specific, B6 advantage/D2 disadvantage peak was present in the expected interval, suggesting that a locus between 50 and 65 Mb was associated with approximately 60 days longer median lifespan in males but not females (Fig. 3). If analyses included all strains, almost identical male-specific strain differences were obtained.

Decrement of survival of male BXD Recombinant Inbred mice carrying D2 alleles at 55 Mb on Chr 13 (at rs13481817)
The female-specific heterozygote advantage locus on the distal Chr X could not be evaluated in the RI panel, which did not include any heterozygotes.
Causes of death before phenotyping
We analyzed pathology data obtained at the time of spontaneous death or scheduled sacrifice, as well as several aspects of physiology of the survivors prior to death to identify causes of death prevalent in this population and to explore genetic linkages to physiological and pathological processes among the survivors. Where available, data on younger age groups (150 and 450 groups) were evaluated because pathological processes often begin long before mortality actually occurs.
Animals that died prior to phenotyping were not genotyped, but were examined at death. This group is useful for evaluating which types of lesions are associated with early death. Among these mice, 102 animals were observed to be moribund prior to behavioral testing (generally between 450 and 700 days of age) Confirmed causes of death in the preliminary gross necropsy report included 14 neoplasias and 10 dental malocclusions. Probable causes of death included 27 other neoplasias and 6 major inflammatory lesions. In the remaining 45 cases, a clear cause of death or moribundity was not ascertained.
We tested for sex differences in cause of death among the probable and confirmed causes. Metastatic neoplasias as a whole (mostly lymphomas) were more common in female than male animals (32 in ♀, 13 in ♂). Pancreatic islet hypertrophy occurred in 12 males and no females. Dental malocclusions occurred in 10 males and no females. Dental malocclusions tended to co-occur with pancreatic islet hypertrophy (8 % prevalence of malocclusions in mice without islet hypertrophy, 56 % prevalence in mice with islet hypertrophy).
Associations of Chr 13 allele with common mouse pathologies
Next, we evaluated which specific types of lesions with at least 2 % prevalence were linked to the mortality loci in the animals that survived to be included in the study (and thus completed phenotyping and genotyping). Mice from all three age groups were included in the analyses, which tested for age differences, sex differences, and genetic effects on each lesion type.
We found no associations between the distal X allele (peaking at marker DXMit135) and any lesion type. However, there were significant associations between the D13Mit125 allele and pancreatic islet hypertrophy, renal tubule cell lipidosis, kidney mineralization, gastric hyperplasia, and hepatic lipidosis in male mice (Table 2).
Association of Chr 13 allele with serum chemistry
To further investigate the hypothesis that the D13Mit125 allele may be involved in glucose and lipid homeostasis, we investigated the relationship between the D13Mit125 allele and a serum chemistry panel that included serum urea nitrogen, glucose, calcium, inorganic phosphorus, cholesterol, total protein, alkaline phosphatase, albumin, aspartate amino transferase, triglycerides, alanine amino transferase, and iron. Animals were tested on three occasions spaced 1 month apart, with blood collection taken after 6 h of fasting during the light phase of the light–dark cycle. Serum data was only available for the age 500 days group of mice (not the 200 or 800 days mice).
Among male mice, the D2 allele at D13Mit125 was significantly associated with lower glucose, higher cholesterol, and higher iron (p at least <0.01, Table 3).
Discussion
Survival locus on Chr 13
There is ample evidence for genetic influences on lifespan in humans and animal models [19]. In many cases, these have been shown to be sex-specific [12, 20, 21]. One approach to identifying loci associated with mortality is to evaluate changes in expected gene frequencies between age groups, as has often been used in human case control studies comparing genotypic frequencies of centenarians to younger populations. In our experimental F2 cross of B6 and D2 inbred mouse lines, one quarter of the progeny is expected be homozygotes for the B6 allele, one half of the population will be heterozygotes, and the remaining quarter of the mice will be homozygotes for the D2 allele. If a deviation from the expected 1:2:1 ratio of genotypic frequencies is observed at birth, this could be due to the effects of this locus on gamete viability, fertilization, implantation, or embryonic mortality. If the expected genotypic frequencies are observed early in life, but then show a distortion at late ages after a substantial proportion of the population has died, we interpret those findings as evidence of genetic effects on age-related mortality. This method revealed a locus on Chr 13 between 35 and 80 megabases that predisposes male carriers of the D2 allele to earlier death than seen with the B6 allele. The expected sex-specific decrement in lifespan with the D2 allele was also observed in a panel of BXD recombinant inbred lines previously studied by [10].
Other prior QTL studies did not identify loci associated with lifespan or duration of survival time on Chr 13 [12, 22]. This is likely due to heterogeneity in genetic background, husbandry, housing, and environmental conditions, diet, etc.
Association of pathological mediators with the Chr 13 QTL
Our exploratory tests for association between available lesions and serum biomarker data prior to the onset of allele specific mortality indicate that the deleterious D2 allele on Chr 13 was associated with increased total lesion load, pancreatic islet hypertrophy, renal and hepatic lipidosis, and gastric hyperplasia. The Chr 13 D2 allele also appeared to have lowered serum glucose and elevated cholesterol levels specifically in male 450 days old mice. These findings support the notion that the Chr 13 locus may be associated with earlier and more frequent onset of these pathologies in the first year and a half of life, which in turn affect mortality risk in the following year. This is consistent with a diabetes-like disease process. In humans, pancreatic islet cell hypertrophy is evident during the development of type 2 diabetes when beta cells increase insulin production to compensate for the emergence of insulin resistance [23].
Compared to B6, D2 mice show impaired glucose homeostasis during fasting [24] and during the weanling transition from a milk diet (ketogenic) to solid chow [25]. The D2 strain is also more sensitive than B6 to the diabetogenic effects of the db leptin receptor mutation [26], presumably because B6 genes improve ability to maintain islet function during long periods of insulin resistance [27, 28]. However, in other contexts, B6 mice are more likely than D2 to spontaneously develop diabetic syndromes, indicating that risk factors exist on both genetic backgrounds [29]. QTL mapping studies indicate that these murine metabolic traits have a complex genetic architecture that is not dominated by any single allele [29–31], much like humans [32, 33].
Prior work identified candidate genes on Chr 13 that might underlie diabetes-related traits, including RASA1, Nnt, and PSK1. RASA1 show strong sequence differences between B6 and D2 strains [34]. Rasche et al. [35] reported that RASA1 was upregulated in the lipidosis-laden kidneys of mice with type 2 diabetes caused by the db/db mutation and [35] reported that RASA1 mutations may affect risk for type 2 diabetes in humans. Nnt is implicated because the B6 strain has a missense mutation which may impair insulin secretion in response to glucose challenge [36–38]. Pcsk1 was considered a candidate gene by [39] in a study identifying a locus on Chr 13 near 72.7 Mb associated with glucose intolerance (slower glucose clearance in B6 than C3H mice; the latter strain is related to D2). Kiliminik et al. [40] reported that Pcsk1 is abnormally expressed in the adult pancreas in obese and diabetic mice. There is one polymorphism (rs33851130) between D2 and B6 strains 3 kb upstream of the Pcsk1 transcriptional start site that has predicted gene expression regulatory potential. Large-scale human GWA studies show robust statistically significant associations of several SNPs in and near PCSK1 with type 2 diabetes-related traits, including proinsulin levels and fasting glucose levels [41, 42]. Further study is needed to determine whether these alleles might affect mortality in B6/D2 crosses.
The elevated blood glucose concentrations in males compared to females (Table 3) could also be indicative of a diabetes-related trait, especially with regards to islet cell lesions. Several lines of previous work in relevant animal models (and humans) indicate that estrogens protect pancreatic islet cells from loss of function and degeneration [43]. With particular reference to this cross, in B6D2F1 hybrid mice—the F1 offspring of B6 dams crossed with D2 sires—males spontaneously develop adult onset, non-obese type II diabetes [44, 45]. Thus, there is a rich literature indicating strong genetic effects on glucose metabolism in the B6 and D2 genetic background, and a male-specific form of diabetes is known to spontaneously occur in hybrids of this strain.
Dental traits
The reported link between a Chr 13 locus and dental malocclusions [46] might provide an alternative or additional explanation of the associations we observe. Dental malocclusions were the only major male-specific cause of death we observed in this mouse population (20 % of males that died before the 750-day phenotyping tests, 0 % of females). In this scenario, difficulty in feeding caused by dental problems would alter energy homeostasis in a manner that could also lead to a variety of lesions and earlier mortality. Of course, diabetes and dental problems are not necessarily mutually exclusive explanations. In humans, diabetes increases risk for periodontal disease [47, 48] and presence of periodontal disease may be a strong predictor of diabetic mortality [49]. In our mice, a diagnosis of pancreatic islet hypertrophy at the time of death was associated with increased rates of fatal dental malocclusions in our sample (for mice that died between age 450 and 800 days of age in our sample, mice with pancreatic islet hypertrophy had a 56 % prevalence of dental malocclusions compared to 8 % in the other mice). Further studies are required to determine whether this association is coincidental due to the linkage disequilibrium among dentition-related and glucose homeostasis-related genes on the same small chromosome, or is indeed due to causal relationships between dentition and islet hypertrophy.
Conclusion
We have exploited allelic imbalance accruing over the lifespan due to mortality to identify a locus associated with male-specific mortality on Chr 13 in a B6/D2 F2 that is associated with several lesions related to energy homeostasis and glucose metabolism. Several candidate genes with biological relevance reside at this locus. We also identified a novel dental pathology that could also play a role in the excess mortality due to the Chr 13 QTL.
References
Herskind AM, McGue M, Holm NV et al (1996) The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870–1900. Hum Genet 97(3):319–323
Miller RA (1999) Kleemeier award lecture: are there genes for aging? J Gerontol A Biol Sci Med Sci 54(7):B297–B307
Willcox BJ, Donlon TA, He Q et al (2008) FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci USA 105(37):13987–13992. doi:10.1073/pnas.0801030105
Anselmi CV, Malovini A, Roncarati R et al (2009) Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res 12(2):95–104. doi:10.1089/rej.2008.0827
Flachsbart F, Caliebe A, Kleindorp R et al (2009) Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci USA 106(8):2700–2705. doi:10.1073/pnas.0809594106
Li Y, Wang WJ, Cao H et al (2009) Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet 18(24):4897–4904. doi:10.1093/hmg/ddp459
Soerensen M, Dato S, Christensen K et al (2010) Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell 9(6):1010–1017. doi:10.1111/j.1474-9726.2010.00627.x
Newman AB, Murabito JM (2013) The epidemiology of longevity and exceptional survival. Epidemiol Rev [Epub ahead of print]
Leduc MS, Hageman RS, Meng Q et al (2010) Identification of genetic determinants of IGF-1 levels and longevity among mouse inbred strains. Aging Cell 9(5):823–836. doi:10.1111/j.1474-9726.2010.00612.x
Lang DH, Gerhard GS, Griffith JW et al (2010) Quantitative trait loci (QTL) analysis of longevity in C57BL/6J by DBA/2J (BXD) recombinant inbred mice. Aging Clin Exp Res 22(1):8–19
Gelman R, Watson A, Bronson R et al (1988) Murine chromosomal regions correlated with longevity. Genetics 118(4):693–704
Jackson AU, Galecki AT, Burke DT et al (2002) Mouse loci associated with life span exhibit sex-specific and epistatic effects. J Gerontol A Biol Sci Med Sci 57(1):B9–B15
Foreman JE, Lionikas A, Lang DH et al (2009) Genetic architecture for hole-board behaviors across substantial time intervals in young, middle-aged and old mice. Genes Brain Behav 8(7):714–727. doi:10.1111/j.1601-183X.2009.00516.x
Lang DH, Conroy DE, Lionikas A et al (2009) Bone, muscle, and physical activity: structural equation modeling of relationships and genetic influence with age. J Bone Miner Res 24(9):1608–1617. doi:10.1359/jbmr.090418
Blizard DA, Lionikas A, Vandenbergh DJ et al (2009) Blood pressure and heart rate QTL in mice of the B6/D2 lineage: sex differences and environmental influences. Physiol Genomics 36(3):158–166. doi:10.1152/physiolgenomics.00035.2008
Johannes F, Blizard DA, Lionikas A et al (2006) QTL influencing baseline hematocrit in the C57BL/6J and DBA/2J lineage: age-related effects. Mamm Genome 17(6):689–699
Foreman JE, Blizard DA, Gerhard G et al (2005) Serum alkaline phosphatase activity is regulated by a chromosomal region containing the alkaline phosphatase 2 gene (Akp2) in C57BL/6J and DBA/2J mice. Physiol Genomics 23(3):295–303
Wang J, Williams RW, Manly KF (2003) WebQTL: web-based complex trait analysis. Neuroinformatics 1(4):299–308
Murabito JM, Yuan R, Lunetta KL (2012) The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. J Gerontol A Biol Sci Med Sci 67(5):470–479. doi:10.1093/gerona/gls089
Nuzhdin SV, Pasyukova EG, Dilda CL et al (1997) Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc Natl Acad Sci USA 94(18):9734–9739
Gems D, Riddle DL (2000) Genetic, behavioral and environmental determinants of male longevity in Caenorhabditis elegans. Genetics 154(4):1597–1610
Henckaerts E, Langer JC, Snoeck HW (2004) Quantitative genetic variation in the hematopoietic stem cell and progenitor cell compartment and in lifespan are closely linked at multiple loci in BXD recombinant inbred mice. Blood 104(2):374–379
Wang Q, Jin T (2009) The role of insulin signaling in the development of β-cell dysfunction and diabetes. Islets 1(2):95–101. doi:10.4161/isl.1.2.9263
Fuller JL, Cooper CW (1967) Saccharin reverses the effect of food deprivation upon fluid intake in mice. Anim Behav 15(4):403–408
Schreiber RA, Ungar AL (1984) Glucose protects DBA/2J mice from audiogenic seizures: correlation with brain glycogen levels. Psychopharmacology 84(1):128–131
Hummel KP, Coleman DL, Lane PW (1972) The influence of genetic background on expression of mutations at the diabetes locus in the mouse. I. C57BL-KsJ and C57BL-6J strains. Biochem Genet 7(1):1–13
Coleman DL (1981) Inherited obesity-diabetes syndromes in the mouse. Prog Clin Biol Res 45:145–158
Anderson AA, Helmering J, Juan T et al (2009) Pancreatic islet expression profiling in diabetes-prone C57BLKS/J mice reveals transcriptional differences contributed by DBA loci, including Plagl1 and Nnt. Pathogenetics 2(1):1. doi:10.1186/1755-8417-2-1
Clee SM, Attie AD (2007) The genetic landscape of type 2 diabetes in mice. Endocr Rev 28(1):48–83
Mu JL, Naggert JK, Svenson KL et al (1999) Quantitative trait loci analysis for the differences in susceptibility to atherosclerosis and diabetes between inbred mouse strains C57BL/6J and C57BLKS/J. J Lipid Res 40(7):1328–1335
Togawa K, Moritani M, Yaguchi H et al (2006) Multidimensional genome scans identify the combinations of genetic loci linked to diabetes-related phenotypes. Hum Mol Genet 15:113–128
Weiss LA, Pan L, Abney M et al (2006) The sex-specific genetic architecture of quantitative traits in humans. Nat Genet 38(2):218–222
Doria A, Patti ME, Kahn CR (2008) The emerging genetic architecture of type 2 diabetes. Cell Metab 8(3):186–200. doi:10.1016/j.cmet.2008.08.006
Blake JA, Bult CJ, Eppig JT et al (2014) The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42(D1):D810–D817
Rasche A, Al-Hasani H, Herwig R (2008) Meta-analysis approach identifies candidate genes and associated molecular networks for type-2 diabetes mellitus. BMC Genomics 9:310. doi:10.1186/1471-2164-9-310
Toye AA, Lippiat JD, Proks P et al (2005) Agenetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48(4):675–686
Aston-Mourney K, Wong N et al (2007) Increased nicotinamide nucleotide transhydrogenase levels predispose to insulin hypersecretion in a mouse strain susceptible to diabetes. Diabetologia 50(12):2476–2485
Wong N, Blair AR, Morahan G et al (2010) The deletion variant of nicotinamide nucleotide transhydrogenase (Nnt) does not affect insulin secretion or glucose tolerance. Endocrinology 151(1):96–102. doi:10.1210/en.2009-0887
Kido Y, Philippe N, Schäffer AA et al (2000) Genetic modifiers of the insulin resistance phenotype in mice. Diabetes 49(4):589–596
Kilimnik G, Kim A, Steiner DF et al (2010) Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of ß-cell regeneration. Islets. 2(3):149–155
Strawbridge RJ, Dupuis J, Prokopenko I et al (2011) Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 60(10):2624–2634. doi:10.2337/db11-0415
Manning AK, Hivert MF, Scott RA et al (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44(6):659–669. doi:10.1038/ng.2274
Liu S, Mauvais-Jarvis F (2010) Minireview: Estrogenic protection of beta-cell failure in metabolic diseases. Endocrinology 151(3):859–864. doi:10.1210/en.2009-1107
Oh YS, Khil LY, Cho KA et al (2008) A potential role for skeletal muscle caveolin-1 as an insulin sensitivity modulator in ageing-dependent non-obese type 2 diabetes: studies in a new mouse model. Diabetologia 51(6):1025–1034. doi:10.1007/s00125-008-0993-0
Oh YS, Lee TS, Cheon GJ et al (2011) Modulation of insulin sensitivity and caveolin-1 expression by orchidectomy in a nonobese type 2 diabetes animal model. Mol Med 17(1–2):4–11. doi:10.2119/molmed.2009.00105
Petznek H, Kappler R, Scherthan H et al (2002) Reduced body growth and excessive incisor length in insertional mutants mapping to mouse Chromosome 13. Mamm Genome 13(9):504–509
Chávarry NG, Vettore MV, Sansone C et al (2009) The relationship between diabetes mellitus and destructive periodontal disease: a meta-analysis. Oral Health Prev Dent 7(2):107–127
Santacroce L, Carlaio RG, Bottalico L (2010) Does it make sense that diabetes is reciprocally associated with periodontal disease? Endocr Metab Immune Disord Drug Targets 10(1):57–70
Saremi A, Nelson RG, Tulloch-Reid M et al (2005) Periodontal disease and mortality in type 2 diabetes. Diabetes Care 28(1):27–32
Acknowledgments
We acknowledge the encouragement of Jerry McClearn Principal Investigator on P01-AG-14731 which supported this research. T32-AG-00276 provided stipends for graduate students. Jeanne Spicer provided superb database management. Thanks to Susan E. Lingenfelter, Kim A. Seymour, David A. Bienus, Gee-Sue Park, and Olivia Nourie for their excellent technical assistance and writing support.
Conflict of interest
All authors declare we have no financial conflicts of interest.
Human and Animal Rights
All animals were maintained according to protocols approved by the Institutional Animal Care and Use Committee of Penn State University.
Informed consent
This article does not report results of human subjects research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gyekis, J.P., Lang, D.H., Vandenbergh, D.J. et al. A Chromosome 13 locus is associated with male-specific mortality in mice. Aging Clin Exp Res 28, 59–67 (2016). https://doi.org/10.1007/s40520-015-0370-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40520-015-0370-z