
Abstract
Purpose of Review
Intestinal mucosal immunity is tightly regulated to ensure effective host defense against invasive microorganisms while limiting the potential for aberrant damage. In inflammatory bowel disease (IBD), an imbalance between effector and regulatory T cell populations results in an uncontrolled inflammatory response to commensal bacteria. Intraepithelial lymphocytes (IEL) are perfectly positioned within the intestinal epithelium to provide the first line of mucosal defense against luminal microbes or rapidly respond to epithelial injury. This review will highlight how IELs promote protective intestinal immunity and discuss the evidence indicating that altered IEL responses contribute to the pathogenesis of IBD.
Recent Findings
Although the role of IELs in mucosal homeostasis has been largely underappreciated, many of the same factors that contribute to the dysregulation of host defense in IBD also adversely affect IELs. For example, IL-23 and the endoplasmic reticulum stress response can enhance IEL lytic activity toward enterocytes. Microbial dysbiosis or defective microbial recognition results in the loss of regulatory IELs, further amplifying these pro-inflammatory effects. Migration of T cells into or within the intraepithelial compartment has a profound effect on their differentiation or effector function demonstrating that IELs are exquisitely sensitive to changes in the local intestinal microenvironment.
Summary
Enhanced mechanistic insight into the regulation of IEL survival, differentiation, and effector function may provide useful tools to modulate IEL surveillance or enhance IEL regulatory function. Elucidation of these processes may result in the development of novel therapeutics to reduce intestinal inflammation and reinforce the mucosal barrier in IBD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) refers to Crohn’s disease and ulcerative colitis, two chronic inflammatory diseases of the gastrointestinal tract that are characterized by an uncontrolled adaptive immune response against intestinal bacteria. Nearly 5 million individuals worldwide are affected by IBD, and the prevalence of disease continues to increase with an estimated 70,000 new diagnoses each year. Our current knowledge indicates that the etiology of IBD is multifactorial, with environmental, microbial, genetic, and immunological components contributing to the pathophysiology of disease [1, 2]. It has been challenging to identify the initiating factor in disease due to the complex regulation of these interrelated factors. However, advances in defining the symbiotic relationship between the immune system and resident microbiota have greatly expanded our understanding of IBD pathophysiology [3, 4]. Uncovering the mechanisms leading to the dysregulation of host defense responses has resulted in the development of new therapeutic strategies to treat IBD [5•, 6].
The intestinal epithelium is a single layer of cells that serves as the physical barrier to separate the mucosal immune system from commensal and pathogenic microbes as well as dietary antigens. Epithelial damage or increased epithelial permeability can disrupt this barrier, leading to activation of the mucosal immune system. Increased intestinal permeability has been observed in Crohn’s disease patients prior to the clinical onset of disease or before relapse, suggesting that barrier dysfunction may trigger disease development [7, 8]. However, compromised barrier function alone is insufficient to cause disease, since increased permeability was observed in an otherwise healthy subset of first-degree relatives of Crohn’s disease patients [9]. Studies of transgenic mice exhibiting enhanced intestinal epithelial permeability show that the microbiota primes the mucosal immune system, thus compensating for compromised barrier function at steady state while increasing susceptibility to experimental colitis [10, 11•]. Yet it remains unclear whether the pathophysiology of IBD stems from an intrinsic barrier defect or if pro-inflammatory cytokines produced by subclinical inflammation enhances epithelial permeability. Regardless of the sequence of events in disease progression, these studies demonstrate how crucial immune surveillance of the intestinal epithelium is for maintenance of mucosal homeostasis and generation of appropriate responses to luminal microbes and antigens.
The intestinal immune system constantly regulates the balance between host defense responses and a state of tolerance toward resident luminal microbes [2, 12]. A large number of intestinal tissue-resident immune cells contribute toward the defense of the epithelial barrier, including T lymphocytes, which are found with highest abundance in the intestinal mucosa relative to anywhere else in the body. This review will focus on intraepithelial lymphocytes (IEL), which are located within the epithelial monolayer just above the basement membrane and between adjacent epithelial cells. Perfectly positioned as first responders to luminal antigens, IELs are antigen-experienced T cells that express either the αβ or γδ T cell receptor (TCR). In this article, we will review the key contributions of different IEL subtypes to intestinal mucosal homeostasis, explore how impaired host-microbe interactions or immune dysregulation that is associated with IBD influences IEL function, and highlight new areas of research that may provide valuable insight into the contribution of IELs in protective intestinal immunity.
Ontogeny and Differentiation of Intestinal IEL Subtypes
In mice, IELs are classified into two main subtypes: “induced” IELs that are phenotypically similar to conventional memory effector T cells and innate-like “natural” IELs that exhibit regulatory properties. Following conventional thymic selection, induced IELs expressing either CD8αβ+ TCRαβ+ or CD4+ TCRαβ+ are recruited to the periphery in response to antigenic stimulation through the upregulation of gut-homing markers [13]. In contrast, natural IELs develop an antigen-experienced phenotype in response to self-antigen during thymic maturation, and then migrate directly to the intestinal epithelium [14, 15]. These IELs express either the αβ or γδ T cell receptor and are typically CD8αα+, but lack CD8αβ or CD4 co-receptors. The role of the thymus in natural IEL development remains controversial, since CD8αα+ IELs can develop extrathymically within cryptopatches or isolated lymphoid follicles in the intestinal mucosa [16,17,18]. Additional studies regarding IEL ontogeny have been reviewed extensively elsewhere [19]. Once IELs traffic to the intestine, these cells become tissue resident and do not return to circulation [20, 21]. The relative frequency of individual IEL subtypes differs based on the region of intestine assessed (Table 1). It is important to consider that the distribution of IEL populations differs between mice and humans. In mice, the number and proportion of IELs can vary based on the strain; for example, BALB/C mice exhibit a lower percentage of γδ T cells than C57BL/6 [22, 26]. IEL number and proportion is also influenced by housing conditions, since these factors are dependent on the level of antigenic stimulation in the intestine [28]. Therefore, with the significant variation in human IEL populations that has been reported in the literature [24, 25, 27], it is difficult to determine relative IEL proportions between mice and humans. This is especially relevant to γδ IELs, in which mouse γδ T cell subsets are functionally characterized by Vγ chain usage, whereas human intestinal γδ T cell subsets are classified by the Vδ chain. However, the overall proportions of αβ versus γδ T cells are similar between mice and humans (Table 1), as are the functional profiles of these IELs [29,30,31]. All IELs possess an antigen-experienced cytolytic effector phenotype; however, the antigenic reactivity of these IEL subtypes is thought to regulate their function within the intestinal epithelium, thus influencing their role in intestinal injury and inflammation [19].
An Imbalance in IEL Cytolytic and Regulatory Functions Contributes to Disease Development
An equilibrium between regulatory and effector lymphocytes is required to ensure appropriate immunological defense against invasive microorganisms while reducing the potential for aberrant inflammation or damage. This delicate balance is disrupted in IBD as naïve cells increasingly differentiate into cytotoxic lymphocytes [19]. Under homeostatic conditions, mucosal dendritic cells sample and present soluble antigens from apoptotic epithelial cells to naïve T cells [32], thus yielding non-responsive, tolerogenic T cells within the intestine. However, increased licensing of CD8+ cytotoxic IELs shifts the ratio of effector/regulatory T cells, leading to a breakdown of mucosal tolerance and subsequent tissue destruction. For example, increased IEL cytolytic activity toward intestinal epithelial cells can result in the villous atrophy that is characteristic of celiac disease [33, 34].
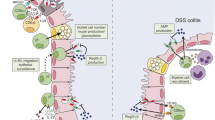
In IBD, an imbalance between regulatory and cytolytic effector cells within the epithelium leads to a dysregulation of mucosal immunity and the generation of a pro-inflammatory microenvironment (Table 2). While epithelial cytolysis results in ulceration, allowing bacterial invasion of the mucosa and enhanced T cell activation, a simultaneous reduction in regulatory cells further amplifies the pro-inflammatory response. This shift toward cytotoxic lymphocyte (CTL) differentiation was observed in the TNFΔARE mouse model, which is histopathologically similar to human IBD due to defective TNF translational regulation [45]. In this model, a reduction of CD8αα+ IELs early in disease is followed by an influx of peripherally activated CD8αβ+ TCRαβ+ lymphocytes into the epithelium [38, 46]. Although the induced IELs in TNFΔARE mice produce more IFNγ and TNF, surprisingly, it is the activation of the unfolded protein response (UPR) that is responsible for enhanced CD8αβ+ IEL granzyme B expression [39]. This occurs as a result of glucose-regulated protein 78 (Grp78)-mediated activation of downstream transcription factors that directly bind to the granzyme B promoter [39]. UPR activation of IEL cytolytic activity is particularly interesting in this context since unresolved ER stress responses have been implicated in risk susceptibility for IBD [47]. While the TNFΔARE model more closely models human disease, it is worth noting that CD8 T cell-mediated epithelial cytolysis has also been implicated in tissue destruction in hapten-induced colitis [44]. Although CD4+ lamina propria lymphocytes are considered to be the primary drivers of intestinal inflammation in IBD, these studies indicate that activation of cytotoxic CD8αβ+ TCRαβ+ IELs also induces epithelial damage.
In humans, CD8+ IELs closely resemble systemic effector memory cells and exhibit cytolytic activity. It is thought that the intestinal microenvironment conditions CD8+ IELs to respond to nonclassical major histocompatibility complex (MHC) class I molecules through the activation of natural killer receptors (NKR). These MHC class I ligands are upregulated in response to epithelial stress, infection, or inflammation (reviewed in [19, 48,49,50]). In mice, NKR are expressed on CD8αα+ TCRαβ+ IELs; however, IELs expressing the CD8αα homodimer have not been identified in humans [48]. Instead, it is thought the activation of antigen-specific conventional CD8αβ+TCRαβ+ IELs or recognition of epithelial stress ligands by these cells induces epithelial cytolysis [48].
IL-23 is critical for the regulation of memory T cell function, Th17 differentiation, and ILC3 activation, and as a result has become one of the most recent targets for IBD immunotherapy [5•, 51]. Colonic CD8+ IELs isolated from IBD patients exhibit higher IL-23R expression compared to healthy controls [52]. Exposure to IL-23 ex vivo enhanced pro-inflammatory cytokine production and lytic activity in activated IELs [52]; however, the effect of IL-23 on specific IEL subtypes and the mechanism by which IL-23 enhanced cytolysis was not evaluated. While it is clear that pro-inflammatory cytokines such as IL-23 can influence the cytolytic potential of these CD8+ IELs, it remains to be determined if (1) increased CTL activity is an intrinsic component of IBD pathogenesis as a result of UPR activation, (2) loss of regulatory IELs during IBD development promotes unchecked CTL activation, or (3) cytolytic activity is enhanced as a secondary response to the inflammatory microenvironment.
Regulatory Phenotype of Natural IELs in Mucosal Homeostasis and Disease
Innate-like CD8αα+ IELs are maintained in a state of partial activation, thus allowing these cells to rapidly respond to stimulation without being self-destructive [53]. CD8αα+ IELs differentiate in response to self-antigen, and thus may specifically target cancerous or infected cells displaying stress ligands. In order to maintain an immunologically quiescent state, CD8αα functions as a TCR co-repressor [53]. Although this repression may be overcome by high levels of antigen stimulation [54], reduced TCR responsiveness results in a limited capacity for IELs to proliferate within the intestine [55]. This anti-proliferative effect is mediated in part by CD8αα binding to the thymus leukemia (TL) antigen, a nonclassical MHC class I molecule expressed on the epithelial surface [56, 57]. Loss of TL expression increases the proliferation of colonic CD8αα+ IELs, but has no effect on small intestinal IELs [58]. This finding suggests that the greater microbial diversity within the colon influences IEL proliferation in the absence of CD8αα-mediated inhibition of T cell activation [58].
A protective role for CD8αα+ IELs in colitis was first demonstrated following adoptive transfer of individual IEL subsets into the CD4+CD45RBhi T cell transfer model [35]. IL-10 production by CD8αα+ IELs was shown to confer protection against disease development by suppressing lamina propria T cell expansion [35, 36, 59], demonstrating that natural IELs have regulatory properties that can limit immune-mediated colitis. Since it is not possible to selectively deplete CD8αα+ TCRαβ+ IELs, much remains unknown about their function; however, gene expression analysis shows that these cells express a variety cytokines and chemokines that may serve to recruit other immune cell populations [60].
Whereas CD4+ effector T cells in the periphery typically exhibit a pro-inflammatory phenotype, CD4+ T cells can reacquire CD8αα once reaching the epithelium [36, 61]. This ability to convert an induced, cytotoxic effector T cell to a regulatory phenotype demonstrates the important influence of the intestinal microenvironment in ensuring an appropriate balance between highly effective protective immunity while limiting the potential for aberrant damage. Although the frequency by which IELs exchange between the epithelial and lamina propria compartments is relatively low, intravital multiphoton microscopy within the intestinal mucosa has begun to provide new insight into how T cell plasticity effects the functional specification of distinct IEL subsets. Recently, the microbiota was shown to mediate the conversion of lamina propria Foxp3+ Tregs to CD4+ CD8αα+ or CD4+ CD8αβ+ IELs through the downregulation of the helper T cell transcription factor ThPOK [62••]. These findings demonstrate a novel mechanism by which the tissue-specific microenvironment drives the differentiation of a presumably stable Treg population into CD4+ IELs. Further investigation is needed to ascertain the extent to which this conversion occurs in an IBD model, and to determine whether pro-inflammatory conditions or microbial dysbiosis influences the effector phenotype of these ex-Treg CD4+ IELs. Although lamina propria CD4+ CD8αα+ T cells were shown to have a regulatory role in IBD [63•], the contribution of CD4+ CD8αα+ IELs to the pathophysiology of IBD remains unknown.
As novel regulatory mechanisms by which the intestinal microenvironment regulates IEL function are identified, it will be important to consider how T cell plasticity will be affected in the context of inflammatory disease. For example, does Foxp3 Treg conversion also occur in the colon? If so, since Clostridia species involved in colonic Treg differentiation are depleted in IBD [4, 64], will changes in microbial composition also affect the generation of this ex-Treg CD4+ IEL population? By understanding the factors that create this imbalance between cytolytic and regulatory IEL populations in IBD, it may become possible to shift these populations back toward equilibrium in an effort to reduce intestinal inflammation.
Commensal Bacteria Influence IEL Development and Function
Genetic variants in NOD2 were the first identified IBD genetic susceptibility factors; since then, genome-wide association studies have discovered that several genes involved in host-microbe interactions are located in the IBD susceptibility loci [1]. As a result, the symbiotic relationship between the microbiome and mucosal immunity has become a main area of focus in efforts to unravel the pathogenesis of IBD. Changes in the composition of the microbiota lead to an overall reduction in bacterial diversity in IBD [3, 4], which has a profound effect on mucosal immunity. Although several commensal species have been shown to directly affect the differentiation of lamina propria lymphocytes [3, 64, 65], significantly less is known regarding specific bacteria that directly influence IEL populations.
Monocolonization of germ-free mice with segmented filamentous bacteria (SFB) increased the total number of small intestinal αβ IELs; however, colonization with both SFB and Clostridia spp. restored total IEL numbers to similar levels observed in conventionalized mice [66]. As expected, each of these strains demonstrated some degree of regional specificity regarding their influence on IEL populations. Clostridia monoassociation increased the proportion of CD8+ IELs relative to CD4+ CD8− in the colon, whereas SFB enhanced the ratio of CD8αβ+ IELs compared to those expressing CD8αα+ [66]. Exposure to two different strains of Lactobacillus reduced epithelial expression of IL-15 and the NKG2D ligand Rae1 (retinoic acid early inducible-1) to alleviate TLR3-mediated increases in CD8αα IEL number and NKG2D expression [67•]. Further, prophylactic administration of a probiotic mixture containing Lactobacillus acidophilus and Bifidobacterium longum conferred protection against TNBS colitis, which was associated with reduced CD4+ T cells and increased γδ T cells in the intraepithelial compartment. Interestingly, the inverse distribution of these cells was observed in the lamina propria in response to TNBS in probiotic-treated mice [68]. Recently, Lactobacillus reuteri was shown to induce CD4+ CD8αα+ IELs, through the generation of AhR ligands that directly activate the T cell leading to ThPOK downregulation [69••]. This report is one of the first to elucidate a specific mechanism by which commensals induce alterations to the IEL compartment, and demonstrated that transplantation of fecal microbiota from mice colonized with L. reuteri could induce IEL differentiation in the transplant recipient [69••]. Based on these findings and those from monocolonization studies, it is possible that the alteration of IEL populations by fecal microbiota transplantation may have potential therapeutic value.
All IELs, with the exception of those that are TCRγδ+ [70, 71], are reduced in gnotobiotic and antibiotic-treated mice [72], indicating that the microbiota is critical for the maintenance of an intact IEL compartment. In support of these findings, several IEL populations fail to survive in mice lacking pattern recognition receptors such as Toll-like receptors (TLR) and nucleotide-binding oligomerization domain (Nod2). Although bacterial recognition through TLR signaling is generally thought to promote mucosal inflammation, several studies have shown that disruption of these innate immune pathways reduces epithelial IL-15 signaling, which is required for CD8+ IEL proliferation and survival [73,74,75]. Impaired bacterial recognition was not sufficient to ablate all IELs, yet the effector function of the remaining IELs was markedly affected [73, 75]. Interestingly, the residual CD8+ and γδ IEL populations in Nod2 knockout mice expressed more IFNγ; this was dependent on MyD88 signaling and the presence of the commensal bacterium, Bacteroides vulgatus [76••]. Enhanced IFNγ production correlated with reduced goblet cell function in the small intestine [76••], yet the contribution of individual IEL subtypes and whether IEL IFNγ production is sufficient to induce this phenotype was not determined. Further identification of the IEL subpopulation(s) responsible for the alterations in goblet cell physiology would be of particular interest since γδ T cells were shown to influence goblet cell number and mucin production and glycosylation [77••]. Pro-inflammatory cytokine production by IELs may impair the protective mucus barrier and contribute to the exacerbation of disease. These findings are consistent with the reduced goblet cell number and altered mucin production observed in Crohn’s disease and ulcerative colitis [78].
While the link between epithelial innate immune recognition and IEL function will be addressed later in this review, the extent to which IEL function depends on luminal microbial recognition by myeloid cells has yet to be explored. Contrary to TLR induction of epithelial IL-15 [73, 74], reduced IEL number in Nod2 knockout mice was attributed to defective IL-15 production by myeloid cells [72]. Since IELs are in close proximity to both luminal sampling antigen-presenting cells and enterocytes, it stands to reason that multiple levels of regulation and crosstalk may inform IEL function and vice versa. With this as a consideration, it remains unclear as to whether the increased susceptibility of TLR or MyD88 knockout mice to experimental colitis [72, 79] is a result of reduced CD8αα+ IEL number, changes in the composition of the microbiota, or defective bacterial recognition in myeloid or epithelial cells. Transfer of WT CD8+ IELs was sufficient to alleviate 2,4,6-trinitrobenzene sulfonic acid (TNBS) colitis in Nod2-deficient mice, suggesting that at least in this model, IEL depletion is a key factor in driving disease [72]. Although bacterial infection leads to increased expression of an intracellular peptidoglycan recognition receptor in CD8+ IELs [80], surprisingly little is known regarding pattern recognition receptor expression or function within specific IEL subsets. Therefore, further studies are needed to determine the extent to which IELs directly respond to microbial antigen either at steady state or in the context of intestinal inflammation. Together, these studies show that in addition to the reduced responsiveness of innate immune cells to invasive microbes, failure to appropriately respond to luminal microbes at steady state also effectively disables the first line of defense by CD8αα+ IEL depletion and may also shift the IEL regulatory response toward a pro-inflammatory phenotype.
Dietary Factors Contributing to IBD Adversely Affect Natural IEL Populations
As IBD diagnoses continue to rise in the industrialized world and in countries previously characterized with a low incidence of IBD [81, 82•], there has been a renewed interest in the influence of the Westernized diet on disease development. Diet not only influences the overall composition of the microbiome [3], but dietary antigens and the production of bacterial metabolites have a profound impact on mucosal immunity [4]. Similar to microbial depletion, animals receiving an elemental antigen diet also fail to develop CD8αα+ IELs [83]. Tryptophan-derived ligands that are produced as metabolites from cruciferous vegetables bind to the aryl hydrocarbon receptor (AhR); this receptor is highly expressed on CD8αα+ and γδ IELs and is required for their survival [84]. As a result, AhR knockout mice demonstrate increased susceptibility to experimental colitis due to an expanded induced colonic IEL population and microbial overgrowth. Mice fed a high-fat diet exhibit an increase in the total number of γδ IELs with no perturbations observed in any other IEL population [85]. Analysis of effector function within total IEL populations demonstrated an increase in TNF, perforin, and granzyme B expression in response to a high-fat diet [86]. While these mice are more susceptible to experimental colitis [86], the relative contribution of IELs to the pathogenesis of disease under these circumstances has not been fully investigated.
Vitamin A is primarily ingested as retinol, which is then converted to retinoid acid by dendritic cells within the gut-associated lymphoid tissue (GALT) [87]. Retinoic acid promotes T cell trafficking into the intestine by enhancing expression of α4β7 integrin and CCR9 [87]. Mice fed a vitamin A-deficient diet exhibited a depleted CD8+ IEL compartment, which may be reflective of a failure to express gut-homing proteins [87, 88]. Interestingly, retinoic acid induced an expansion of Lactobacillus in the microbiota [89], suggesting that the effect observed on IEL homeostasis may also be attributed to changes in microbiota composition.
Commensal bacteria also contribute to the processing of vitamin D and influence the expression of vitamin D receptor (VDR) [4]. Vitamin D deficiency has been shown to be predictive of IBD relapse [82•], and VDR polymorphisms have been identified in CD and UC patients [90, 91]. Further investigation of the role of vitamin D in intestinal inflammation showed that while mice lacking vitamin D or VDR do not develop spontaneous inflammation, both of these strains show an exacerbation in experimental colitis [92]. The increase in disease susceptibility coincides with a 50% reduction in CD8αα+ IELs due to defective gut homing, [93] and impaired IL-10 production among the remaining IELs, thus increasing the inflammatory response to commensal bacteria [94].
With the steady increase in IBD diagnoses [81], understanding the environmental factors that predispose individuals to IBD becomes even more essential. Studies show that a diet high in fat and low in fruits and vegetables may be one of the environmental factors that contribute to the development of IBD [82•, 95]. As this area of investigation continues to expand, identification of the effect of diet and bacterial metabolites on IEL populations and function will be increasingly important to our understanding of the causal relationship between dietary changes and mucosal immunity and how this influences intestinal homeostasis.
γδ IELs Protect Against Experimental Colitis
γδ T cells are far less common than αβ T cells within the peripheral blood and lymphoid tissues, yet γδ T cells represent a large proportion of the lymphocytes located at mucosal surfaces. In mice, CD8αα+ γδ IELs are the predominant IEL subset in the small intestine, whereas a smaller proportion of colonic IELs are CD4− CD8− TCRγδ+ [22]. Unlike the αβ TCR, the γδ TCR does not require antigen presentation by MHC, but can be stimulated by non-MHC-like self or microbial antigens (reviewed in depth in [96, 97]). Elimination of the need for MHC processing and presentation permits a more rapid response to challenge, thus allowing γδ IELs to bridge innate and adaptive immune responses.
Adoptive transfer of CD8αα+ TCRαβ+ IELs reduced the severity of injury and inflammation in experimental colitis [35]; however, the inability to specifically deplete this subset has made it difficult to pinpoint their precise function. In contrast, genetic or antibody-mediated depletion of γδ T cells showed that loss of γδ T cells results in more severe disease in response to TNBS [42, 43] and dextran sulfate sodium (DSS) [98], as well as in TNFΔARE mice [37], demonstrating a protective role for γδ IELs in intestinal injury. Further characterization of γδ IELs showed that these cells limit intestinal inflammation through multiple mechanisms. Increased IL-10 and TGFβ production by adoptively transferred γδ T cells was sufficient to reduce disease severity and mortality in TNBS colitis [43, 59]. Further, DSS treatment of Tcrd knockout mice resulted in lesions dominated by monocytes rather than granulocytes, indicating that γδ T cells promote neutrophil infiltration to limit the extent of mucosal injury [98]. These findings were supported by gene expression analysis of colonic γδ IEL following DSS treatment, which revealed that the microbiota is required to stimulate γδ IEL production of pro-inflammatory cytokines, chemokines, and the antimicrobial peptide (AMP) RegIIIγ in response to injury [99]. Further, commensal bacteria induced γδ IEL signaling in response to mucosal injury through both MyD88-dependent and MyD88-independent pathways [99]. Taken together, these signals likely help regulate both the magnitude and timing of mucosal immune responses to rapidly neutralize invading microbes and clear damaged cells without causing further mucosal damage.
γδ IELs Prevent Acute Bacterial Translocation Across the Epithelium
γδ IELs limit the acute translocation of invasive commensal and pathogenic bacteria within the first hours after exposure [71, 100••]. At steady state, small intestinal γδ IEL RegIIIγ production requires commensal-induced epithelial MyD88 signaling [71], again highlighting the crucial role of microbial recognition in IEL-mediated host defense. Although human γδ T cells respond directly to microbial products through TLR stimulation and the recognition of bacterial phosphoantigens (reviewed in [101]), whether γδ IELs are also capable of directly recognizing luminal microbes has yet to be determined.
In response to Salmonella typhimurium, γδ IELs limit bacterial invasion by stimulating AMP production by Paneth cells in an IL-22-dependent manner [102]. The generation of AMPs is necessary to prevent commensal overgrowth and spatially segregate the microbiota from the apical surface of intestinal epithelium through the formation of an exclusion barrier [103]. Since a high degree of functional redundancy exists among antimicrobial responses to ensure proper host defense, it remains unclear as to whether AMP expression induced either directly or indirectly by γδ IELs is required to confer protection against bacterial invasion.
The mechanisms regulating the rapid, innate-like response of γδ IELs to microbial challenge at the epithelial barrier have not been well-characterized. Once presumed to be sessile within the epithelial compartment [50], timelapse intravital microscopy revealed that γδ IELs are highly motile within the villous epithelium [104]. γδ IELs continuously survey the epithelium by migrating along the basement membrane and into the lateral intercellular space between adjacent epithelial cells [104]. Epithelial retention within the lateral intercellular space is mediated in part by direct interaction of CD103 with epithelial E-cadherin, disruption of which reduces γδ IEL dwell time and enhances migratory speed of these cells [104]. Further, migration into the lateral intercellular space is essential for γδ IEL-mediated protection against microbial translocation, as shown by an increase in acute invasion of either S. typhimurium or Toxoplasma gondii in mice with γδ IELs deficient in occludin, a junction protein critical for γδ T cell motility [100••, 104]. Interestingly, γδ IELs were shown to preferentially migrate toward sites of mucosal pathogen adherence [100••], suggesting that innate signaling pathways regulate γδ IEL surveillance of the villous epithelium. These host-microbe interactions may direct the migration of these sentinels to sites of microbial invasion as a means to deliver a precise and localized effector response. The microbial cues regulating γδ IEL migration are not restricted to enteric pathogens. Antibiotic treatment significantly reduces γδ IEL migration into the lateral intercellular space (Edelblum et al., unpublished observation), indicating that commensal bacteria modulate basal γδ IEL migration. Additional studies are required to identify the signals involved in mediating γδ IEL surveillance and to determine if and how γδ IEL retention within a defined location in the epithelial monolayer triggers a localized antimicrobial response.
The patrolling behavior of γδ IELs emphasizes the importance of direct ligand/receptor interactions between γδ IELs and epithelial cells. Although there is only one IEL for every 5–10 epithelial cells, the migration of γδ IELs results in the constant and dynamic interaction of these cells with neighboring enterocytes. It is likely that intestinal injury or inflammation will alter cell surface ligand or receptor expression, to influence or regulate γδ IEL function. For example, pathological levels of tumor necrosis factor (TNF) induce occludin internalization from the plasma membrane [105], thus reducing its availability on the epithelial cell surface and inhibiting γδ IEL motility and surveillance [104]. Impaired γδ IEL surveillance increases susceptibility to infection, thus it would be interesting to determine whether defective migration triggers disease development in susceptible hosts by increasing the likelihood of invasion by commensal pathobionts.
Consistent with the role of γδ IELs in maintaining epithelial integrity, activated γδ IELs produce keratinocyte growth factor (KGF) to stimulate epithelial regeneration following DSS treatment [41, 106]. KGF production is impaired in DSS-treated mice deficient for CD100, which is a γδ T cell co-stimulatory molecule that binds to plexin B2 on intestinal epithelial cells [107•]. This γδ IEL ligand/receptor pair was initially identified in the skin; therefore, the conservation of function between ligand/receptor interactions in the skin and gut provides valuable insight into γδ IEL/epithelial crosstalk and the mechanisms by which epithelial cells regulate γδ IEL development and function. It will be interesting to determine how these interactions translate from mice into humans, and the extent to which these signals are affected in the context of IBD.
Location of γδ T Cells May Dictate Function in Intestinal Inflammation
Despite the large number of studies that demonstrate a protective role for γδ IELs in experimental colitis, several reports also suggest that γδ T cells may promote disease pathogenesis. Antibody-mediated depletion of γδ T cells partially reduced the severity of colitis in TCRα knockout mice [40], which develop spontaneous colitis resembling ulcerative colitis between 16 and 20 weeks of age [108]. Disease development is driven by the selection and expansion of a unique CD4+ TCRββ+ population that produces IL-4 to drive a TH2 inflammatory response [109, 110]. In the absence of αβ T cells, dysregulated γδ T cells expand within the colonic lamina propria to induce a pro-inflammatory IL-17 response [108]. Further supporting a role for γδ T cells in disease pathogenesis, adoptive transfer of IL-17+ or CD103+α4β7hi γδ T cells increased the severity of disease in a T cell transfer models of colitis [111, 112••].
Differences in Vγ subsets and genetic knockout mice used in the various mouse models of experimental colitis may account for the seemingly contradictory conclusions drawn between these studies and those that demonstrate a protective role for γδ T cells. While none of these experimental colitis models fully mimic the multifactorial disease process involved in IBD, taken as a whole, the information gained from these studies strongly indicates that γδ T cell subtype or localization may predict their role in intestinal inflammation. The majority of the studies indicating a pathogenic role for γδ T cells exhibit a pro-inflammatory γδ T cell phenotype localized to the lamina propria, not the intraepithelial compartment. Further, IL-17 production is restricted to γδ T cells in the periphery and lamina propria, which are comprised of Vγ subsets that are distinct from Vγ7+ IELs that do not express IL-17 [50]. Although the data overwhelmingly indicate that γδ IELs confer protection against disease development, further investigation into the frequency and dynamics of γδ T cell migration between the intraepithelial compartment and lamina propria during active inflammation is warranted. It is reasonable to hypothesize that disruption of the epithelial barrier during severe inflammation would adversely affect resident T cells. Therefore, to gain a better appreciation of the role of IELs in IBD, continued investigation is required to determine how increased epithelial permeability or ulceration impacts IEL number and/or function.
Addressing the Differences Between Mice and Men: γδ IELs in IBD
Despite the numerous studies of γδ T cell function in mice, relatively little is known regarding the role of γδ IELs in IBD. Investigations of γδ T cells within the intestinal mucosa of IBD patients have proven contradictory and inconclusive, with various studies showing increases, decreases, or no change in the number or proportion of γδ IELs present (reviewed by Catalan-Serra et al. 2017 [113•]). It should also be noted that a number of these studies focused broadly on all γδ T cells in the mucosa rather than distinguishing between their precise location (i.e. lamina propria vs. intraepithelial) or the various γδ TCR subtypes (i.e., Vδ1+ vs. Vδ2+); this may account for some of the discrepancies between these studies. Additionally, observations regarding γδ IEL number are difficult to assess in isolation, as no studies have addressed whether γδ T cells in the intestinal epithelium are contributing to or regulating the inflammation observed in IBD patients.
The main obstacles involved in performing these studies are obtaining a sufficient number of IELs from a tissue biopsy for functional study and successfully maintaining these IELs ex vivo. As a result of these challenges, the most well-characterized population of human γδ T cells is circulating Vγ9/Vδ2 T cells that respond to endogenous and microbial phosphoantigen [114]. Vγ9/Vδ2 T cells are recruited from the periphery into the lamina propria in IBD patients where they can exhibit both pro-inflammatory and protective functions [115, 116••]. Interestingly, azathioprine (AZA) treatment selectively depletes this subtype in Crohn’s disease patients [117]. Although the mechanism by which AZA reduces Vδ2+ T cell number has yet to be determined, this effect could not be attributed solely to impaired purine biosynthesis since similar results were not observed in patients treated with methotrexate [117]. However, its effect on Vδ1+ T cells, which constitute the majority of human γδ IELs, remains unclear. These CD8+ Vδ1+ cells display a cytotoxic Th1 phenotype and produce IFNγ similar to murine γδ IELs [118]. Relatively little is known regarding the role of Vδ1+(Vδ2−) IELs in IBD, although recent studies demonstrate that butrophylin-like genes (BTNL3 and 8) can selectively regulate TCR activation in Vγ4+Vδ2− IELs [119••]. Butrophylins are transmembrane proteins related to the immunoglobulin superfamily expressed by intestinal epithelial cells that shape and regulate local γδ T cell populations [119••]. Gene expression analysis of biopsies from ulcerative colitis patients showed increased BTN3 and decreased BTNL8 expression [120••]. Based on the function of these genes, these changes in expression correlate with a suppressive effect on T cell activation [120••]; however, the effect that this has on γδ IEL activation remains to be determined.
Effect of IBD Therapeutics on IEL Function
Historically, IBD therapy has broadly focused on anti-inflammatory drugs, antibiotic use, and immune suppression to reduce the destructive inflammation associated with disease pathology. Current and emerging therapies favor a more targeted approach including biologics designed to specifically inhibit key cytokines, such as TNF or IL-12p40, which is associated with both IL-12 and IL-23 (reviewed by M. Neurath [5•]). Alternatively, the use of monoclonal antibodies against α4β7 or CD103 (α3β7) integrin prevents the trafficking of activated T cells to the gut. While these targeted therapies may be useful in limiting the recruitment and activation of both lamina propria CD4 T cells and conventional cytotoxic IELs, how these molecules affect natural IEL populations remains unclear.
More recently, clinical trials of Janus kinases (JAK) inhibitors showed promising results in inducing clinical remission in IBD patients [5•]. Inhibition of JAK signaling, which is activated downstream of several cytokine receptors, may be an effective means to block multiple pro-inflammatory signaling pathways. While these inhibitors may reduce the severity of active disease by suppressing pro-inflammatory responses, it is necessary to consider that IL-2- and IL-15-mediated IEL survival is also JAK-dependent. Although biologic agents are highly efficacious when administered during active disease, biologic therapy is generally continued in an effort to maintain clinical remission and prevent disease relapse. As a result, continued use of JAK inhibitors as maintenance therapy will likely adversely affect overall IEL number and function, thus compromising this first line of defense.
As translational studies begin to investigate the functional role of IELs in IBD, careful consideration must be given to how current therapies affect IEL function in the context of both active disease and remission. While effective against the dysregulated immune response associated with IBD, long-term immunosuppressive therapy presents its own risks including increased susceptibility to infection and impaired detection of cancerous cells. Ultimately, improved understanding of the mechanisms regulating IEL activation and motility may provide us with useful tools to modulate natural IEL surveillance and regulatory function as a means to reinforce the mucosal barrier. As an alternative to generalized immune suppression, novel approaches to amplify the frontline of defense in conjunction with targeted anti-inflammatory biologic therapies may prevent disease relapse while reducing the unwanted side effects of many of today’s current treatments.
Conclusion
Although IELs are often underappreciated by epithelial biologists and mucosal immunologists alike, these unique cells integrate tightly regulated signals from the microbiota, intestinal epithelium, and lamina propria to provide an immediate response to pathogens while also promoting mucosal tolerance. From studies in mice, it is clear that the same microbial- and diet-driven host immune interactions that are associated with the development of IBD directly influence IEL development and function. Previous studies in the field have been hindered by technical challenges in evaluating IEL function; however, recent methodological innovations, including intravital microscopy [62••, 100••, 104], ex vivo IEL culture [119••], and IEL co-culture with 3D intestinal stem cell cultures (enteroids) [121•, 122], are increasing the feasibility of IEL studies in both mice and men. As our understanding of IEL biology and interactions with the mucosal microenvironment continues to evolve, these studies will provide insight into the contribution of IELs to mucosal homeostasis and may lead to novel therapeutic approaches for the treatment of IBD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24. https://doi.org/10.1038/nature11582.
Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140(6):1729–37. https://doi.org/10.1053/j.gastro.2011.02.012.
Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14(1):20–32. https://doi.org/10.1038/nrmicro3552.
Basson A, Trotter A, Rodriguez-Palacios A, Cominelli F. Mucosal interactions between genetics, diet, and microbiome in inflammatory bowel disease. Front Immunol. 2016;7:290. https://doi.org/10.3389/fimmu.2016.00290.
• Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14(5):269–78. https://doi.org/10.1038/nrgastro.2016.208. A recent review of T cell signaling pathways currently being targeted for IBD therapeutic development.
Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. 2017;152(2):327–39 e4. https://doi.org/10.1053/j.gastro.2016.10.012.
Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341(8858):1437–9.
D'Inca R, Di Leo V, Corrao G, Martines D, D'Odorico A, Mestriner C, et al. Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am J Gastroenterol. 1999;94(10):2956–60.
May GR, Sutherland LR, Meddings JB. Is small intestinal permeability really increased in relatives of patients with Crohn’s disease? Gastroenterology. 1993;104(6):1627–32.
Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136(2):551–63. https://doi.org/10.1053/j.gastro.2008.10.081.
• Edelblum KL, Sharon G, Singh G, Odenwald MA, Sailer A, Cao S, et al. The microbiome activates CD4 T-cell-mediated immunity to compensate for increased intestinal permeability. Cell Mol Gastroenterol Hepatol. 2017;4(2):285–97. https://doi.org/10.1016/j.jcmgh.2017.06.001. This study is the first demonstration that the microbiota stimulates protective immunity against acute bacterial invasion in response to tight junction-mediated increases in intestinal permeability.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23. https://doi.org/10.1038/nri2515.
Cheroutre H. Starting at the beginning: new perspectives on the biology of mucosal T cells. Annu Rev Immunol. 2004;22:217–46. https://doi.org/10.1146/annurev.immunol.22.012703.104522.
Leishman AJ, Gapin L, Capone M, Palmer E, MacDonald HR, Kronenberg M, et al. Precursors of functional MHC class I- or class II-restricted CD8alphaalpha(+) T cells are positively selected in the thymus by agonist self-peptides. Immunity. 2002;16(3):355–64.
Gangadharan D, Lambolez F, Attinger A, Wang-Zhu Y, Sullivan BA, Cheroutre H. Identification of pre- and postselection TCRalphabeta+ intraepithelial lymphocyte precursors in the thymus. Immunity. 2006;25(4):631–41. https://doi.org/10.1016/j.immuni.2006.08.018.
Suzuki K, Oida T, Hamada H, Hitotsumatsu O, Watanabe M, Hibi T, et al. Gut cryptopatches: direct evidence of extrathymic anatomical sites for intestinal T lymphopoiesis. Immunity. 2000;13(5):691–702.
Oida T, Suzuki K, Nanno M, Kanamori Y, Saito H, Kubota E, et al. Role of gut cryptopatches in early extrathymic maturation of intestinal intraepithelial T cells. J Immunol. 2000;164(7):3616–26.
Nonaka S, Naito T, Chen H, Yamamoto M, Moro K, Kiyono H, et al. Intestinal gamma delta T cells develop in mice lacking thymus, all lymph nodes, Peyer's patches, and isolated lymphoid follicles. J Immunol. 2005;174(4):1906–12.
Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol. 2011;11(7):445–56. https://doi.org/10.1038/nri3007.
Chennupati V, Worbs T, Liu X, Malinarich FH, Schmitz S, Haas JD, et al. Intra- and intercompartmental movement of gammadelta T cells: intestinal intraepithelial and peripheral gammadelta T cells represent exclusive nonoverlapping populations with distinct migration characteristics. J Immunol. 2010;185(9):5160–8. https://doi.org/10.4049/jimmunol.1001652.
Sugahara S, Shimizu T, Yoshida Y, Aiba T, Yamagiwa S, Asakura H, et al. Extrathymic derivation of gut lymphocytes in parabiotic mice. Immunology. 1999;96(1):57–65.
Camerini V, Panwala C, Kronenberg M. Regional specialization of the mucosal immune system. Intraepithelial lymphocytes of the large intestine have a different phenotype and function than those of the small intestine. J Immunol. 1993;151(4):1765–76.
Bonneville M, Itohara S, Krecko EG, Mombaerts P, Ishida I, Katsuki M, et al. Transgenic mice demonstrate that epithelial homing of gamma/delta T cells is determined by cell lineages independent of T cell receptor specificity. J Exp Med. 1990;171(4):1015–26.
Jarry A, Cerf-Bensussan N, Brousse N, Selz F, Guy-Grand D. Subsets of CD3+ (T cell receptor alpha/beta or gamma/delta) and CD3- lymphocytes isolated from normal human gut epithelium display phenotypical features different from their counterparts in peripheral blood. Eur J Immunol. 1990;20(5):1097–103. https://doi.org/10.1002/eji.1830200523.
Lundqvist C, Baranov V, Hammarstrom S, Athlin L, Hammarstrom ML. Intra-epithelial lymphocytes. Evidence for regional specialization and extrathymic T cell maturation in the human gut epithelium. Int Immunol. 1995;7(9):1473–87.
Suzuki H. Differences in intraepithelial lymphocytes in the proximal, middle, distal parts of small intestine, cecum, and colon of mice. Immunol Investig. 2009;38(8):780–96. https://doi.org/10.3109/08820130903258800.
Hirata I, Berrebi G, Austin LL, Keren DF, Dobbins WO III. Immunohistological characterization of intraepithelial and lamina propria lymphocytes in control ileum and colon and in inflammatory bowel disease. Dig Dis Sci. 1986;31(6):593–603.
Cerf-Bensussan N, Brousse N, Jarry A, Goulet O, Revillon Y, Ricour C, et al. Role of in vivo activated T cells in the mechanisms of villous atrophy in humans: study of allograft rejection. Digestion. 1990;46(Suppl 2):297–301.
Guy-Grand D, Cuenod-Jabri B, Malassis-Seris M, Selz F, Vassalli P. Complexity of the mouse gut T cell immune system: identification of two distinct natural killer T cell intraepithelial lineages. Eur J Immunol. 1996;26(9):2248–56. https://doi.org/10.1002/eji.1830260942.
Das H, Groh V, Kuijl C, Sugita M, Morita CT, Spies T, et al. MICA engagement by human Vgamma2Vdelta2 T cells enhances their antigen-dependent effector function. Immunity. 2001;15(1):83–93.
Ebert EC. IL-15 converts human intestinal intraepithelial lymphocytes to CD94 producers of IFN-gamma and IL-10, the latter promoting Fas ligand-mediated cytotoxicity. Immunology. 2005;115(1):118–26. https://doi.org/10.1111/j.1365-2567.2005.02132.x.
Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD, et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. 2000;191(3):435–44.
Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21(3):357–66. https://doi.org/10.1016/j.immuni.2004.06.020.
Setty M, Discepolo V, Abadie V, Kamhawi S, Mayassi T, Kent A, et al. Distinct and synergistic contributions of epithelial stress and adaptive immunity to functions of intraepithelial killer cells and active celiac disease. Gastroenterology. 2015;149(3):681–691 e10. https://doi.org/10.1053/j.gastro.2015.05.013.
Poussier P, Ning T, Banerjee D, Julius M. A unique subset of self-specific intraintestinal T cells maintains gut integrity. J Exp Med. 2002;195(11):1491–7.
Das G, Augustine MM, Das J, Bottomly K, Ray P, Ray A. An important regulatory role for CD4+CD8 alpha alpha T cells in the intestinal epithelial layer in the prevention of inflammatory bowel disease. Proc Natl Acad Sci U S A. 2003;100(9):5324–9. https://doi.org/10.1073/pnas.0831037100.
Kuhl AA, Pawlowski NN, Grollich K, Loddenkemper C, Zeitz M, Hoffmann JC. Aggravation of intestinal inflammation by depletion/deficiency of gammadelta T cells in different types of IBD animal models. J Leukoc Biol. 2007;81(1):168–75. https://doi.org/10.1189/jlb.1105696.
Kontoyiannis D, Boulougouris G, Manoloukos M, Armaka M, Apostolaki M, Pizarro T, et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J Exp Med. 2002;196(12):1563–74.
Chang JS, Ocvirk S, Berger E, Kisling S, Binder U, Skerra A, et al. Endoplasmic reticulum stress response promotes cytotoxic phenotype of CD8alphabeta+ intraepithelial lymphocytes in a mouse model for Crohn’s disease-like ileitis. J Immunol. 2012;189(3):1510–20. https://doi.org/10.4049/jimmunol.1200166.
Kawaguchi-Miyashita M, Shimada S, Kurosu H, Kato-Nagaoka N, Matsuoka Y, Ohwaki M, et al. An accessory role of TCRgammadelta (+) cells in the exacerbation of inflammatory bowel disease in TCRalpha mutant mice. Eur J Immunol. 2001;31(4):980–8.
Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A. 2002;99(22):14338–43. https://doi.org/10.1073/pnas.212290499.
Hoffmann JC, Peters K, Henschke S, Herrmann B, Pfister K, Westermann J, et al. Role of T lymphocytes in rat 2,4,6-trinitrobenzene sulphonic acid (TNBS) induced colitis: increased mortality after gammadelta T cell depletion and no effect of alphabeta T cell depletion. Gut. 2001;48(4):489–95.
Inagaki-Ohara K, Chinen T, Matsuzaki G, Sasaki A, Sakamoto Y, Hiromatsu K, et al. Mucosal T cells bearing TCRgammadelta play a protective role in intestinal inflammation. J Immunol. 2004;173(2):1390–8.
Nancey S, Holvoet S, Graber I, Joubert G, Philippe D, Martin S, et al. CD8+ cytotoxic T cells induce relapsing colitis in normal mice. Gastroenterology. 2006;131(2):485–96. https://doi.org/10.1053/j.gastro.2006.05.018.
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–98.
Apostolaki M, Manoloukos M, Roulis M, Wurbel MA, Muller W, Papadakis KA, et al. Role of beta7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF-dependent Crohn's disease. Gastroenterology. 2008;134(7):2025–35. https://doi.org/10.1053/j.gastro.2008.02.085.
Kaser A, Martinez-Naves E, Blumberg RS. Endoplasmic reticulum stress: implications for inflammatory bowel disease pathogenesis. Curr Opin Gastroenterol. 2010;26(4):318–26. https://doi.org/10.1097/MOG.0b013e32833a9ff1.
Jabri B, Ebert E. Human CD8+ intraepithelial lymphocytes: a unique model to study the regulation of effector cytotoxic T lymphocytes in tissue. Immunol Rev. 2007;215:202–14. https://doi.org/10.1111/j.1600-065X.2006.00481.x.
Cheroutre H. IELs: enforcing law and order in the court of the intestinal epithelium. Immunol Rev. 2005;206:114–31. https://doi.org/10.1111/j.0105-2896.2005.00284.x.
Swamy M, Jamora C, Havran W, Hayday A. Epithelial decision makers: in search of the ‘epimmunome’. Nat Immunol. 2010;11(8):656–65. https://doi.org/10.1038/ni.1905.
Fuchs A, Colonna M. Innate lymphoid cells in homeostasis, infection, chronic inflammation and tumors of the gastrointestinal tract. Curr Opin Gastroenterol. 2013;29(6):581–7. https://doi.org/10.1097/MOG.0b013e328365d339.
Liu Z, Yadav PK, Xu X, Su J, Chen C, Tang M, et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. 2011;89(4):597–606. https://doi.org/10.1189/jlb.0810456.
Cheroutre H, Lambolez F. The thymus chapter in the life of gut-specific intra epithelial lymphocytes. Curr Opin Immunol. 2008;20(2):185–91. https://doi.org/10.1016/j.coi.2008.03.009.
Cawthon AG, Lu H, Alexander-Miller MA. Peptide requirement for CTL activation reflects the sensitivity to CD3 engagement: correlation with CD8alphabeta versus CD8alphaalpha expression. J Immunol. 2001;167(5):2577–84.
Sydora BC, Mixter PF, Holcombe HR, Eghtesady P, Williams K, Amaral MC, et al. Intestinal intraepithelial lymphocytes are activated and cytolytic but do not proliferate as well as other T cells in response to mitogenic signals. J Immunol. 1993;150(6):2179–91.
Leishman AJ, Naidenko OV, Attinger A, Koning F, Lena CJ, Xiong Y, et al. T cell responses modulated through interaction between CD8alphaalpha and the nonclassical MHC class I molecule, TL. Science. 2001;294(5548):1936–9. https://doi.org/10.1126/science.1063564.
Teitell M, Mescher MF, Olson CA, Littman DR, Kronenberg M. The thymus leukemia antigen binds human and mouse CD8. J Exp Med. 1991;174(5):1131–8.
Olivares-Villagomez D, Mendez-Fernandez YV, Parekh VV, Lalani S, Vincent TL, Cheroutre H, et al. Thymus leukemia antigen controls intraepithelial lymphocyte function and inflammatory bowel disease. Proc Natl Acad Sci U S A. 2008;105(46):17931–6. https://doi.org/10.1073/pnas.0808242105.
Hoffmann JC, Pawlowski NN, Grollich K, Loddenkemper C, Zeitz M, Kuhl AA. Gammadelta T lymphocytes: a new type of regulatory T cells suppressing murine 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced colitis. Int J Color Dis. 2008;23(10):909–20. https://doi.org/10.1007/s00384-008-0535-8.
Shires J, Theodoridis E, Hayday AC. Biological insights into TCRgammadelta+ and TCRalphabeta+ intraepithelial lymphocytes provided by serial analysis of gene expression (SAGE). Immunity. 2001;15(3):419–34.
Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol. 2013;14(3):281–9. https://doi.org/10.1038/ni.2523.
•• Sujino T, London M, Hoytema van Konijnenburg DP, Rendon T, Buch T, Silva HM, et al. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science. 2016;352(6293):1581–6. https://doi.org/10.1126/science.aaf3892. This report is the first to demonstrate the conversion of lamina propria Foxp3 + Tregs into Foxp3 - CD4 + CD8 + IELs.
• Sarrabayrouse G, Bossard C, Chauvin JM, Jarry A, Meurette G, Quevrain E, et al. CD4CD8alphaalpha lymphocytes, a novel human regulatory T cell subset induced by colonic bacteria and deficient in patients with inflammatory bowel disease. PLoS Biol. 2014;12(4):e1001833. https://doi.org/10.1371/journal.pbio.1001833. This report identifies a protective role for CD4 + CD8 + T cells in IBD patients.
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331(6015):337–41. https://doi.org/10.1126/science.1198469.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98. https://doi.org/10.1016/j.cell.2009.09.033.
Umesaki Y, Setoyama H, Matsumoto S, Imaoka A, Itoh K. Differential roles of segmented filamentous bacteria and clostridia in development of the intestinal immune system. Infect Immun. 1999;67(7):3504–11.
• Tada A, Zelaya H, Clua P, Salva S, Alvarez S, Kitazawa H, et al. Immunobiotic Lactobacillus strains reduce small intestinal injury induced by intraepithelial lymphocytes after Toll-like receptor 3 activation. Inflamm Res. 2016;65(10):771–83. https://doi.org/10.1007/s00011-016-0957-7. This study demonstrates that commensal Lactobacillus reduces the licensing of IEL cytolytic activity in response to TLR3 activation.
Roselli M, Finamore A, Nuccitelli S, Carnevali P, Brigidi P, Vitali B, et al. Prevention of TNBS-induced colitis by different Lactobacillus and Bifidobacterium strains is associated with an expansion of gammadeltaT and regulatory T cells of intestinal intraepithelial lymphocytes. Inflamm Bowel Dis. 2009;15(10):1526–36. https://doi.org/10.1002/ibd.20961.
•• Cervantes-Barragan L, Chai JN, Tianero MD, DiLuccia B, Ahern PP, Merriman J et al. Lactobacillus reuteri induces gut intraepithelial CD4+CD8alphaalpha+ T cells. Science. 2017. https://doi.org/10.1126/science.aah5825. This report shows that colonization or fecal transfer of L. reuteri can induce the differentiation of CD4+ CD8+ IELs through an AhR-dependent mechanism.
Kawaguchi M, Nanno M, Umesaki Y, Matsumoto S, Okada Y, Cai Z, et al. Cytolytic activity of intestinal intraepithelial lymphocytes in germ-free mice is strain dependent and determined by T cells expressing gamma delta T-cell antigen receptors. Proc Natl Acad Sci U S A. 1993;90(18):8591–4.
Ismail AS, Severson KM, Vaishnava S, Behrendt CL, Yu X, Benjamin JL, et al. Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci U S A. 2011;108(21):8743–8. https://doi.org/10.1073/pnas.1019574108.
Jiang W, Wang X, Zeng B, Liu L, Tardivel A, Wei H, et al. Recognition of gut microbiota by NOD2 is essential for the homeostasis of intestinal intraepithelial lymphocytes. J Exp Med. 2013;210(11):2465–76. https://doi.org/10.1084/jem.20122490.
Qiu Y, Pu A, Zheng H, Liu M, Chen W, Wang W, et al. TLR2-dependent signaling for IL-15 production is essential for the homeostasis of intestinal intraepithelial lymphocytes. Mediat Inflamm. 2016;2016:4281865. https://doi.org/10.1155/2016/4281865.
Kaneko M, Mizunuma T, Takimoto H, Kumazawa Y. Development of TCR alpha beta CD8 alpha alpha intestinal intraepithelial lymphocytes is promoted by interleukin-15-producing epithelial cells constitutively stimulated by gram-negative bacteria via TLR4. Biol Pharm Bull. 2004;27(6):883–9.
Yu Q, Tang C, Xun S, Yajima T, Takeda K, Yoshikai Y. MyD88-dependent signaling for IL-15 production plays an important role in maintenance of CD8 alpha alpha TCR alpha beta and TCR gamma delta intestinal intraepithelial lymphocytes. J Immunol. 2006;176(10):6180–5.
•• Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. 2014;41(2):311–24. https://doi.org/10.1016/j.immuni.2014.06.015. This is the first report linking pro-inflammatory changes in IEL effector function to an expansion of a specific commensal B. vulgatus as a result of Nod2 deficiency.
•• Kober OI, Ahl D, Pin C, Holm L, Carding SR, Juge N. Gammadelta T-cell-deficient mice show alterations in mucin expression, glycosylation, and goblet cells but maintain an intact mucus layer. Am J Physiol Gastrointest Liver Physiol. 2014;306(7):G582–93. https://doi.org/10.1152/ajpgi.00218.2013. This report demonstrates that γδ T cell-deficiency adversely affects goblet cell number and mucus production.
Sheng YH, Hasnain SZ, Florin TH, McGuckin MA. Mucins in inflammatory bowel diseases and colorectal cancer. J Gastroenterol Hepatol. 2012;27(1):28–38. https://doi.org/10.1111/j.1440-1746.2011.06909.x.
Fukata M, Arditi M. The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol. 2013;6(3):451–63. https://doi.org/10.1038/mi.2013.13.
Lee J, Geddes K, Streutker C, Philpott DJ, Girardin SE. Role of mouse peptidoglycan recognition protein PGLYRP2 in the innate immune response to Salmonella enterica serovar Typhimurium infection in vivo. Infect Immun. 2012;80(8):2645–54. https://doi.org/10.1128/IAI.00168-12.
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54 e42; quiz e30. https://doi.org/10.1053/j.gastro.2011.10.001.
• Lewis JD, Abreu MT. Diet as a trigger or therapy for inflammatory bowel diseases. Gastroenterology. 2017;152(2):398–414 e6. https://doi.org/10.1053/j.gastro.2016.10.019. This recent review discusses the impact of diet and bacterial metabolism on IBD development and therapies.
Menezes JS, Mucida DS, Cara DC, Alvarez-Leite JI, Russo M, Vaz NM, et al. Stimulation by food proteins plays a critical role in the maturation of the immune system. Int Immunol. 2003;15(3):447–55.
Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147(3):629–40. https://doi.org/10.1016/j.cell.2011.09.025.
Franco Robles E, Pérez Vázquez V, Ramírez Emiliano J, González Amaro R, López BS. High fat diet induces alterations to intraepithelial lymphocyte and cytokine mRNA in the small intestine of C57BL/6 mice. Royal Society of Chemistry. 2017;7(9):5322–30. https://doi.org/10.1039/C6RA24689C.
Ma X, Torbenson M, Hamad AR, Soloski MJ, Li Z. High-fat diet modulates non-CD1d-restricted natural killer T cells and regulatory T cells in mouse colon and exacerbates experimental colitis. Clin Exp Immunol. 2008;151(1):130–8. https://doi.org/10.1111/j.1365-2249.2007.03530.x.
Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–38. https://doi.org/10.1016/j.immuni.2004.08.011.
Iwata M. Retinoic acid production by intestinal dendritic cells and its role in T-cell trafficking. Semin Immunol. 2009;21(1):8–13. https://doi.org/10.1016/j.smim.2008.09.002.
Lee H, Ko G. Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome. Sci Rep. 2016;6:25835. https://doi.org/10.1038/srep25835.
Simmons JD, Mullighan C, Welsh KI, Jewell DP. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut. 2000;47(2):211–4.
Dresner-Pollak R, Ackerman Z, Eliakim R, Karban A, Chowers Y, Fidder HH. The BsmI vitamin D receptor gene polymorphism is associated with ulcerative colitis in Jewish Ashkenazi patients. Genet Test. 2004;8(4):417–20. https://doi.org/10.1089/gte.2004.8.417.
Cantorna MT. Vitamin D and autoimmunity: is vitamin D status an environmental factor affecting autoimmune disease prevalence? Proc Soc Exp Biol Med. 2000;223(3):230–3.
Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes. 2002;51(5):1367–74.
Yu S, Bruce D, Froicu M, Weaver V, Cantorna MT. Failure of T cell homing, reduced CD4/CD8alphaalpha intraepithelial lymphocytes, and inflammation in the gut of vitamin D receptor KO mice. Proc Natl Acad Sci U S A. 2008;105(52):20834–9. https://doi.org/10.1073/pnas.0808700106.
Amre DK, D'Souza S, Morgan K, Seidman G, Lambrette P, Grimard G, et al. Imbalances in dietary consumption of fatty acids, vegetables, and fruits are associated with risk for Crohn’s disease in children. Am J Gastroenterol. 2007;102(9):2016–25. https://doi.org/10.1111/j.1572-0241.2007.01411.x.
Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13(2):88–100. https://doi.org/10.1038/nri3384.
Adams EJ, Gu S, Luoma AM. Human gamma delta T cells: evolution and ligand recognition. Cell Immunol. 2015;296(1):31–40. https://doi.org/10.1016/j.cellimm.2015.04.008.
Tsuchiya T, Fukuda S, Hamada H, Nakamura A, Kohama Y, Ishikawa H, et al. Role of gamma delta T cells in the inflammatory response of experimental colitis mice. J Immunol. 2003;171(10):5507–13.
Ismail AS, Behrendt CL, Hooper LV. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J Immunol. 2009;182(5):3047–54. https://doi.org/10.4049/jimmunol.0802705.
•• Edelblum KL, Singh G, Odenwald MA, Lingaraju A, El Bissati K, McLeod R, et al. Gammadelta intraepithelial lymphocyte migration limits transepithelial pathogen invasion and systemic disease in mice. Gastroenterology. 2015;148(7):1417–26. https://doi.org/10.1053/j.gastro.2015.02.053. This is the first report demonstrating that γδ IEL motility and surveillance of the epithelium is required for protection against acute microbial invasion.
Kabelitz D, Lettau M, Janssen O. Immunosurveillance by human gammadelta T lymphocytes: the emerging role of butyrophilins. F1000Res. 2017;6. https://doi.org/10.12688/f1000research.11057.1.
Walker CR, Hautefort I, Dalton JE, Overweg K, Egan CE, Bongaerts RJ, et al. Intestinal intraepithelial lymphocyte-enterocyte crosstalk regulates production of bactericidal angiogenin 4 by Paneth cells upon microbial challenge. PLoS One. 2013;8(12):e84553. https://doi.org/10.1371/journal.pone.0084553.
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–8. https://doi.org/10.1126/science.1209791.
Edelblum KL, Shen L, Weber CR, Marchiando AM, Clay BS, Wang Y, et al. Dynamic migration of gammadelta intraepithelial lymphocytes requires occludin. Proc Natl Acad Sci U S A. 2012;109(18):7097–102. https://doi.org/10.1073/pnas.1112519109.
Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR II, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189(1):111–26. https://doi.org/10.1083/jcb.200902153.
Boismenu R, Havran WL. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science. 1994;266(5188):1253–5.
• Meehan TF, Witherden DA, Kim CH, Sendaydiego K, Ye I, Garijo O, et al. Protection against colitis by CD100-dependent modulation of intraepithelial gammadelta T lymphocyte function. Mucosal Immunol. 2014;7(1):134–42. https://doi.org/10.1038/mi.2013.32. This is the first demonstration that a direct ligand/receptor interaction between γδ IELs and epithelial cells is required to promote epithelial regeneration following DSS-induced injury.
Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75(2):274–82.
Mizoguchi A, Mizoguchi E, Chiba C, Spiekermann GM, Tonegawa S, Nagler-Anderson C, et al. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J Exp Med. 1996;183(3):847–56.
Takahashi I, Iijima H, Katashima R, Itakura M, Kiyono H. Clonal expansion of CD4+ TCRbetabeta+ T cells in TCR alpha-chain- deficient mice by gut-derived antigens. J Immunol. 1999;162(3):1843–50.
Do JS, Fink PJ, Li L, Spolski R, Robinson J, Leonard WJ, et al. Cutting edge: spontaneous development of IL-17-producing gamma delta T cells in the thymus occurs via a TGF-beta 1-dependent mechanism. J Immunol. 2011;184(4):1675–9. https://doi.org/10.4049/jimmunol.0903539.
•• Do JS, Kim S, Keslar K, Jang E, Huang E, Fairchild RL, et al. Gammadelta T cells coexpressing gut homing alpha4beta7 and alphaE integrins define a novel subset promoting intestinal inflammation. J Immunol. 2017;198(2):908–15. https://doi.org/10.4049/jimmunol.1601060. This study shows that a subset of inflammatory γδ T cells within the lamina propria and mesenteric lymph node promotes chronic intestinal inflammation.
• Catalan-Serra I, Sandvik AK, Bruland T, Andreu-Ballester JC. Gammadelta T cells in Crohn’s disease: a new player in the disease pathogenesis? J Crohns Colitis. 2017; https://doi.org/10.1093/ecco-jcc/jjx039. This review provides a detailed overview of the current knowledge regarding the role of γδ T cells in the pathogenesis of Crohn’s disease and the potential of these cells as a therapeutic target for immunotherapy.
Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood. 2012;120(11):2269–79. https://doi.org/10.1182/blood-2012-05-430470.
McCarthy NE, Bashir Z, Vossenkamper A, Hedin CR, Giles EM, Bhattacharjee S, et al. Proinflammatory Vdelta2+ T cells populate the human intestinal mucosa and enhance IFN-gamma production by colonic alphabeta T cells. J Immunol. 2013;191(5):2752–63. https://doi.org/10.4049/jimmunol.1202959.
•• Tyler CJ, McCarthy NE, Lindsay JO, Stagg AJ, Moser B, Eberl M. Antigen-presenting human gammadelta T cells promote intestinal CD4+ T cell expression of IL-22 and mucosal release of calprotectin. J Immunol. 2017;198(9):3417–25. https://doi.org/10.4049/jimmunol.1700003. This report demonstrates that Vγ9/Vδ2 T cells promote local barrier defense through the production of antimicrobial proteins.
McCarthy NE, Hedin CR, Sanders TJ, Amon P, Hoti I, Ayada I, et al. Azathioprine therapy selectively ablates human Vdelta2(+) T cells in Crohn’s disease. J Clin Invest. 2015;125(8):3215–25. https://doi.org/10.1172/JCI80840.
Deusch K, Luling F, Reich K, Classen M, Wagner H, Pfeffer K. A major fraction of human intraepithelial lymphocytes simultaneously expresses the gamma/delta T cell receptor, the CD8 accessory molecule and preferentially uses the V delta 1 gene segment. Eur J Immunol. 1991;21(4):1053–9. https://doi.org/10.1002/eji.1830210429.
•• Di Marco BR, Roberts NA, Dart RJ, Vantourout P, Jandke A, Nussbaumer O, et al. Epithelia use butyrophilin-like molecules to shape organ-specific gammadelta T cell compartments. Cell. 2016;167(1):203–18 e17. https://doi.org/10.1016/j.cell.2016.08.030. This is the first demonstration that epithelial butyrophilin expression shapes local intestinal γδ T cell development and function.
•• Lebrero-Fernandez C, Wenzel UA, Akeus P, Wang Y, Strid H, Simren M, et al. Altered expression of Butyrophilin (BTN) and BTN-like (BTNL) genes in intestinal inflammation and colon cancer. Immun Inflamm Dis. 2016;4(2):191–200. https://doi.org/10.1002/iid3.105. This report provides the first indication that butryophilin gene expression is altered in ulcerative colitis.
• Rogoz A, Reis BS, Karssemeijer RA, Mucida D. A 3-D enteroid-based model to study T-cell and epithelial cell interaction. J Immunol Methods. 2015;421:89–95. https://doi.org/10.1016/j.jim.2015.03.014. This is the first report demonstrating IEL migration in a novel murine IEL/enteroid co-culture model.
Nozaki K, Mochizuki W, Matsumoto Y, Matsumoto T, Fukuda M, Mizutani T, et al. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J Gastroenterol. 2016;51(3):206–13. https://doi.org/10.1007/s00535-016-1170-8.
Acknowledgements
The authors would like to thank Dr. Tessa Bergsbaken for her critical review and thoughtful suggestions regarding the manuscript.
Funding
This work is supported by funding from the National Institutes of Health K01 DK093627, R03 DK106484 and the Feldstein Medical Foundation (KLE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immunology and Inflammation
Rights and permissions
About this article

Cite this article
Hu, M.D., Edelblum, K.L. Sentinels at the Frontline: the Role of Intraepithelial Lymphocytes in Inflammatory Bowel Disease. Curr Pharmacol Rep 3, 321–334 (2017). https://doi.org/10.1007/s40495-017-0105-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-017-0105-2