
Abstract
Purpose of Review
Hepatic ischemia-reperfusion injury (IRI), an inevitable event during liver transplantation, represents a major risk factor for the primary graft dysfunction as well as the development of acute and chronic rejection. Neutrophils, along macrophages, are pivotal in the innate immune-driven liver IRI, whereas the effective neutrophil-targeting therapies remain to be established. In this review, we summarize progress in our appreciation of the neutrophil biology and discuss neutrophil-based therapeutic perspectives.
Recent Findings
New technological advances enable to accurately track neutrophil movements and help to understand molecular mechanisms in neutrophil function, such as selective recruitment to IR-stressed tissue, formation of neutrophil extracellular traps, or reverse migration into circulation. In addition to pro-inflammatory and tissue-destructive functions, immune regulatory and tissue-repairing phenotype associated with distinct neutrophil subsets have been identified.
Summary
Newly recognized and therapeutically attractive neutrophil characteristics warrant comprehensive preclinical and clinical attention to target IRI in transplant recipients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Liver transplantation (LT) has become the standard of care for patients with end-stage liver disease and those with liver malignancies [1]. Hepatic ischemia-reperfusion injury (IRI), an inevitable event during LT, represents a major risk factor for the primary graft dysfunction as well as the development of acute and chronic rejection [2, 3]. Hence, minimizing IRI is important not only for improving clinical outcomes but also for successful use of marginal liver grafts and expansion of donor organ pool available for transplantation. Despite its obvious clinical importance, however, the mechanisms accounting for liver IRI are only partially understood and effective preventive or therapeutic strategies remain to be established.
In the initial stage of liver IRI, ischemic insult renders hepatic cells sensitive to glycogen consumption, oxygen deprivation, pH changes, and adenosine triphosphate (ATP) depletion [4]. These are followed by enhanced production of reactive oxygen species (ROS), higher intracellular calcium concentration, and organelle damage, leading to the initial parenchymal cell death [5]. The reperfusion itself disturbs liver metabolism and evokes inflammatory cascades leading to aggravated hepatocellular damage. Innate immune activation plays a central role in this inflammatory response via cytotoxic mechanisms and dynamic cross-talk with adaptive immune cell repertoires, ultimately converting immunologically quiescent hepatic milieu into an inflammatory organ [6, 7].
In unstressed human liver, Kupffer cells (liver-resident macrophages) account for about 15% of total hepatocellular population and 80–90% of whole body macrophages [8]. In IRI–LT pathophysiology, both Kupffer cells (donor-origin) and liver-infiltrating bone marrow-derived macrophages (recipient-origin) play dominant roles in priming innate immune responses [9,10,11], with the majority of studies focusing on macrophage regulation [12, 13]. On the other hand, neutrophils are dominant immune cells in the steady-state blood circulation (50–70% in human, 10–25% in mouse [14, 15]), constantly patrolling and serving as the first line of defense against invading pathogens [16]. Likewise, in IR-stressed blood-perfused liver, neutrophils are recruited to the injury site, contributing to sterile inflammation and enhancing the hepatocellular damage. Indeed, despite being long considered as “non-specialized” innate effector cells, neutrophil infiltration into hepatic sinusoidal lumen is now considered as one of reliable biomarkers of liver IRI [17, 18].
In this review, we first summarize progress in our appreciation of the neutrophil biology, and then discuss therapeutic prospects of their targeting for the treatment of inflammatory states, such as IRI in LT recipients.
Neutrophil Activation and Migration
Neutrophils, the largest circulating fraction of leukocytes, are continuously generated from myeloid precursors in the bone marrow in a process of “granulopoiesis” (daily production reaches up to 2 × 1011 cells) [16, 19]. The crucial signals for neutrophil activation are provided by danger-associated molecular patterns (DAMPs), i.e., endogenous molecules constitutively expressed in nuclear, cytoplasm, and extracellular matrix under basal conditions. Although indispensable for homeostasis maintenance, once released in response to tissue injury, DAMPs are detected by and become critical triggers of the inflammatory cell activation. A growing number of DAMPs have been identified to date, such as ATP, histone, high mobility group box 1 (HMGB1), with complementary pattern recognition receptors (PRRs) crucial in cell recognition and signaling pathways (representative are listed in Table 1). The initial parenchymal cell damage results in the release of DAMPs from damaged/dead/moribund cells that stimulate local sentinel Kupffer cells and liver sinusoidal endothelial cells (LSECs). Kupffer cells sense neighboring cell death by receiving DAMPs signals and produce IL1β, which up-regulates intercellular adhesion molecule-1 (ICAM-1) on LSECs. Neutrophils are then recruited via integrin αMβ2 (Mac1)-dependent adhesion to endothelial ICAM-1. This neutrophil adhesion mechanism requires ATP-induced activation of P2X7 receptor and NOD-like receptor pyrin domain-containing-3 (NLRP3) inflammasome [20]. Indeed, mice depleted of Kupffer cells by clodronate showed reduced caspase, IL1, and neutrophil recruitment, whereas impaired P2X7R signaling was accompanied by equivalently impaired neutrophil accumulation in a liver thermal injury model [21]. Activated Kupffer cells, along with parenchymal cells, also release chemokines detected by G protein coupled receptors (e.g., CXCR2) on neutrophils and thereby recruit them to the site of inflammatory damage. In addition, LSECs themselves can sense DAMPs via TLR9 to release IL1β and IL18 [22] (Fig. 1a). Because of the unusual hepatic microvasculature, circulating neutrophils can move directly to integrin-mediated adhesion devoid of selectin-mediated rolling. In line with this scenario, Mac-1 or ICAM-1 neutralizing antibodies effectively alleviated liver IRI [23, 24].

Putative mechanisms of neutrophil recruitment in liver ischemia-reperfusion injury. a Liver-resident Kupffer cells and liver sinusoidal endothelial cells (LSECs) sense initial danger-associated molecular patterns (DAMPs) via pattern recognition receptors (PRRs) to trigger intercellular adhesion molecule-1 (ICAM-1) expression on endothelial cells and neutrophil adhesion by integrin αMβ2 (Mac-1) signaling. b Neutrophils move on the LSECs luminal side toward the damaged site, guided by Kupffer cell-derived chemokine (CXCL1/CXCL2) gradient. c Neutrophils detect mitochondrial N-formyl peptides (FMIT) via formyl peptide receptor 1 (FPR1) and migrate underneath LSECs toward the injury site. d Neutrophils express and secret matrix metalloproteinase 9 (MMP9) to degrade extracellular matrix (ECM) during migration
Histone and HMGB1 are among the most tested DAMPs in the model of liver IRI. The levels of circulating histones are significantly increased in liver IRI, and anti-histone neutralizing antibodies suppress TLR9/MyD88 signaling while decreasing inflammatory hepatic damage [25]. Treatment with HMGB1 neutralizing antibody alleviated IRI in WT but not TLR4-deficient mouse recipients [26]. Likewise, recombinant thrombomodulin (HMGB1 inactivator) mitigated liver IRI in WT but not TLR4-knockout mouse [27•]. Since broad spectrum of immune cells express TLRs, the HMGB1–TLR4 inflammatory axis is unlikely to be exclusive for neutrophil regulation. Indeed, hepatocyte-specific HMGB1 deficient mice showed decreased hepatic necrosis and neutrophil accumulation, whereas the number of their macrophages remained unchanged in acetaminophen-induced liver injury model [28]. In contrast, hepatocyte-specific HMGB1-deficient mice exhibited exaggerated liver IR-damage, implying HMGB1 is essential for hepatocyte resistance against hypoxia [29]. Clearly, extracellular DAMPs are promising therapeutic targets to manage liver IRI by alleviating innate, including neutrophil, cell responses.
Following adhesion to the endothelium, neutrophils are guided by Kupffer cell-derived chemokines (CXCL1, CXCL2), bind to CXCR2 on the luminal surface of LSECs, and form the chemotactic gradient at the injury site [20] (Fig. 1b). Consistent with such a mechanism, reparixin (an allosteric CXCR2 antagonist) decreased neutrophil infiltration and mitigated liver IRI [30]. In addition, CXCL1 blocking antibody alleviated hepatic infiltration in necrotic cell-induced neutrophil mobilization model [31], whereas in a carbon tetrachloride (CCl4)-induced acute liver injury, defective CXCL2 expression in TLR2-knockout or S100A9-knockout mice was accompanied by suppressed hepatic neutrophil recruitment [32]. In the following dynamic stage, neutrophils moving on the LSECs luminal side migrate underneath the sinusoidal wall to the damaged site. Mitochondrial N-formyl peptides (FMIT), cleavage products of mitochondrial proteins, are critically involved in this process serving as chemoattractant DAMPs. Damaged cells release FMIT and create a gradient that attracts neutrophils through neutrophil-specific formyl peptide receptor 1 (FPR1) [33]. Honda et al. reported that cyclosporine H (FPR1 antagonist) decreased the number and crawling velocity of neutrophils while alleviating liver IRI and inhibiting the accumulation of neutrophils into laser irradiation-induced necrotic areas [34•]. Interestingly, at this time point, neutrophils are attracted by two opposing chemoattractant gradients, CXCL2 mostly anchored on the luminal LSECs side and FMIT emanated from parenchymal damaged cells. The final signaling via FMIT-FPR1 overrides the CXCL1/CXCL2-CXCR2 axis, making neutrophils to preferentially migrate toward the FMIT while ignoring the CXCL2 attraction (Fig. 1c). Such a chemotactic hierarchy has been consistently observed in pathogen-triggered infection where bacterial formylated peptides are dominant to endogenous chemoattractant [35], as well as in sterile inflammation where neutrophils preferentially migrated toward injured cells rather than soluble chemokines [21]. Indeed, Marques et al. demonstrated that CXCR2 and FPR1 antagonists alleviated neutrophil migration in acetaminophen-induced liver damage, while simultaneous CXCR2 and FPR1 antagonism achieved maximum protection [36]. Hence, both CXCL2 and FMIT are critically involved in the hepatic neutrophil migration in a dynamic and sequential manner.
The extracellular matrix (ECM), a network of proteins and sugars in all solid organs, is the essential scaffold and structural cell support as well as tissue regulator of cell adhesion, migration, differentiation, proliferation, and survival. Once neutrophils migrate across LSEC, they move toward damaged site while simultaneously degrading ECM. This process is dominated by matrix metalloproteinases (MMPs), zinc-dependent endopeptidases involved in the breakdown of ECM, and tissue inhibitor of metalloproteinases (TIMPs), which proteolytically regulate MMPs activity. MMP2 and MMP9 are the most prominent in the pathophysiology of liver IRI [37]. In liver IRI, MMP9 is predominantly expressed by Ly6G-positive neutrophils, while neutrophils are the only cells in the body which can release MMP9 free of its endogenous inhibitor TIMPs, and therefore are uniquely capable of delivering highly active MMP9 to digest its fibronectin and collagen type IV substrates [38]. Fibronectin, a major ECM component of the subendothelial space of Disse in normal liver, interacts with integrin receptor α4β1 on activated neutrophils to induce MMP9 [39] (Fig. 1d). Hamada et al. demonstrated that MMP9 deficient neutrophils were impaired in their ability to migrate across fibronectin in a transwell culture system, whereas genetic disruption or exogenous blocking with MMP9 antibody decreased neutrophil infiltration and cytoprotection in a mouse hepatic IRI model [40]. Meantime, IR-insult markedly increases TIMP1 expression, an endogenous MMP9 inhibitor. Indeed, TIMP1 deficient mice had markedly increased MMP9 activity and massive neutrophil infiltration in their IR-stressed livers, which has led to 60% death. In contrast, virus-mediated gene transfer of TIMP1 increased TIMP1 expression primarily on parenchymal cells, suppressed gelatinolytic activity of neutrophil MMP9, attenuated neutrophil infiltration, and alleviated the hepatocellular damage [41, 42]. Furthermore, LT patients experiencing acute rejection had elevated serum MMP9 activity at 1 week, implying its role as a potential biomarker in clinical LT [43].
Neutrophil-Associated Tissue Injury
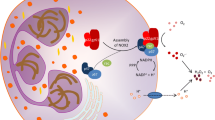
Neutrophils recruited or activated in liver sinusoids do not cause hepatic damage per se, as they can exert cytotoxicity only after migrating across the endothelium and in close proximity to the parenchymal cells [17]. Tissue damage is mediated primarily by ROS and proteases, with Kupffer cells being principal ROS producers early after reperfusion, and neutrophil oxidative burst becoming the main source of ROS in the later IRI phase. Indeed, by 6 h of reperfusion (the peak of hepatocellular damage in a murine IRI–LT model), infiltrating Ly6G-positive neutrophils elaborated large amounts of ROS metabolite (4-hydroxynonenal) [11]. Neutrophils generate ROS through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which consists of a Rho GTPase, two membrane-bound phox (phagocytic oxidase) proteins (p22phox and gp91phox), and three cytosolic phox proteins (p40phox, p47phox, and p67phox). Patients encoded with hypofunctional mutations in gp91phox or p47phox were protected from transiently induced upper limb IRI [44], while apocynin (gp91phox inhibitor) suppressed ROS metabolite (8-hydroxy-2-deoxyguanosine) and alleviated liver IRI in methionine/choline-deficient diet conditioned rats [45]. Meanwhile, neutrophils contain large quantities of serine proteases in the azurophilic granules, such as neutrophil elastase, proteinase 3, and cathepsin G. After arriving at the damaged site, neutrophils degranulate and release them to exert, in combination with ROS, their effector functions. Neutrophil elastase, a 29 kDa chymotrypsin-like serine protease which degrades ECMs causes organ damage, whereas sivelestat (neutrophil elastase inhibitor) is widely used in treating acute lung injury in humans. We and others have reported that sivelestat inhibited adhesion and migration of neutrophils to vascular endothelium and protected mouse livers against IRI [46, 47]. Furthermore, a randomized clinical study in patients that underwent hepatic resection revealed sivelestat treatment reduced post-operative serum HMGB1 and IL6 levels [48]. Myeloperoxidase (MPO), a heme protein synthesized during myeloid differentiation, is often used as a neutrophil activity marker. In the presence of physiological chloride concentrations, MPO reacts with hydrogen peroxide to catalyze formation of hypochlorous acid/hypochlorite and other oxidizing species [49], and thus, it is also considered as a cytotoxic molecule in liver pathophysiology. However, its cytotoxicity in liver IRI remains to be rigorously re-evaluated as a recent study demonstrated MPO was required for hepatoprotection in endotoxin-induced inflammation [50•]. With recent data showing efficacy and tolerability of AZD3241 (an orally absorbed irreversible human MPO inhibitor) in Parkinson patients [51], the thorough evaluation of MPO in liver LT settings seems warranted.
Activated neutrophils can enhance inflammatory tissue damage by releasing neutrophil extracellular traps (NETs), i.e., extracellular scaffolds of DNA fibers decorated with histone, granule-derived antimicrobial peptides, and enzymes, such as neutrophil erastase, cathepsin G, and MPO [52]. Generation of ROS by NADPH oxidase and activation of protein-arginine deiminase 4 (PAD4), an enzyme that converts arginine to citrulline on histones, are essential steps for chromatin decondensation in the NET formation. NE and MPO translocated into the nucleus further unfold chromatin, leading to the nuclear membrane break, chromatin release into cytosol where it is further decorated with granular and cytosolic proteins, and the emission of these traps into extracellular space [53]. NETs function was originally described as the efficient means to immobilize, catch, and eliminate pathogens, whereas recent studies have recognized its critical involvement in non-infectious inflammatory states, including IR-stress. Indeed, biopsies obtained from human kidney transplant recipients with post-transplant acute tubular necrosis exhibited increased NETs formation [54•], while bronchoalveolar lavage fluid collected from human lung transplant recipients with primary graft dysfunction contained extensive NETs [55]. Furthermore, several NET-targeting agents successfully attenuated IRI in murine models, including PAD4 inhibitors (YW3-56, YW4-03), which reduced NETs formation and decreased severity of liver IRI [56•]; PAD inhibitor (Cl-amidine), which suppressed NETs and alleviated renal IRI [54•]; or DNase I combined with recombinant tissue-type plasminogen activator, which inhibited NETs and alleviated myocardial IRI [57]. Additional functional aspects of NETs are detailed below.
Anti-inflammatory and Tissue-Repairing Neutrophil Functions
In addition to pro-inflammatory and tissue-destructive functions, recent studies have identified previously unappreciated novel role of neutrophils to control inflammation and resolve tissue damage. First, neutrophils retain the potential to inhibit T cell cytotoxicity. Arginase-1 (Arg1, a tertiary granule content) is a manganese-containing enzyme expressed in mouse myeloid cells, such as macrophages. However, in humans, it has been detected selectively in neutrophils [58, 59], whereas activated neutrophils release Arg1 during degranulation process, which inhibit T cell proliferation and expansion [60, 61]. Neutrophil elastase (an azurophilic granule content) cleaves CD2, CD4, and CD8 on peripheral blood T lymphocytes, leading to reduced IL2 production and suppressed T cell cytotoxicity [62]. Neutrophil-derived ROS may also suppress T cells via inactivation of coffilin, an actin-remodeling protein, which impairs formation of the immune synapse and cell activation [63]. Moreover, as regulatory T cells (Treg) are less sensitive to inhibition by ROS [64], production of ROS by neutrophils effectively creates a Treg dominant environment [65].
Several lines of evidence indicate that neutrophil can limit inflammation response in other innate immune cells. Selective neutrophil ablation or genetic MPO deletion in endotoxin-challenged mice unexpectedly enhanced inflammation responses and increased mortality, indicating that neutrophils and neutrophil-derived MPO can contribute to host resistance by limiting endotoxin-driven innate immune pathology [50•]. Cathepsin G (an azurophilic granule content) cleaves NKp46 and impairs NKp46-mediated responses of NK cells, including IFNγ production and cell degranulation [66]. Neutrophil-derived microvesicles cause macrophages to generate lipid pro-resolving mediators and TGFβ, leading to counter inflammatory response [67]. Dead neutrophils secret cytosolic protein annexin A1 (AnxA1), which limits neutrophil migration by interacting with formyl peptide receptor 2 (FPR2) [68, 69] and prompts neutrophil apoptosis [70]. Clearance of apoptotic neutrophils by macrophage efferocytosis may then switch macrophages to anti-inflammatory IL10/TGFβ dominant phenotype, leading to tissue-repairing environment as well as Treg dominated immune tolerance.
Analogous to the concept of pro-inflammatory (M1) and anti-inflammatory (M2) macrophage phenotypes [71], tissue-specific microenvironment may promote neutrophil polarization into two distinct phenotypes [72]. Thus, N1 neutrophils are anti-tumoral/pro-inflammatory, having increased TNFα expression and reduced arginase/CXCR4/MMP-9/VEGF levels, whereas N2 neutrophils are pro-tumoral/anti-inflammatory and characterized by decreased TNFα and increased arginase/CXCR4/MMP-9/VEGF expression [73, 74]. Several cytokines may contribute to N1/N2 polarization, with TGFβ promoting neutrophils to acquire N2 phenotype by simultaneously inhibiting N1 neutrophil generation [72]. Endogenous IFNβ imprints neutrophils to N1 phenotype, which restrict tumor angiogenesis and enhance inflammatory cytotoxicity [75]. A recent study demonstrated that analogous to M1/M2 macrophage polarization, LPS and IFNγ drove peripheral blood neutrophils toward N1 phenotype while IL4 polarized toward N2 phenotype [76]. In addition, murine infarcted-myocardium was infiltrated by day 1 with N1 dominant neutrophils expressing pro-inflammatory markers, whereas N2 neutrophils increased by day 7 concomitantly with enhanced levels of anti-inflammatory markers, supporting putative role of N2 neutrophils in the resolution of inflammation and promoting tissue repair [76]. Noteworthy, rosiglitazone (peroxisome proliferator-activated receptor-γ agonist) treatment in a mouse brain stroke model shifted neutrophil population toward N2 phenotype (31% vs. 77%), suppressed inflammation, enhanced neutrophil clearance, and alleviated brain damage [77]. Although macrophage M1/M2 plasticity is broadly accepted [78], neutrophils generally have short life-span (6–8 h in blood stream), their plasticity remains unknown. Future studies need to elucidate whether N1/N2 polarization is dominated by re-programming of existing neutrophils or environmental influence on de novo neutrophil populations. By virtue of technological advances and efforts to identify neutrophil heterogeneity, additional anti-inflammatory neutrophil subsets have been recognized. Studies by Tirouvanziam et al. identified a CD63+/MHC-II+/CD80+/CD294+ human neutrophil subset in mature cystic fibrosis airway, which retained anabolic/pro-survival phenotype and suppressed T cell function [60, 79]. Pillay et al. found a subset of mature human CD16bright/CD62Ldim neutrophils in endotoxin-challenged blood, which suppressed T cell proliferation via neutrophil MAC-1 dependent manner [80].
Neutrophils may also contribute to tissue-repairing processes following infectious or sterile inflammatory organ damage [81]. Clinical observations support this concept, as patients with leukocyte adhesion deficiency type 1 (an autosomal recessive disorder characterized by defects in neutrophil adhesion ability) experience delayed wound healing [82]. In the beginning of tissue repair process, neutrophils acting as professional phagocytes remove tissue debris. Indeed, in a thermal hepatic injury model, neutrophil depletion resulted in far more debris remaining at the injury site [83•]. Next, neutrophil apoptosis shifts the phagocytosing macrophages to the resolution phase (M2), which governs tissue-repairing environment. M2 macrophages secrete IL1Ra, IL10, TGFβ, and VEGF, leading to fibroblast differentiation into myofibroblasts, synthesis of interstitial fibrillar collagens by myofibroblasts, and expression of MMPs/TIMPs that control ECM turnover [84]. Since neutrophils can release MMP9 free of its endogenous inhibitors (TIMPs), they are capable of delivering highly active MMP9, a key proangiogenic factor degrading ECM, to release ECM-bounded VEGF and other growth factors [85]. A series of recent studies have deepened our understanding of that process. For instance, Christoffersson et al. identified a CD11b+/Gr1+/CXCR4high neutrophil subset in a syngeneic mouse pancreatic islet transplant model, which was recruited via VEGF-A depending pathway. It contained higher amount of MMP9 than those recruited to an inflammatory stimulus, and contributed to revascularization via MMP9 dependent mechanism [86]. They also found an analogous CD49d+/VEGFR1high/CXCR4high neutrophil population in mouse and human, which was recruited by hypoxia-induced VEGF-A [87]. In a liver thermal injury model, limited period of neutrophil depletion (~ 48 h) resulted in delayed revascularization at 7 days and the presence of non-healing injury area at 4 weeks, suggesting the requirement for neutrophil in liver homeostasis [83•]. Likewise, in a transplantation model of bioengineered U-graft, which contained an unassembled suspension of vascular cells embedded in a hydrogel, host-derived alternatively polarized neutrophils (N2) contributed to graft revascularization [88•].
Termination of Neutrophil Effector Functions
In a successful response to an acute injury, it is crucial to prevent tissue damage by promoting the local resolution of inflammation through the removal of neutrophils from the stressed site [89]. Apoptosis, a highly organized programmed cell death program, is considered a favorable neutrophil death mechanism because the orderly cell elimination limits uncontrolled release of toxic neutrophil contents and DAMPs, which potentially may cause further tissue damage [90]. Morphologically, apoptotic neutrophils show the condensation of chromatin and its migration to the nuclear periphery, fragmentation of nuclear DNA, and the blebbing of cell membranes leading to apoptotic body formation. As discussed, engulfment of apoptotic neutrophils by macrophages occurs in the resolution phase of inflammation and repair from tissue injury by turning off production of pro-inflammatory cytokines and launching an anti-inflammatory transcriptional program characterized by the release of IL10 and TGFβ [84].
In contrast to the organized apoptotic cell death, neutrophil necrosis is a traumatic and highly detrimental event resulting in disintegration of the nuclear envelope and cytoplasmic membranes. Neutrophils unavoidably undergo necrosis if the insult is too severe or the step-by-step apoptosis procedure fails to be achieved in a timely fashion, a process called a “secondary necrosis.” Necrotic neutrophil passively releases toxic effector molecules as well as DAMPs leading to further damage and inflammation in neighboring tissues [90].
NETosis is the process by which neutrophils produce and release NETs. During NETosis, intracellular proteins become exposed to the extracellular space, which results in the potential presentation of autoantigens to the host immune system and the release of DAMPs that amplify ongoing immune reactions [53]. In this inflammatory aspect, the NETosis seems closer to necrosis than apoptosis. On the other hand, the function of NETosis to promote coagulation and thrombosis have been now recognized [91, 92], whereas a recent study from McDonald et al. has revealed NET–platelet–thrombin axis that promotes intravascular coagulation and microvascular dysfunction in endotoxin-induced liver injury model [93•]. Considering neutrophils preferentially undergo necrosis/NETosis rather than apoptosis if the stress/infection is severe [94], the function of NETosis to block blood flow to/from the severely damaged site seems to be favorable by averting the spread of DAMPs/pathogens to remote organs and preventing distant organ injury. In this context, Nakazawa et al. revealed NETs inhibitor alleviated acute kidney injury while concomitantly reducing serum NETs levels and mitigating remote organ injury in the lung, liver, heart, and brain [54•]. In contrast, homozygous PAD4 knockout in MRSA challenged mice was accompanied by decreased NETs and reduced lung injury despite higher bacterial counts, increased levels of inflammatory cytokines, and equivalent overall survival as compared to WT counterparts. The heterozygous PAD4 knockout mice unexpectedly showed improved survival as compared with WT counterparts or homozygous knockout mice [95•]; in a mouse model of peritonitis, degradation of NETs by DNase attenuated organ damage only when combined with antibiotics [96]; in an experimental model of a gout (acute sterile inflammatory reaction to monosodium urate crystals), NETosis-deficient mice developed exacerbated and chronic disease that could be reduced by adoptive transfer of aggregated NETs [97]. Therefore, the influence of NETs inhibition depends on disease etiology and severity of the inflammation response. Because of increased incidence of acute respiratory distress syndrome (ARDS) and acute kidney injury in LT patients [98, 99], it is important to evaluate potential clinical value of NETs reducing therapeutic strategies.
It has long been accepted that apoptosis is the most common and preferred cell death program to terminate neutrophil activity. However, recent evidence suggests that neutrophils do not necessarily die in an inflamed site, and instead can leave the site of tissue damage in a process termed as a “reverse migration.” This phenomenon was first visualized in zebrafish larvae in which most of the recruited neutrophils leave a damaged site and traffic to a distal site [100, 101]. A recent study combining intravital imaging and photoactivation techniques in a mouse thermal hepatic injury model demonstrated that neutrophils recruited to the stressed tissue neither underwent apoptosis, necrosis, nor phagocytosis by monocytes, but instead reversely migrated to the circulation, became arrested within the lung without causing local tissue injury, and then homed to the bone marrow where they ultimately underwent apoptosis [83•]. In contrast, in hepatic IRI or acute pancreatitis model, activated neutrophils were redistributed to produce remote organ injury [102,103,104]. It is noteworthy that neutrophils undergoing transendothelial migration in vitro expressed specific marker, ICAM1high/CXCR1low, which was resistant to apoptosis and produced more ROS [102], whereas patients with acute pancreatitis who developed acute lung injury had more ICAM1high/CXCR1low expressing neutrophils in their circulation [104]. This implies the potential for neutrophil reprograming or existence of active/inactive neutrophil subsets after reverse migration. However, there is no rigorous evidence as to why and how reversely migrating neutrophils can be toxic in some cases but inactive in other pathology states.
Neutrophil-Targeted Therapies
Neutrophils have long been recognized as important players in liver IRI, while great majority of studies focused on their pro-inflammatory and tissue-destructive functions. Indeed, neutrophil-targeting strategies mostly narrowed at reducing their target organ infiltration. Table 2 lists representative preclinical studies on neutrophil targeting in liver IRI [23, 24, 30, 34•, 40, 45, 47, 56•, 105,106,107].
It is important to keep in mind that LT recipients are immunocompromised while neutrophils constitute the forefront of pathogen host defense. Hence, therapies limiting neutrophil sequestration and/or effector functions may in turn increase the risk of infections in transplant recipients. Despite preclinical studies showing therapeutic efficacy, global neutrophil depletion is unlikely to be clinically feasible. More specific maneuvers to inhibit neutrophil adhesion, migration, and effector functions are required, though it remains to be determined whether and how such therapies can achieve optimal balance between effective host immune surveillance while controlling cytodestruction.
Albeit non-specifically targeting neutrophil functions, the reduction of extracellular DAMPs such as HMGB1 or histone seems favorable, because DAMPs are the immune-activating signals released from host cells but not from pathogens. Of note, one of the HMGB1 inactivators (ART-123, recombinant thrombomodulin), originally shown to mitigate liver IRI in TLR4 depending manner [27•], is now used clinically for disseminated intravascular coagulation [108, 109]. In agreement with recent data [54•, 56•], inhibition of NETs formation by specifically targeting highly destructive neutrophil function seems biologically beneficial. However, futures studies need to evaluate potential risks, as this maneuver may spread abundant DAMPs or pathogens, which otherwise should be isolated inside the NETs areas. Since DNase has been successfully applied for the treatment of cystic fibrosis patients and the NETs digestion is considered to be a part of its beneficial effects [110, 111], these investigations are clearly warranted.
Although a number of recent studies revealed neutrophil immunoregulatory and tissue-repairing capabilities, little is known about these functions in liver IRI. This might be due to the overwhelming focus on the “toxic” side of the neutrophil biology. Although T cells, particularly of CD4+ phenotype, are indispensable for the activation/regulation of sterile inflammation in liver IRI [112, 113], it seems that the presence of CD4+ T cells per se rather than Ag-specific de novo T cell activation is needed for immune regulation [114]. Hence, the ability of neutrophils to reduce T cell activation may be insufficient to significantly impact IRI outcomes. In addition, the severe hepatocellular damage produced in currently used murine IRI models may lead to a failure in detecting discrete immunomodulatory and/or tissue-repairing neutrophil functions. Future studies need to focus on neutrophil involvement in the resolution of inflammation and recreation of tissue homeostasis. Experimental evidence indicates the anti-inflammatory (N2) neutrophil polarization by TGFβ, IL4 or rosiglitazone [75,76,77]. Although the neutrophil plasticity has not been rigorously proven, the N1 to N2 phenotype shifting should be of a potential therapeutic interest.
Neutrophil apoptosis is a favorable way of its functional termination, leading to an immunomodulatory shift of phagocytosing Kupffer cells and prevention of the DAMPs burst, while excessive induction of neutrophil apoptosis may cause unnecessary immune suppression. Indeed, ectoine, a natural cell protectant, improved inflammatory resolution in a sterile inflammation lung model by preferentially prompting apoptosis in activated but not inactivated neutrophils, reducing neutrophil numbers and improving resolution of the lung pathology [115]. Furthermore, a recent randomized clinical trial in elderly patients highlight the efficacy of inhaled ectoine in neutrophilic lung inflammation [116]. Studies by Christoffersson et al. have identified VEGF-A-induced CXCR4high unique neutrophil subset, which retained the ability of revascularization in a pancreatic islet transplant model [86, 87], while a study by Wang et al. [83•] implied CXCR4 expressing non-inflammatory neutrophil subset capable of homing back to bone marrow in the “reverse migration” mechanism [117]. Thus, one may envision a great potential for future regimens shifting neutrophils into tissue-resolving CXCR4-positive dominant population.
Conclusion
Due to technological advances, a remarkable progress has been recently made in our appreciation of the neutrophil biology, especially its immune regulatory and tissue-repairing functions. Live-cell and deep-tissue imaging combined with single cell flow cytometry analyses [118] allow to accurately track neutrophil functions and interactions in real-time. Unlike LysM reporter mice, unable to distinguish between neutrophil and monocyte/macrophage lineages, a newly developed murine strain with a more specific Ly6G promoter [119] has enabled to investigate in vivo neutrophil functions more selectively and unequivocally. Since efficient strategies against hepatic IRI have not been yet developed, newly discovered and therapeutically attractive neutrophil functions, as discussed in this review, warrant future comprehensive preclinical and clinical attention in transplant recipients.
Abbreviations
- AnxA1:
-
Annexin A1
- ARDS:
-
Acute respiratory distress syndrome
- ATP:
-
Adenosine triphosphate
- CCl4:
-
Carbon tetrachloride
- DAMPs:
-
Danger-associated molecular patterns
- ECM:
-
Extracellular matrix
- FMIT:
-
Mitochondrial N-formyl peptides
- FPR1:
-
Formyl peptide receptor 1
- FPR2:
-
Formyl peptide receptor 2
- HMGB1:
-
High mobility group box 1
- ICAM-1:
-
Intercellular adhesion molecule-1
- IRI:
-
Ischemia-reperfusion injury
- LSEC:
-
Liver sinusoidal endothelial cell
- LT:
-
Liver transplantation
- Mac1:
-
Integrin αMβ2
- MMP:
-
Matrix metalloproteinase
- MPO:
-
Myeloperoxidase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NET:
-
Neutrophil extracellular trap
- NLRP3:
-
NOD-like receptor pyrin domain-containing-3
- PAD4:
-
Protein-arginine deiminase 4
- PRR:
-
Pattern recognition receptor
- ROS:
-
Reactive oxygen species
- TIMP:
-
Tissue inhibitor of metalloproteinase
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Dutkowski P, Linecker M, DeOliveira ML, Mullhaupt B, Clavien PA. Challenges to liver transplantation and strategies to improve outcomes. Gastroenterology. 2015;148(2):307–23. https://doi.org/10.1053/j.gastro.2014.08.045.
Busuttil RW, Tanaka K. The utility of marginal donors in liver transplantation. Liver Transpl. 2003;9(7):651–63. https://doi.org/10.1053/jlts.2003.50105.
Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10(2):79–89. https://doi.org/10.1038/nrgastro.2012.225.
Selzner M, Selzner N, Jochum W, Graf R, Clavien PA. Increased ischemic injury in old mouse liver: an ATP-dependent mechanism. Liver Transpl. 2007;13(3):382–90. https://doi.org/10.1002/lt.21100.
Guan LY, Fu PY, Li PD, Li ZN, Liu HY, Xin MG, et al. Mechanisms of hepatic injury and protective effects of nitric oxide. World J Gastrointest Surg. 2014;6(7):122–8. https://doi.org/10.4240/wjgs.v6.i7.122.
Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2011;11(8):1563–9. https://doi.org/10.1111/j.1600-6143.2011.03579.x.
Nakamura K, Kageyama S, Ke B, Fujii T, Sosa RA, Reed EF, et al. Sirtuin 1 attenuates inflammation and hepatocellular damage in liver transplant ischemia-reperfusion: from mouse-to-human. Liver Transpl. 2017;23:1282–93. https://doi.org/10.1002/lt.24821.
Vollmar B, Menger MD. The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev. 2009;89(4):1269–339. https://doi.org/10.1152/physrev.00027.2008.
Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, et al. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol Ther. 2009;17(1):65–72. https://doi.org/10.1038/mt.2008.237.
Nakamura K, Kageyama S, Yue S, Huang J, Fujii T, Ke B, et al. Heme oxygenase-1 regulates sirtuin-1 - autophagy pathway in liver transplantation: from mouse-to-human. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2017;18:1110–21. https://doi.org/10.1111/ajt.14586.
Kageyama S, Hirao H, Nakamura K, Ke B, Zhang M, Ito T, et al. Recipient HO-1 inducibility is essential for post-transplant hepatic HO-1 expression and graft protection: from bench-to-bedside. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2018. https://doi.org/10.1111/ajt.15043.
Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa R, et al. Macrophage HO-1–SIRT1–p53 Axis regulates sterile inflammation in liver ischemia–reperfusion injury. J Hepatol. 2017;67:1232–42.
Kageyama S, Nakamura K, Ke B, Busuttil RW, Kupiec-Weglinski JW. Serelaxin induces Notch1 signaling and alleviates hepatocellular damage in orthotopic liver transplantation. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2018;18:1755–63. https://doi.org/10.1111/ajt.14706.
Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–8.
Doeing DC, Borowicz JL, Crockett ET. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin Pathol. 2003;3(1):3. https://doi.org/10.1186/1472-6890-3-3.
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–75. https://doi.org/10.1038/nri3399.
Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA. Neutrophils--a key component of ischemia-reperfusion injury. Shock. 2013;40(6):463–70. https://doi.org/10.1097/shk.0000000000000044.
Kageyama S, Nakamura K, Fujii T, Ke B, Sosa RA, Reed EF, et al. Recombinant relaxin protects liver transplants from ischemia damage via hepatocyte glucocorticoid receptor: from bench-to-bedside. Hepatology. 2018. https://doi.org/10.1002/hep.29787.
Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33(5):657–70. https://doi.org/10.1016/j.immuni.2010.11.011.
Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143(5):1158–72. https://doi.org/10.1053/j.gastro.2012.09.008.
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–6. https://doi.org/10.1126/science.1195491.
Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119(2):305–14. https://doi.org/10.1172/jci35958.
Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ, Smith CW. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology. 1993;17(5):915–23.
Nakano H, Kuzume M, Namatame K, Yamaguchi M, Kumada K. Efficacy of intraportal injection of anti-ICAM-1 monoclonal antibody against liver cell injury following warm ischemia in the rat. Am J Surg. 1995;170(1):64–6.
Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology. 2011;54(3):999–1008. https://doi.org/10.1002/hep.24501.
Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–43. https://doi.org/10.1084/jem.20042614.
• Kadono K, Uchida Y, Hirao H, Miyauchi T, Watanabe T, Iida T, et al. Thrombomodulin attenuates inflammatory damage due to liver ischemia and reperfusion injury in mice in Toll-like receptor 4-dependent manner. Am J Transpl. 2017;17(1):69–80. https://doi.org/10.1111/ajt.13991 This study documents the efficacy of recombinant thrombomodulin (HMGB1 inactivator) to mitigate liver IRI via TLR4-dependent signaling.
Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125(2):539–50. https://doi.org/10.1172/jci76887.
Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology. 2014;59(5):1984–97. https://doi.org/10.1002/hep.26976.
de Oliveira THC, Marques PE, Poosti F, Ruytinx P, Amaral FA, Brandolini L, et al. Intravital microscopic evaluation of the effects of a CXCR2 antagonist in a model of liver ischemia reperfusion injury in mice. Front Immunol. 2017;8:1917. https://doi.org/10.3389/fimmu.2017.01917.
Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-alpha promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis. 2018;9(3):323. https://doi.org/10.1038/s41419-018-0377-4.
Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2014;60(4):782–91. https://doi.org/10.1016/j.jhep.2013.12.005.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. https://doi.org/10.1038/nature08780.
• Honda M, Takeichi T, Hashimoto S, Yoshii D, Isono K, Hayashida S, et al. Intravital imaging of neutrophil recruitment reveals the efficacy of FPR1 blockade in hepatic ischemia-reperfusion injury. J Immunol. 2017;198(4):1718–28. https://doi.org/10.4049/jimmunol.1601773 This study revealed the efficacy of FPR1 antagonist to inhibit neutrophil migration and alleviate liver IRI.
Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159(1):91–102. https://doi.org/10.1083/jcb.200202114.
Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. 2012;56(5):1971–82. https://doi.org/10.1002/hep.25801.
Kato H, Duarte S, Liu D, Busuttil RW, Coito AJ. Matrix metalloproteinase-2 (MMP-2) gene deletion enhances MMP-9 activity, impairs PARP-1 degradation, and exacerbates hepatic ischemia and reperfusion injury in mice. PLoS One. 2015;10(9):e0137642. https://doi.org/10.1371/journal.pone.0137642.
Li J, Zhang YP, Kirsner RS. Angiogenesis in wound repair: angiogenic growth factors and the extracellular matrix. Microsc Res Tech. 2003;60(1):107–14. https://doi.org/10.1002/jemt.10249.
Moore C, Shen XD, Gao F, Busuttil RW, Coito AJ. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am J Pathol. 2007;170(2):567–77. https://doi.org/10.2353/ajpath.2007.060456.
Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47(1):186–98. https://doi.org/10.1002/hep.21922.
Duarte S, Hamada T, Kuriyama N, Busuttil RW, Coito AJ. TIMP-1 deficiency leads to lethal partial hepatic ischemia and reperfusion injury. Hepatology. 2012;56(3):1074–85. https://doi.org/10.1002/hep.25710.
Duarte S, Matian P, Ma S, Busuttil RW, Coito AJ. Adeno-associated virus-mediated gene transfer of tissue inhibitor of metalloproteinases-1 impairs neutrophil extracellular trap formation and ameliorates hepatic ischemia and reperfusion injury. Am J Pathol. 2018;188(8):1820–32. https://doi.org/10.1016/j.ajpath.2018.05.002.
Kuyvenhoven JP, Verspaget HW, Gao Q, Ringers J, Smit VT, Lamers CB, et al. Assessment of serum matrix metalloproteinases MMP-2 and MMP-9 after human liver transplantation: increased serum MMP-9 level in acute rejection. Transplantation. 2004;77(11):1646–52.
Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, et al. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation. 2010;121(21):2310–6. https://doi.org/10.1161/circulationaha.108.814731.
Kimura K, Shirabe K, Yoshizumi T, Takeishi K, Itoh S, Harimoto N, et al. Ischemia-reperfusion injury in fatty liver is mediated by activated NADPH oxidase 2 in rats. Transplantation. 2016;100(4):791–800. https://doi.org/10.1097/tp.0000000000001130.
Sakai S, Tajima H, Miyashita T, Nakanuma S, Makino I, Hayashi H, et al. Sivelestat sodium hydrate inhibits neutrophil migration to the vessel wall and suppresses hepatic ischemia-reperfusion injury. Dig Dis Sci. 2014;59(4):787–94. https://doi.org/10.1007/s10620-013-2963-8.
Uchida Y, Freitas MC, Zhao D, Busuttil RW, Kupiec-Weglinski JW. The inhibition of neutrophil elastase ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. 2009;15(8):939–47. https://doi.org/10.1002/lt.21770.
Tsujii S, Okabayashi T, Shiga M, Takezaki Y, Sugimoto T, Kobayashi M, et al. The effect of the neutrophil elastase inhibitor sivelestat on early injury after liver resection. World J Surg. 2012;36(5):1122–7. https://doi.org/10.1007/s00268-012-1501-8.
Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77(5):598–625. https://doi.org/10.1189/jlb.1204697.
• Reber LL, Gillis CM, Starkl P, Jonsson F, Sibilano R, Marichal T, et al. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J Exp Med. 2017;214(5):1249–58. https://doi.org/10.1084/jem.20161238 This study demonstrates the unexpected requirement for neutrophils and neutrophil-derived MPO in hepatoprotection against endotoxin-induced liver injury and highlights a novel immunoregulatory neutrophil function.
Jucaite A, Svenningsson P, Rinne JO, Cselenyi Z, Varnas K, Johnstrom P, et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson’s disease. Brain. 2015;138(Pt 9):2687–700. https://doi.org/10.1093/brain/awv184.
Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189(6):2689–95. https://doi.org/10.4049/jimmunol.1201719.
Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23(3):279–87. https://doi.org/10.1038/nm.4294.
• Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol. 2017;28(6):1753–68. https://doi.org/10.1681/asn.2016080925 Important study showing the efficacy of NETs inhibiting therapy not only to reduce kidney IRI but also prevent remote (distal) organ damage.
Sayah DM, Mallavia B, Liu F, Ortiz-Munoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med. 2015;191(4):455–63. https://doi.org/10.1164/rccm.201406-1086OC.
• Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–14. https://doi.org/10.1002/hep.27841 This study highlights the mechanism of NETs formation and efficacy of NETs inhibition in liver IRI.
Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308(5):H500–9. https://doi.org/10.1152/ajpheart.00381.2014.
Luckner-Minden C, Fischer I, Langhans CD, Schiller M, Kropf P, Muller I, et al. Human eosinophil granulocytes do not express the enzyme arginase. J Leukoc Biol. 2010;87(6):1125–32. https://doi.org/10.1189/jlb.1109741.
Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105(6):2549–56. https://doi.org/10.1182/blood-2004-07-2521.
Ingersoll SA, Laval J, Forrest OA, Preininger M, Brown MR, Arafat D, et al. Mature cystic fibrosis airway neutrophils suppress T cell function: evidence for a role of arginase 1 but not programmed death-ligand 1. J Immunol. 2015;194(11):5520–8. https://doi.org/10.4049/jimmunol.1500312.
Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015;75(2):275–83. https://doi.org/10.1158/0008-5472.can-14-1491.
Doring G, Frank F, Boudier C, Herbert S, Fleischer B, Bellon G. Cleavage of lymphocyte surface antigens CD2, CD4, and CD8 by polymorphonuclear leukocyte elastase and cathepsin G in patients with cystic fibrosis. J Immunol. 1995;154(9):4842–50.
Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity. 2008;29(3):404–13. https://doi.org/10.1016/j.immuni.2008.06.016.
Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood. 2009;113(15):3542–5. https://doi.org/10.1182/blood-2008-09-181040.
Nicolas-Avila JA, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity. 2017;46(1):15–28. https://doi.org/10.1016/j.immuni.2016.12.012.
Valayer A, Brea D, Lajoie L, Avezard L, Combes-Soia L, Labas V, et al. Neutrophils can disarm NK cell response through cleavage of NKp46. J Leukoc Biol. 2017;101(1):253–9. https://doi.org/10.1189/jlb.3AB0316-140RR.
Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15):e60–72. https://doi.org/10.1182/blood-2012-04-423525.
Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. 2002;8(11):1296–302. https://doi.org/10.1038/nm786.
Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, Perretti M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood. 2008;112(6):2512–9. https://doi.org/10.1182/blood-2008-02-140533.
Scannell M, Flanagan MB, de Stefani A, Wynne KJ, Cagney G, Godson C, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178(7):4595–605.
Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64. https://doi.org/10.1038/nri1733.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94. https://doi.org/10.1016/j.ccr.2009.06.017.
Piccard H, Muschel RJ, Opdenakker G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit Rev Oncol Hematol. 2012;82(3):296–309. https://doi.org/10.1016/j.critrevonc.2011.06.004.
Selders GS, Fetz AE, Radic MZ, Bowlin GL. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen Biomater. 2017;4(1):55–68. https://doi.org/10.1093/rb/rbw041.
Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120(4):1151–64. https://doi.org/10.1172/jci37223.
Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110(1):51–61. https://doi.org/10.1093/cvr/cvw024.
Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke. 2013;44(12):3498–508. https://doi.org/10.1161/strokeaha.113.002470.
Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–40. https://doi.org/10.1002/jcp.26429.
Tirouvanziam R, Gernez Y, Conrad CK, Moss RB, Schrijver I, Dunn CE, et al. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci U S A. 2008;105(11):4335–9. https://doi.org/10.1073/pnas.0712386105.
Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012;122(1):327–36. https://doi.org/10.1172/jci57990.
Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371(3):531–9. https://doi.org/10.1007/s00441-017-2785-7.
Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med. 2000;343(23):1703–14. https://doi.org/10.1056/nejm200012073432307.
• Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. 2017;358(6359):111–6. https://doi.org/10.1126/science.aam9690 Important study visualizing the non-inflammatory reverse migration of neutrophils and their homing back to the bone marrow.
Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. 2013;5(5):661–74. https://doi.org/10.1002/emmm.201202382.
Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104(51):20262–7. https://doi.org/10.1073/pnas.0706438104.
Christoffersson G, Vagesjo E, Vandooren J, Liden M, Massena S, Reinert RB, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood. 2012;120(23):4653–62. https://doi.org/10.1182/blood-2012-04-421040.
Massena S, Christoffersson G, Vagesjo E, Seignez C, Gustafsson K, Binet F, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood. 2015;126(17):2016–26. https://doi.org/10.1182/blood-2015-03-631572.
• Lin RZ, Lee CN, Moreno-Luna R, Neumeyer J, Piekarski B, Zhou P, et al. Host non-inflammatory neutrophils mediate the engraftment of bioengineered vascular networks. Nat Biomed Eng. 2017;1. https://doi.org/10.1038/s41551-017-0081 This study documents the requirement for neutrophils to improve the engraftment of bioengineered tissues.
Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10(6):427–39. https://doi.org/10.1038/nri2779.
Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Murai M. Neutrophil cell death in response to infection and its relation to coagulation. J Intensive Care. 2013;1(1):13. https://doi.org/10.1186/2052-0492-1-13.
Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. 2015;36(22):1405–14. https://doi.org/10.1093/eurheartj/ehv007.
Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. 2014;73(10):1854–63. https://doi.org/10.1136/annrheumdis-2013-203430.
• McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129(10):1357–67. https://doi.org/10.1182/blood-2016-09-741298 This study focuses on NETs, which promote intravascular coagulation and microvascular dysfunction.
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18(9):1386–93. https://doi.org/10.1038/nm.2847.
• Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3(3). https://doi.org/10.1172/jci.insight.98178 Interesting study highlighting the ability of excessive NETs inhibition to cause pathogen spread and increase inflammation.
Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One. 2016;11(2):e0148142. https://doi.org/10.1371/journal.pone.0148142.
Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20(5):511–7. https://doi.org/10.1038/nm.3547.
Zhao W, Ge X, Sun K, Agopian VG, Wang Y, Yan M, et al. Acute respiratory distress syndrome after orthotopic liver transplantation. J Crit Care. 2016;31(1):163–7. https://doi.org/10.1016/j.jcrc.2015.09.028.
Rahman S, Davidson BR, Mallett SV. Early acute kidney injury after liver transplantation: predisposing factors and clinical implications. World J Hepatol. 2017;9(18):823–32. https://doi.org/10.4254/wjh.v9.i18.823.
Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. 2006;80(6):1281–8. https://doi.org/10.1189/jlb.0506346.
Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, Chimen M, et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci Transl Med. 2014;6(225):225ra29. https://doi.org/10.1126/scitranslmed.3007672.
Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12(8):761–9. https://doi.org/10.1038/ni.2062.
Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity. 2015;42(6):1075–86. https://doi.org/10.1016/j.immuni.2015.05.010.
Wu D, Zeng Y, Fan Y, Wu J, Mulatibieke T, Ni J, et al. Reverse-migrated neutrophils regulated by JAM-C are involved in acute pancreatitis-associated lung injury. Sci Rep. 2016;6:20545. https://doi.org/10.1038/srep20545.
Loi P, Yuan Q, Torres D, Delbauve S, Laute MA, Lalmand MC, et al. Interferon regulatory factor 3 deficiency leads to interleukin-17-mediated liver ischemia-reperfusion injury. Hepatology. 2013;57(1):351–61. https://doi.org/10.1002/hep.26022.
Liu A, Fang H, Yang Y, Sun J, Fan H, Liu S, et al. The fibrin-derived peptide bbeta15-42 attenuates liver damage in a rat model of liver ischemia/reperfusion injury. Shock. 2013;39(4):397–403. https://doi.org/10.1097/SHK.0b013e31828c2b75.
Palumbo T, Nakamura K, Lassman C, Kidani Y, Bensinger SJ, Busuttil R, et al. Bruton tyrosine kinase inhibition attenuates liver damage in a mouse warm ischemia and reperfusion model. Transplantation. 2017;101(2):322–31. https://doi.org/10.1097/tp.0000000000001552.
Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115(5):1267–74. https://doi.org/10.1172/jci22782.
Nakamura K, Hatano E, Miyagawa-Hayashino A, Okuno M, Koyama Y, Narita M, et al. Soluble thrombomodulin attenuates sinusoidal obstruction syndrome in rat through suppression of high mobility group box 1. Liver Int. 2014;34(10):1473–87. https://doi.org/10.1111/liv.12420.
Papayannopoulos V, Staab D, Zychlinsky A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One. 2011;6(12):e28526. https://doi.org/10.1371/journal.pone.0028526.
Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. https://doi.org/10.1038/ncomms7673.
Uchida Y, Ke B, Freitas MC, Ji H, Zhao D, Benjamin ER, et al. The emerging role of T cell immunoglobulin mucin-1 in the mechanism of liver ischemia and reperfusion injury in the mouse. Hepatology. 2010;51(4):1363–72. https://doi.org/10.1002/hep.23442.
Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW, et al. T-cell immunoglobulin mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology. 2010;139(6):2195–206. https://doi.org/10.1053/j.gastro.2010.07.003.
Shen X, Wang Y, Gao F, Ren F, Busuttil RW, Kupiec-Weglinski JW, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology. 2009;50(5):1537–46. https://doi.org/10.1002/hep.23153.
Sydlik U, Peuschel H, Paunel-Gorgulu A, Keymel S, Kramer U, Weissenberg A, et al. Recovery of neutrophil apoptosis by ectoine: a new strategy against lung inflammation. Eur Respir J. 2013;41(2):433–42. https://doi.org/10.1183/09031936.00132211.
Unfried K, Kramer U, Sydlik U, Autengruber A, Bilstein A, Stolz S, et al. Reduction of neutrophilic lung inflammation by inhalation of the compatible solute ectoine: a randomized trial with elderly individuals. Int J Chron Obstruct Pulmon Dis. 2016;11:2573–83. https://doi.org/10.2147/copd.s115061.
Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125(3):281–8. https://doi.org/10.1111/j.1365-2567.2008.02950.x.
Fairchild RL. Turning on the lights inside neutrophils. J Immunol. 2016;197(3):681–2. https://doi.org/10.4049/jimmunol.1600985.
Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods. 2015;12(5):445–52. https://doi.org/10.1038/nmeth.3322.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Immunology
Rights and permissions
About this article

Cite this article
Nakamura, K., Kageyama, S. & Kupiec-Weglinski, J.W. The Evolving Role of Neutrophils in Liver Transplant Ischemia-Reperfusion Injury. Curr Transpl Rep 6, 78–89 (2019). https://doi.org/10.1007/s40472-019-0230-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40472-019-0230-4