
Abstract
The complement system plays a dual role in the body, either as a first-line defense barrier when balanced between activation and inhibition or as a potential driver of complement-associated injury or diseases when unbalanced or over-activated. C4b-binding protein (C4BP) was the first circulating complement regulatory protein identified and it functions as an important complement inhibitor. C4BP can suppress the over-activation of complement components and prevent the complement system from attacking the host cells through the binding of complement cleavage products C4b and C3b, working in concert as a cofactor for factor I in the degradation of C4b and C3b, and consequently preventing or reducing the assembly of C3 convertase and C5 convertase, respectively. C4BP, particularly C4BP α-chain (C4BPα), exerts its unique inhibitory effects on complement activation and opsonization, systemic inflammation, and platelet activation and aggregation. It has long been acknowledged that crosstalk or interplay exists between the complement system and platelets. Our unpublished preliminary data suggest that circulating C4BPα exerts its antiplatelet effects through inhibition of both complement activity levels and complement-induced platelet reactivity. Plasma C4BPα levels appear to be significantly higher in patients sensitive to, rather than resistant to, clopidogrel, and we suggest that a plasma C4BPα measurement could be used to predict clopidogrel resistance in the clinical settings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The complement inhibitor C4b-binding protein alpha may contribute to platelet inhibition through its complement inhibitory effects. |
Plasma C4b-binding protein alpha levels may be a promising biomarker to predict clopidogrel resistance in patients with coronary artery disease. |
Altered C4b-binding protein alpha expression levels may also be used to predict other severe clinical conditions. |
1 Introduction
The complement system is an important component of the innate host defense system, through recognition of invading microbes (such as bacteria and viruses) and uncleared cell debris (such as apoptotic and necrotic cells, misfolded proteins, and released DNA), against microbial invasion (infections) and for enhanced opsonization, inflammation, cytolysis, phagocytosis, or clearance in addition to enhanced adaptive immunity [1, 2]. As a “complemented” component for antibodies, complement is a double-edged sword, playing a dual role, either as a first-line barrier of the normal physiological defense or doing harm when activation and inhibition are unbalanced or the system prefers to be over-activated. It is well recognized that there are more than 40 proteins that can be counted as family members of the natural complement system [1], including C1q, C3, C4, C4b-binding protein (also known as C4BP), and C5.
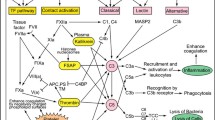
Complement activation is the result of an aggressive proteolytical cascade, working in concert with one another but under tight control of several complement inhibitors, and thus, the final outcome of any scenario will generally depend on the balance between complement activation and inhibition, and which complement pathway(s) and components are over-activated [1, 2]. The complement system can be activated via three major routes: the classical, lectin, and alternative pathways (see Fig. 1 for details) [1,2,3]. For example, the classical pathway is initiated with complement component C1 as its initiator or initial recognition molecule and is activated in turn by the cleavage of a certain complement component (such as C2 and C4) in a cascade-like manner. However, all complement activation pathways converge at the formation of the two distinct subtypes of complement C3 convertases; one is C4b2a for the classical and lectin pathways, and the other is C3bBbP for the alternative pathway (see Fig. 1) [1,2,3]. The C3 convertases initiate the cleavage of complement component C3 to C3a (a major anaphylatoxin) and C3b (a complement activation product) [1,2,3]. As the complement cascades proceed, two distinct complement C5 convertases (C4b2a3b and C3bBbBbP) are formed, facilitating the cleavage of complement component C5 to C5a (another important anaphylatoxin) and C5b (complement activation product) [1,2,3]. Ultimately, deposition of activated C5b on the surface of target cells triggers the formation of the common complement terminal complex C5b-9 (termed membrane attack complex, or MAC), leading to the formation of a cytolytic pore and consequently, lysis of opsonized cells or microbes [1,2,3].

Adapted from Garred et al. [1]
Three major pathways of complement activation cascade. The complement system is primarily activated via the classical (CP), lectin (LP), and alternative (AP) pathways. Under normal physiological situations, the complement system is well balanced between activation and inhibition, both of which are fine turned by an array of intrinsic complement inhibitors, including C1 inhibitor (C1-INH), C4BP, factor H (FH), factor I (FI), CD35, CD46, CD55, CD59, and clusterin. From these cascades, elimination of C1q and/or C3 would prevent and/or reduce the assembly of the C3-convertases and C5-convertases, as well as the generation of C3a and C5a, and even suppress the almost entire complement system. If this is the case, blocking the cleavage of C5 would be more specific and precise than blocking the cleavage of C3 because C3 is common for or shared with all complement cascades and that C5 is downstream of C3. Further, C5, C5a, and C5aR would be important therapeutic targets for upstream imbalance and over-activation of the complement system. Of them, C5a is the most potent activator of an inflammatory response and the most pathogenetic mediator. If C5a is the only mediator or cytokine, suppression of the C5a/C5aR axis would be scientifically sound to keep the C5b-9 intact and the complement system activated constitutively for essential innate immunity, such as necessary opsonization, phagocytosis, or efficient defense against microbial invasion or injury. C1-INH is the first regulatory protein in the classical and lectin complement pathways with a broad-spectrum activity of serine protease inhibitor. C4BP is the second regulator in the classical pathway, and serves as a cofactor for FI in the cleavage and inactivation of C4b, and, to a lesser extent, C3b, in addition to accelerating the decay of C3 convertase. FH, as the most important complement regulatory protein present in the alternative pathway, is similar to C4BP of the classical pathway, primarily binding and inactivating C3b. FI is responsible for cleavage and inactivation of C3b to iC3b (an opsonin). CD35 (complement receptor 1, CR1) is a cofactor for FI that cleaves C3b to iC3b and enhances inactivation of C3b through binding C3b, C4b, and, to a lesser extent, C1q, respectively, and thus, a very efficient C3 inhibitor. CD36 (membrane cofactor protein, MCP) and CD55 (decay-accelerating factor, DAF) are a protective regulator of C3b activity on the cell surface, inhibiting at the C3 level. Bb (a cleavage product of factor B) is one of the cleavage fragments of factor B present in the alternative pathway (Ba and Bb). P, abbreviated for properdin, is the only one regulatory protein that enhances complement activation for its amplification loop as a positive regulator or activator, in addition to enabling more C3 cleavage and activation due to extension of the C3 convertase’s half-life. iC3b, or inactive C3b, is generated from the cleavage and inactivation of C3b. CD59 can bind C8 and C9, inhibiting the C8 and C9 insertion into the cell membrane, preventing final assembly of the common C5b-9 complex, and consequently blocking the formation of lytic pore and lysis of self-cells as well as destruction. Clusterin, one of the 5 most abundant transcripts in human platelets, can bind and inactivate the final MAC complex (C5b-9) at the cytolytic stage of the complement activation.
To minimize or avoid the occurrence of uncontrolled complement activation that will lead to severe damage of the host cells and tissues, complement activation needs to be tightly regulated by the fluid-phase (soluble) and membrane-bound complement inhibitors, including C4BP, C1 inhibitor (C1-INH), factor H (FH), factor I (FI), and clusterin (see Fig. 1). Of these, C4BP is a soluble complement inhibitor of the classical and lectin complement activation pathways [3]. In this mini-review, we briefly summarize and discuss current knowledge and future perspectives of the complement system and C4BP, we also highlight the possible use of C4BPα as a novel biomarker to predict clopidogrel resistance and for the prognosis of other severe clinical manifestations in future patient care.
2 C4BP: Past and Present
The C4BP is one of the largest plasma glycoproteins (570 kDa) and circulates in the blood at estimated plasma levels of 150–200 µg/mL [4,5,6,7,8], it is rapidly cleared from the blood with an elimination half-life of 10 hours [3]. As a circulating plasma protein, it is synthesized predominantly in the liver [9,10,11,12,13], and to a lesser extent, in the pancreas [3], epididymal epithelial cells [10], and the fetal lung [14], and is secreted into the blood. Structurally, C4BP is a large heterogeneous polymeric glycoprotein, and exists in several isoforms having different combinations of 75-kDa polypeptide (α-chain) and 40-kDa polypeptide (β-chain) [3]. The α-chains and β-chains contain eight [9, 15] and three [15] identical complement control protein (CCP) repeats, respectively (see Fig. 2a). In humans, the major isoform of C4BP (75–80% of C4BP in plasma) refers to C4BP α7/β1, and other less abundant forms include α6/β1, α7/β0 (lacking β-chain, and seven α-chains present only), or α6/β0 [15]. Of these, the C4BP α7/β1 molecule comprises seven identical α-chains and one unique β-chain (see Fig. 2a), with all the α-chains being covalently linked to one another in their C-terminal extensions where the two cysteine residues link the different chains by interchain disulfide bridges [9, 16] to form intracellular polymerization of the molecule. The C4BP molecule is assembled in the endoplasmic reticulum [17]. In contrast, the β-chain subunit is not required for polymerization of the α-chains [18], but contains a high-affinity binding site for the vitamin K-dependent protein S (also known as PS) [19, 20] and all β-chain-containing C4BP molecules (α7/β1 and α6/β1) in plasma circulate in complex with PS [5] (see Fig. 2a), which helps localize the C4BP-PS complex to the cell surface through binding of the Gla domain of the PS to negatively charged phospholipids of cell membrane [15, 18]. All C4BP molecules display a spider-like or octopus-like shape [5, 16, 19, 21], with each of the extended tentacles being formed by an α-chain, as visualized by a high-resolution electronic microscopy [5]. The isoform pattern is based on the differential expression of the two genes encoding α-chains and β-chains (C4BPA and C4BPB). The expression is dependent on a variety of factors [22], for example, during the acute-phase reactions (such as sepsis, trauma, or systemic inflammation), the synthesis of the α-chains, rather than of the β-chains, increases, and as a result, the plasma levels of the isoforms of C4BP lacking a β-chain (i.e., α7/β0 and α6/β0) increase, but the levels of β-chain-containing C4BP isoforms (i.e., α7/β1 and α6/β1) remain relatively stable [23]. One of the acute-phase reactants, plasma C4BP, (predominantly C4BPα), was found to be significantly elevated during inflammation [6, 23,24,25,26] and surgery [27], consistent with enhanced secretion of C4BP in HepG2 cells upon stimulation by interleukin-6 (IL)-6 and tumor necrosis factor alpha (TNFα) [19, 28], both of which are known to be modulators of acute-phase reactants [29, 30]. Moreover, C4BP lacking the β-chain (i.e., C4BPα) was significantly elevated in response to inflammation [3]. When incubated with necrotic cells in the presence of C4BP-deficient serum, THP-1-derived macrophages released excessive TNFα, which was reversed by purified C4BP [31]. These data demonstrated that C4BPα is an acute-phase reactant protein in the body, and that C4BPα suppresses the proinflammatory potential of necrotic cells and complement involved, exhibiting an anti-inflammatory effect.

Major structure of human C4BP (a) and C4BPα (b). The representative structure model of human C4BP is composed of seven identical α-chains and a single unique β-chain. As for each α-chain, it is composed of eight complement control protein (CCP) repeats (or domains), in which CCP1-3 repeats bind C4b, and CCP1-4 repeats bind C3b. As for the β-chain, CCP1 binds protein S (PS)
C4BP, through direct binding to activated complement cleavage fragment C4b and further restricting its function, decreases the assembly of the C4b2a complex (one of the two distinct C3 convertases) of the classical and lectin pathways and subsequently the assembly of the C4b2a3b complex (one of the two distinct C5 convertases), and consequently, the assembly of MAC (see Fig. 1 for details). In addition, acting as a cofactor to FI (a serine proteinase) in the proteolytical inactivation of soluble and membrane-bound C4b [21, 32,33,34,35], C4BP prevents the assembly of the classical C3 convertase [36]; and acting as a cofactor to FI in the cleavage of activated complement fragment C3b in the liquid phase, C4BP also suppresses the alternative pathways of the complement activation cascade [37], which is regulated mainly by FH, another soluble complement inhibitor [2]. Further, C4BP increases the dissociation of the enzyme complexes, accelerating the natural decay of the classical C3 convertase (C4b2a complex) [34, 38]. Clearly, C4BP has an important inhibitory effect on complement activation, resulting in significant decreases in the activity levels of C3 and C5 convertases, C3b/iC3b deposition and associated opsonization, release of complement-derived anaphylatoxins (C3a and C5a), and the formation of MAC, and consequently, leading to attenuated lysis and clearance of dying cells, misfolded proteins, and released DNA fragments, as well as C3a-induced and C5a-induced systemic inflammatory reactions [1, 2].
3 C4BPα: Pointers to Complement Inhibition
As a peptide encoded by the gene C4BPA that is localized at 1q32 [39, 40], C4BPα contributes to complement inhibition [37, 41,42,43] when the CCP1-3 domains of C4BPα bind the activated C4b fragments [36, 43,44,45] (see Fig. 2a). Upon binding of C4b to the α-chains, C4b is converted to a substrate for FI, which cleaves and inhibits C4b when it is bound to C4BP [46]. Moreover, the CCP1-4 domains of C4BPα are the binding sites of C3b for its subsequent proteolysis [15] (see Fig. 2a), leading to a decrease in the unbound form of C3b in the blood [37]. Therefore, C4BPα expression levels can negatively regulate the assembly of all C3-convertases and C5-convertases through binding to and degradation of complement activated fragments C4b and C3b (see Figs. 1 and 2). These observations demonstrated that C4BPα exerts its inhibitory effects on the entire complement cascades, directly and indirectly.
4 Crosstalk Between C4BPα and Platelets
It is well known that there are complex interactions between coagulation and complement systems [47,48,49,50,51]. Blood coagulation can activate the complement system through a platelet-dependent mechanism [52, 53]. Platelet activation leads to complement activation [54, 55], and conversely, activated complement components (such as C3a) can induce platelet activation and aggregation [56,57,58], such as in the trauma setting.
4.1 Direct Effects of the Complement Components on Platelet Activation and Aggregation
As discussed above, all three complement pathways converge at the cleavage of C3 to C3a and C3b (see Fig. 1), and complement C3 is thought to be at the center of the complement activation cascade. Therefore, the effects of C3 on platelets can be demonstrated in C3-deficient mice [59]. When compared with wild-type (WT) littermates, C3-deficient mice exhibited pronounced prolongation of tail-cut bleeding time, attenuation of platelet aggregation induced by the PAR4 (protease-activated receptor 4) peptide (but not by ADP or collagen), delay of thrombus formation in arterioles, and a reduction in thrombus stability, but an increase in embolism events [59, 60]. Further, platelets isolated from WT mice aggregate less in C3-deficient plasma than in normal plasma, but the addition of plasma from WT mice or plasma-purified C3 can restore attenuated aggregation of C3-deficient platelets [59]. However, clinical studies have demonstrated that complement activation in healthy donor or trauma sera can trigger significant platelet aggregation in respective platelets when incubated with activated healthy donor or trauma sera (non-activated vs activated) through an increase in SFLLRN (a thrombin receptor-activating peptide)-mediated [Ca2+]i flux in platelets, but that C3a-depleted or C4d-depleted trauma sera lead to a marked reduction in platelet aggregation (depleted vs non-depleted) [58]. These results demonstrated that C3 activation can enhance platelet activation and aggregation, and consequently thrombus formation, but that inhibition of C3 can suppress platelet aggregation and increase the bleeding risk. In other words, C3 is crucial for direct platelet activation, aggregation, and iC3b deposition on the platelet surface as well as fibrin formation in vivo.
4.2 Platelets as a Target of the Complement System via C3aR and C5aR
Platelets contain complement C3 and C5 proteins and messenger RNA that originate from megakaryocytes [61]. C3 and C5 stored in the granules of platelets can be translocated to the platelet surface or even released into the blood upon platelet activation and degranulation [61] (see Fig. 3 for details). Activated platelets can induce activation of the complement cascades in the blood, further potentiating complement activation. When C3 is cleaved to C3a and C3b by C3 convertases, C3 is activated. When added into the gel-filtered or washing human platelet preparations at a concentration of 2 × 10-12 M or 10 µM, C3a or ADP alone failed to induce platelet activation or aggregation; however, concomitant use of C3a and ADP induces platelet aggregation in the gel-filtered platelets synergistically [62]. The anaphylatoxin receptors, C3a receptor (C3aR) and C5a receptors (C5aR1 and C5aR2), are expressed on the surface of platelets [63,64,65], which all exert their effects through distinct signaling pathways [63, 64, 66,67,68,69,70,71,72,73,74,75,76] as summarized in Fig. 3. Via these receptors, platelets bind C3a and C5a present in plasma (hepatocytes as their main source) and consequently, platelets are activated. Further, using C3aR–/– mice or C3aR–/– with re-injection of C3a, C3a–/– mice were less prone to experimental myocardial infarction and stroke, with the bleeding time shortened by C3a after tail injury and thrombosis [63]. Platelet-derived C3aR contributed to experimental stroke or myocardial infarction in mice through the formation of platelet C3aR-mediated thrombosis [63]. After reconstituting C3aR–/– mice with C3aR+/+ platelets or platelet depletion, all the observed changes were confirmed to be a result of the presence or absence of platelet C3aR [63]. Consistent with findings obtained from mice, there was a strong positive correlation between C3aR expression levels and activated GP IIb/IIIa (glycoprotein IIb/IIIa, or integrin αIIbβ3) in platelets of 501 patients with coronary artery disease [63]. These results demonstrated that the C3a/C3aR axis on platelets facilitates platelet activation and thrombus formation.

Main platelet receptors and enzymes that are expressed on the platelet surface when platelets (particularly immature platelets) are activated or opsonized. Platelets bind complement components from plasma, and initiate and enhance plasmatic complement activation. For details, platelets contain complement C3 and C5 proteins and RNA molecules, all of which originate from megakaryocytes directly. Completment receptors, such as C3aR, C5aR, and C1qR, are expressed on the platelet surface. When platelets are activated, P-selectin (CD62P) on the platelet surface can bind C3b, leading to increased assembly of C3 convertase in combination with factor B, and consequently to triggering of the assembly of MAC (C5b-9) on the platelet surface. In addition, collagen and fibrinogen in plasma bind the platelet surface receptors GPVI and GPIIb/IIIa, respectively, activating such platelets. C3 and C5 stored in the α and dense granules of the platelets can be translocated to the platelet surface or even released into the blood upon platelet activation and degranulation. Moreover, platelets also bind C3a and C5a present in plasma (hepatocytes as their main source) via complement receptors (C3aR, C5aR1, and C5aR2) expressed on the platelet surface and consequently, platelets are activated. Activated platelets can induce activation of the complement cascades in the blood, further enhancing complement activation. Platelets also express some complement regulatory molecules, such as factor H, CD55 (decay-accelerating factor), and CD59, all of which can help prevent excessive complement activation on the platelet surface. In addition to suppression of binding of cluster of differentiation ligand 40 (CD40L) to CD40, C4BPα can decrease the assembly of the C3 convertase and C5 convertase in the blood, and decrease the generation of C3a and C5a, and the assembly of C5b-9. All information and evidence presented in this figure are retrieved from, but not limited to, the references [63, 64, 66,67,68,69,70,71,72,73,74,75,76]. ADP adenosine diphosphate, Akt serine-threonine kinase, protein kinase B, or PKB, AP-1 activator protein-1, a transcription factor, CD cluster of differentiation, CXCL C-X-C motif chemokine ligand, CXCR C-X-C chemokine receptor, ERK extracellular signal-regulated kinase, or extracellular stimuli response kinase, IL-1β interleukin-1 beta, IL-6 interleukin-6, GP glycoprotein, MAPK mitogen-activated protein kinase, NOX NADPH oxidase, NFκB nuclear factor κB, P2Y purinergic receptor family, PDI protein disulfide isomerase, PI3K phosphoinositol 3-kinase, Rap1b a small GTPase, abundantly expressed in platelets, Rap1b-GTP active form, Rap1b-GDP inactive form, PAR protease-activatd receptor, ROS reactive oxygen species, Spa-1 signal-induced proliferation-associated gene-1, a Rap1 GTPase-activating protein, STAT3 signal transduction and transcription activator 3, TNFα tumor necrosis factor alpha; TNFR tumor necrosis factor receptor, vWF von Willebrand factor
In line with the fact that the activation of complement component C5 is C3 dependent (see Fig. 1), initial membrane insertion of downstream non-lytic complement component C5b-7 is C5 dependent [60]. In addition to the generation of anaphylatoxin C5a (an endogenous ligand for C5aR), proteolytic activation of C5 also generates C5b that initiates MAC formation. The formation of the MAC then enhances platelet activation and subsequent hemostatic responses [56], which are suppressed by clusterin [77]. However, compared with WT mice, C5-deficent mice have no apparent defect in platelet or leukocyte interactions at the flow-restricted vessel wall, but their fibrin deposition is abnormal, which is similar to the effect seen in mice treated with rutin, an inhibitor of protein disulfide isomerase [60].
As summarized in Fig. 3, in addition to the presence of the intrinsic capacity of stimulated platelets to activate complement, such as C3b-bound P-selectin (CD62P) that leads to the generation of C3a and the assembly of C5b-9 [54], platelets also become the targets of complement activation in the settings that induce complement activation on the platelet surface when exposed to plasma or serum, a weak platelet aggregation agonist (ADP or epinephrine), a potent agonist (arachidonic acid, and thrombin or its receptor activation peptide), or even shear stress alone or in combination with more than one stimulus or agonist [49]. Thus, inhibition of the generation of anaphylactic peptides C3a and C5a (by recombinant or endogenous C4BPα) as well as a blockade of C3aR and C5aR expressed on platelets (by eculizumab or ravulizumab) can exert their antiplatelet effects, suppressing platelet activation and aggregation as well as thrombus formation [1, 63].
5 C4BPα as a Novel Biomarker to Predict Attenuated Platelet Response to Clopidogrel
As discussed above, C4BPα exhibits its inhibitory effects on complement cascades through prevention of the assembly of C3-convertases and C5-convertases and by facilitating the decay of these two convertases (see Fig. 1), decreasing the formation of the complement cleavage products C3a, C3b, iC3b, C5a, and C5b, suppressing the deposition of C3b, iC3b, C4d, and C5b-9 on the surface of cells, including activated or opsonized platelets. Thus, it is not difficult to understand that C4BPα exerts its antiplatelet effects to some extent through suppression of C3a-induced platelet aggregation in humans [57, 62]. However, C4BPα also exhibits certain anti-inflammatory effects, which is strongly supported by a dramatic increase in TNFα levels released when complement is over-activated, or when complement inhibition is impaired in the presence of C4BP-deficient sera [31]. Further, proinflammatory cytokines, including TNFα, IL-1β, and IL-6, are generally associated with the suppression of most (if not all) drug-metabolizing cytochrome P450 enzymes at the protein expression and/or catalytical activity levels across assay systems [78,79,80], which are known to be responsible for the metabolic activation of clopidogrel (an inactive prodrug itself) in the liver [81, 82]. It might be inferred that patients could exhibit more potent platelet inhibition of clopidogrel because of an increased formation of clopidogrel active metabolite (predominantly H4) in the liver when their plasma C4BPα levels are elevated and/or plasma levels of proinflammatory cytokines, such as TNFα and IL-6, are decreased. However, in addition to impaired metabolic activation of clopidogrel, altered function of platelet ADP receptor P2Y12 itself may also contribute to clopidogrel resistance in patients with type 2 diabetes mellitus or in those of Caucasian descent [83]. In addition, clinical research studies indicated that C4BP negatively regulates the classical complement pathway in human atherosclerotic lesions, and may be involved in the clean-up of apoptotic cells and cell debris in the arterial intima [84], suggesting a beneficial effect on coronary artery disease as an endogenous protein. Accordingly, it was hypothesized that plasma C4BPα levels could be used to predict clopidogrel resistance in clinical settings. In a cohort of 88 patients undergoing coronary stenting, then receiving clopidogrel at a maintenance dose of 75 mg/day for at least 6 months to prevent in-stent thrombosis and/or recurrent ischemic events, clopidogrel resistance was defined with ADP-induced whole-blood platelet aggregation of 10 ohm (Ω) or above. Then, subgrouping of patients sensitive or resistant to clopidogrel was performed, and the assay was evaluated with an area under the receiver operating characteristic curve (also known as AUC) of 0.852, sensitivity (true-positive) of 88.6%, and specificity (true-negative) of 75%, respectively (unpublished data). C4BPα was patented in 2019 as a novel biomarker [85], and the plasma C4BPα assay is being explored for validation for future clinical use. In terms of the nature of its quantitative measures, circulating C4BPα would become a promising biomarker to predict clopidogrel resistance in patients when taking clopidogrel [85], although the causes that lead to clopidogrel resistance seem to be inclusive [86,87,88,89,90].
6 C4BPα as a Biomarker to Predict Other Severe or Life-Threatening Clinical Conditions
Increasing evidence demonstrated that, in addition to predicting clopidogrel resistance, C4BPα can be used as a potential biomarker in other clinical settings, including but not limited to, non-alcoholic fatty liver disease [91], non-small cell lung cancer [92], pancreatic cancer [93], lymph node metastasis in pancreatic ductal adenocarcinoma [94], ovarian clear cell carcinoma [95], relapse in pediatric/adolescent Hodgkin lymphoma [96], latent tuberculosis infection [97], spinal dural arteriovenous fistula [98], and serum resistance to or complement immune evasion of certain infections [99, 100]. Of these, possible explanations were that triglycerid content might increase with complement activation because of down-regulation of C4BPA [91], but that resistance of cancer cells to humoral and phagolytic immune responses might increase with C4BPA expression levels at the earlier stage of disease [92,93,94]. As these data have all been derived from pilot studies, further clinical evaluation and validation studies need to be performed in their respective settings before they will be translated to patient care.
Recently, evidence demonstrated that the severe acute respiratory syndrome coronavirus 2 spike protein induces dysfunction of lung endothelial cells and formation of thromboinflammation, which are dependent on C3a/C3aR signaling [101]. As discussed above, in addition to respective suppression of the assembly of C3-convertase and C5-convertase, C4BPα suppresses the complement system by multiple known and unknown mechanisms, and consequently, decreases, in turn, the assembly of the common membrane attack complex (C5b-9), the formation of cytolytic pore on the membrane, and lysis of opsonized microbes, including severe acute respiratory syndrome coronavirus 2 viruses. Theoretically, elevated plasma C4BPα levels exhibit an attenuated virus killing or viral clearance (eradication) effect through suppression of complement defense capacity. However, C4BPα can decrease the formation of C3a, C5a, and proinflammatory cytokines, including TNFα, and thus, suppresses the “cytokine storm” that frequently occurs in patients infected with severe acute respiratory syndrome coronavirus 2 in a complement activation-independent and/or-dependent manner [102,103,104,105].
7 Conclusions and Future Perspectives
As an abundant complement inhibitor in the blood, C4BP, particularly C4BPα, exerts its unique innate immune regulatory functions in the suppression of complement activation, opsonization, complement deposition, generation of the anaphylactic peptides C3a and C5a, platelet activation and aggregation, microbial killing, inflammation, oxidative stress, and more to be explored. In clinical practice, many diseases are associated with rampant complement activation, including coronary artery disease and stroke. Thus, suppression of complement activation will be an effective therapeutic strategy. Emerging novel small-molecule drugs and biologics (referred to as complement-targeted drugs), including C3 or C5 inhibitors, as well as C3aR or C5aR antagonists, are being developed, modified, evaluated, or validated to reverse complement dysfunction or over-activation [1]. In fact, “Complement Revolution” came of age when eculizumab, a monoclonal antibody that inhibits the cleavage of C5, was marketed in 2007 [1]. Thereafter, the major complement-targeted therapy strategy is inhibition of a C3 or C5 or blockade of C3aR or C5aR [105], such as ravulizumab (a monoclonal antibody that inhibits the cleavage of C5), BDB-001 (anti-C5a monoclonal antibody), AMY-101 (the first C3 inhibitor), and ALXN1210 (a long-acting C5 inhibitor). There is a wide individual variation in plasma C4BPα levels owing to the nature of its acute-phase reactants, and therefore, plasma C4BPα levels may be a promising biomarker to predict alterations in platelet response to clopidogrel in patients with coronary artery disease or ischemic stroke and beyond.
References
Garred P, Tenner AJ, Mollnes TE. Therapeutic targeting of the complement system: from rare diseases to pandemics. Pharmacol Rev. 2021;73:792–827. https://doi.org/10.1124/pharmrev.120.000072.
Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90. https://doi.org/10.1016/j.it.2008.11.003.
Ermert D, Blom AM. C4b-binding protein: the good, the bad and the deadly. Novel functions of an old friend. Immunol Lett. 2016;169:82–92. https://doi.org/10.1016/j.imlet.2015.11.014.
Marcovina SM, Zoppo A, Vigano-Angelo S, Di CG, D’Angelo A. Determination of serum levels of complement component C4b-binding protein: influence of age and inflammation. Int J Clin Lab Res. 1991;21:171–5. https://doi.org/10.1007/BF02591638.
Dahlback B, Smith CA, Muller-Eberhard HJ. Visualization of human C4b-binding protein and its complexes with vitamin K-dependent protein S and complement protein C4b. Proc Natl Acad Sci U S A. 1983;80:3461–5. https://doi.org/10.1073/pnas.80.11.3461.
Barnum SR, Dahlback B. C4b-binding protein, a regulatory component of the classical pathway of complement, is an acute-phase protein and is elevated in systemic lupus erythematosus. Complement Inflamm. 1990;7:71–7. https://doi.org/10.1159/000463131.
Dahlback B. Purification of human C4b-binding protein and formation of its complex with vitamin K-dependent protein S. Biochem J. 1983;209:847–56. https://doi.org/10.1042/bj2090847.
Zoller B, de Garcia FP, Dahlback B. Evaluation of the relationship between protein S and C4b-binding protein isoforms in hereditary protein S deficiency demonstrating type I and type III deficiencies to be phenotypic variants of the same genetic disease. Blood. 1995;85:3524–31.
Chung LP, Bentley DR, Reid KB. Molecular cloning and characterization of the cDNA coding for C4b-binding protein, a regulatory protein of the classical pathway of the human complement system. Biochem J. 1985;230:133–41. https://doi.org/10.1042/bj2300133.
Nonaka MI, Hishikawa Y, Moriyama N, Koji T, Ogata RT, Kudo A, et al. Complement C4b-binding protein as a novel murine epididymal secretory protein. Biol Reprod. 2003;69:1931–9. https://doi.org/10.1095/biolreprod.103.020289.
Aso T, Okamura S, Matsuguchi T, Sakamoto N, Sata T, Niho Y. Genomic organization of the alpha chain of the human C4b-binding protein gene. Biochem Biophys Res Commun. 1991;174:222–7. https://doi.org/10.1016/0006-291x(91)90509-6.
Morris KM, Aden DP, Knowles BB, Colten HR. Complement biosynthesis by the human hepatoma-derived cell line HepG2. J Clin Invest. 1982;70:906–13. https://doi.org/10.1172/jci110687.
de Cordoba SR, Sanchez-Corral P, Rey-Campos J. Structure of the gene coding for the alpha polypeptide chain of the human complement component C4b-binding protein. J Exp Med. 1991;173:1073–82. https://doi.org/10.1084/jem.173.5.1073.
Ithier MC, Parobchak N, Yadava S, Cheng J, Wang B, Rosen T. Fetal lung C4BPA induces p100 processing in human placenta. Sci Rep. 2019;9:5519. https://doi.org/10.1038/s41598-019-42078-0.
Dahlback B. C4b-binding protein: a forgotten factor in thrombosis and hemostasis. Semin Thromb Hemost. 2011;37:355–61. https://doi.org/10.1055/s-0031-1276584.
Hillarp A, Dahlback B. Cloning of cDNA coding for the beta chain of human complement component C4b-binding protein: sequence homology with the alpha chain. Proc Natl Acad Sci U S A. 1990;87:1183–7. https://doi.org/10.1073/pnas.87.3.1183.
Kask L, Hillarp A, Ramesh B, Dahlback B, Blom AM. Structural requirements for the intracellular subunit polymerization of the complement inhibitor C4b-binding protein. Biochemistry. 2002;41:9349–57. https://doi.org/10.1021/bi025980.
Blom AM, Villoutreix BO, Dahlback B. Complement inhibitor C4b-binding protein: friend or foe in the innate immune system? Mol Immunol. 2004;40:1333–46. https://doi.org/10.1016/j.molimm.2003.12.002.
Hillarp A, Dahlback B. Novel subunit in C4b-binding protein required for protein S binding. J Biol Chem. 1988;263:12759–64.
Hillarp A, Hessing M, Dahlback B. Protein S binding in relation to the subunit composition of human C4b-binding protein. FEBS Lett. 1989;259:53–6. https://doi.org/10.1016/0014-5793(89)81492-9.
Scharfstein J, Ferreira A, Gigli I, Nussenzweig V. Human C4-binding protein. I. Isolation and characterization. J Exp Med. 1978;148:207–22. https://doi.org/10.1084/jem.148.1.207.
Sanchez-Corral P, Criado GO, de Cordoba SR. Isoforms of human C4b-binding protein. I. Molecular basis for the C4BP isoform pattern and its variations in human plasma. J Immunol. 1995;155:4030–6.
de Garcia FP, Alim RI, Hardig Y, Zoller B, Dahlback B. Differential regulation of alpha and beta chains of C4b-binding protein during acute-phase response resulting in stable plasma levels of free anticoagulant protein S. Blood. 1994;84:815–22.
Criado GO, Sanchez-Corral P, de Cordoba SR. Isoforms of human C4b-binding protein. II. Differential modulation of the C4BPA and C4BPB genes by acute phase cytokines. J Immunol. 1995;155:4037–43.
Saeki T, Hirose S, Nukatsuka M, Kusunoki Y, Nagasawa S. Evidence that C4b-binding protein is an acute phase protein. Biochem Biophys Res Commun. 1989;164:1446–51. https://doi.org/10.1016/0006-291x(89)91832-9.
Funakoshi M, Sasaki J, Arakawa K. Proline-rich protein is a glycoprotein and an acute phase reactant. Biochim Biophys Acta. 1988;963:98–108. https://doi.org/10.1016/0005-2760(88)90342-6.
Criado-Garcia O, Gonzalez-Rubio C, Lopez-Trascasa M, Pascual-Salcedo D, Munuera L, de Cordoba SR. Modulation of C4b-binding protein isoforms during the acute phase response caused by orthopedic surgery. Haemostasis. 1997;27:25–34. https://doi.org/10.1159/000217430.
Moffat GJ, Tack BF. Regulation of C4b-binding protein gene expression by the acute-phase mediators tumor necrosis factor-alpha, interleukin-6, and interleukin-1. Biochemistry. 1992;31:12376–84. https://doi.org/10.1021/bi00164a012.
Castell JV, Gomez-Lechon MJ, David M, Hirano T, Kishimoto T, Heinrich PC. Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett. 1988;232:347–50. https://doi.org/10.1016/0014-5793(88)80766-x.
Perlmutter DH, Dinarello CA, Punsal PI, Colten HR. Cachectin/tumor necrosis factor regulates hepatic acute-phase gene expression. J Clin Invest. 1986;78:1349–54. https://doi.org/10.1172/JCI112721.
Trouw LA, Nilsson SC, Goncalves I, Landberg G, Blom AM. C4b-binding protein binds to necrotic cells and DNA, limiting DNA release and inhibiting complement activation. J Exp Med. 2005;201:1937–48. https://doi.org/10.1084/jem.20050189.
Fujita T, Gigli I, Nussenzweig V. Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. J Exp Med. 1978;148:1044–51. https://doi.org/10.1084/jem.148.4.1044.
Fujita T, Nussenzweig V. The role of C4-binding protein and beta 1H in proteolysis of C4b and C3b. J Exp Med. 1979;150:267–76. https://doi.org/10.1084/jem.150.2.267.
Gigli I, Fujita T, Nussenzweig V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc Natl Acad Sci USA. 1979;76:6596–600. https://doi.org/10.1073/pnas.76.12.6596.
Nagasawa S, Stroud RM. Purification and characterization of a macromolecular weight cofactor for C3b-inactivator, C4bC3bINA-cofactor, of human plasma. Mol Immunol. 1980;17:1365–72. https://doi.org/10.1016/0161-5890(80)90005-x.
Fukui A, Yuasa-Nakagawa T, Murakami Y, Funami K, Kishi N, Matsuda T, et al. Mapping of the sites responsible for factor I-cofactor activity for cleavage of C3b and C4b on human C4b-binding protein (C4bp) by deletion mutagenesis. J Biochem. 2002;132:719–28. https://doi.org/10.1093/oxfordjournals.jbchem.a003279.
Blom AM, Kask L, Dahlback B. CCP1-4 of the C4b-binding protein alpha-chain are required for factor I mediated cleavage of complement factor C3b. Mol Immunol. 2003;39:547–56. https://doi.org/10.1016/s0161-5890(02)00213-4.
Daha MR, van Es LA. Relative resistance of the F-42-stabilized classical pathway C3 convertase to inactivation by C4-binding protein. J Immunol. 1980;125:2051–4.
Andersson A, Dahlback B, Hanson C, Hillarp A, Levan G, Szpirer J, et al. Genes for C4b-binding protein alpha- and beta-chains (C4BPA and C4BPB) are located on chromosome 1, band 1q32, in humans and on chromosome 13 in rats. Somat Cell Mol Genet. 1990;16:493–500. https://doi.org/10.1007/BF01233199.
Lublin DM, Liszewski MK, Post TW, Arce MA, Le Beau MM, Rebentisch MB, et al. Molecular cloning and chromosomal localization of human membrane cofactor protein (MCP): evidence for inclusion in the multigene family of complement-regulatory proteins. J Exp Med. 1988;168:181–94. https://doi.org/10.1084/jem.168.1.181.
Blom AM, Webb J, Villoutreix BO, Dahlback B. A cluster of positively charged amino acids in the C4BP alpha-chain is crucial for C4b binding and factor I cofactor function. J Biol Chem. 1999;274:19237–45. https://doi.org/10.1074/jbc.274.27.19237.
Blom AM, Zadura AF, Villoutreix BO, Dahlback B. Positively charged amino acids at the interface between alpha-chain CCP1 and CCP2 of C4BP are required for regulation of the classical C3-convertase. Mol Immunol. 2000;37:445–53. https://doi.org/10.1016/s0161-5890(00)00059-6.
Blom AM, Kask L, Dahlback B. Structural requirements for the complement regulatory activities of C4BP. J Biol Chem. 2001;276:27136–44. https://doi.org/10.1074/jbc.M102445200.
Hardig Y, Hillarp A, Dahlback B. The amino-terminal module of the C4b-binding protein alpha-chain is crucial for C4b binding and factor I-cofactor function. Biochem J. 1997;323:469–75. https://doi.org/10.1042/bj3230469.
Ogata RT, Mathias P, Bradt BM, Cooper NR. Murine C4b-binding protein. Mapping of the ligand binding site and the N-terminus of the pre-protein. J Immunol. 1993;150:2273–80.
Blom AM. Structural and functional studies of complement inhibitor C4b-binding protein. Biochem Soc Trans. 2002;30:978–82. https://doi.org/10.1042/bst0300978.
Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71–9. https://doi.org/10.1007/978-0-387-78952-1_6.
Verschoor A, Langer HF. Crosstalk between platelets and the complement system in immune protection and disease. Thromb Haemost. 2013;110:910–9. https://doi.org/10.1160/TH13-02-0102.
Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. 2010;47:2170–5. https://doi.org/10.1016/j.molimm.2010.05.009.
Zimmerman TS, Muller-Eberhard HJ. Complement-induced platelet protein alterations. Science. 1973;180:1183–5. https://doi.org/10.1126/science.180.4091.1183.
Boyce S, Eren E, Lwaleed BA, Kazmi RS. The activation of complement and its role in the pathogenesis of thromboembolism. Semin Thromb Hemost. 2015;41:665–72. https://doi.org/10.1055/s-0035-1556732.
Kalowski S, Howes EL Jr, Margaretten W, McKay DG. Effects of intravascular clotting on the activation of the complement system: the role of the platelet. Am J Pathol. 1975;78:525–36.
Siraganian RP. Platelet requirement in the interaction of the complement and clotting systems. Nat New Biol. 1972;239:208–10. https://doi.org/10.1038/newbio239208a0.
del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–9. https://doi.org/10.1084/jem.20041497.
Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. 2006;4:2035–42. https://doi.org/10.1111/j.1538-7836.2006.02065.x.
Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol Today. 1991;12:338–42. https://doi.org/10.1016/0167-5699(91)90012-I.
Polley MJ, Nachman R. The human complement system in thrombin-mediated platelet function. J Exp Med. 1978;147:1713–26. https://doi.org/10.1084/jem.147.6.1713.
Atefi G, Aisiku O, Shapiro N, Hauser C, Dalle LJ, Flaumenhaft R, et al. Complement activation in trauma patients alters platelet function. Shock. 2016;46:83–8. https://doi.org/10.1097/SHK.0000000000000675.
Gushiken FC, Han H, Li J, Rumbaut RE, Afshar-Kharghan V. Abnormal platelet function in C3-deficient mice. J Thromb Haemost. 2009;7:865–70. https://doi.org/10.1111/j.1538-7836.2009.03334.x.
Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129:2291–302. https://doi.org/10.1182/blood-2016-11-749879.
Arbesu I, Bucsaiova M, Fischer MB, Mannhalter C. Platelet-borne complement proteins and their role in platelet-bacteria interactions. J Thromb Haemost. 2016;14:2241–52. https://doi.org/10.1111/jth.13495.
Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med. 1983;158:603–15. https://doi.org/10.1084/jem.158.2.603.
Sauter RJ, Sauter M, Reis ES, Emschermann FN, Nording H, Ebenhoch S, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation. 2018;138:1720–35. https://doi.org/10.1161/CIRCULATIONAHA.118.034600.
Sauter RJ, Sauter M, Obrich M, Emschermann FN, Nording H, Patzelt J, et al. Anaphylatoxin receptor C3aR contributes to platelet function, thrombus formation and in vivo haemostasis. Thromb Haemost. 2019;119:179–82. https://doi.org/10.1055/s-0038-1676349.
Patzelt J, Mueller KA, Breuning S, Karathanos A, Schleicher R, Seizer P, et al. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis. 2015;238:289–95. https://doi.org/10.1016/j.atherosclerosis.2014.12.002.
Yadav MK, Maharana J, Yadav R, Saha S, Sarma P, Soni C, et al. Molecular basis of anaphylatoxin binding, activation, and signaling bias at complement receptors. Cell. 2023;186:4956-73.e21. https://doi.org/10.1016/j.cell.2023.09.020.
Wang Y, Liu W, Xu Y, He X, Yuan Q, Luo P, et al. Revealing the signaling of complement receptors C3aR and C5aR1 by anaphylatoxins. Nat Chem Biol. 2023;19:1351–60. https://doi.org/10.1038/s41589-023-01339-w.
Ghosh M, Rana S. The anaphylatoxin C5a: structure, function, signaling, physiology, disease, and therapeutics. Int Immunopharmacol. 2023;118: 110081. https://doi.org/10.1016/j.intimp.2023.110081.
Bamberg CE, Mackay CR, Lee H, Zahra D, Jackson J, Lim YS, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem. 2010;285:7633–44. https://doi.org/10.1074/jbc.M109.092106.
Zarantonello A, Revel M, Grunenwald A, Roumenina LT. C3-dependent effector functions of complement. Immunol Rev. 2023;313:120–38. https://doi.org/10.1111/imr.13147.
Luo S, Xu H, Gong X, Shen J, Chen X, Wu Z. The complement C3a–C3aR and C5a–C5aR pathways promote viability and inflammation of human retinal pigment epithelium cells by targeting NF-κB signaling. Exp Ther Med. 2022;24:493. https://doi.org/10.3892/etm.2022.11420.
Li XX, Woodruff TM. Protocol for cell-based screening assay to measure ERK1/2 phosphorylation as a readout for complement receptor activation. STAR Protoc. 2023;4: 102758. https://doi.org/10.1016/j.xpro.2023.102758.
Wang Y, Zhang H, He YW. The complement receptors C3aR and C5aR are a new class of immune checkpoint receptor in cancer immunotherapy. Front Immunol. 2019;10:1574. https://doi.org/10.3389/fimmu.2019.01574.
Shu C, Zha H, Long H, Wang X, Yang F, Gao J, et al. C3a–C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J Exp Clin Cancer Res. 2020;39:11. https://doi.org/10.1186/s13046-019-1515-2.
Yin H, Stojanovic A, Hay N, Du X. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. 2008;111:658–65. https://doi.org/10.1182/blood-2007-04-085514.
Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Immunol. 2000;165:5406–9. https://doi.org/10.4049/jimmunol.165.10.5406.
Rosenberg ME, Silkensen J. Clusterin: physiologic and pathophysiologic considerations. Int J Biochem Cell Biol. 1995;27:633–45. https://doi.org/10.1016/1357-2725(95)00027-m.
Stipp MC, Acco A. Involvement of cytochrome P450 enzymes in inflammation and cancer: a review. Cancer Chemother Pharmacol. 2021;87:295–309. https://doi.org/10.1007/s00280-020-04181-2.
Frye RF, Schneider VM, Frye CS, Feldman AM. Plasma levels of TNF-α and IL-6 are inversely related to cytochrome P450-dependent dryg metabolism in patients with congestive heart failure. J Card Fail. 2002;8:315–9. https://doi.org/10.1054/jcaf.2002.127773.
Yu Y, Henrich C, Wang D. Assessment of the drug-drug interaction potential for therapeutic proteins with pro-inflammatory activities. Clin Transl Sci. 2023;16:922–36. https://doi.org/10.1111/cts.13507.
Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38:92–9. https://doi.org/10.1124/dmd.109.029132.
Xie HG, Zou JJ, Hu ZY, Zhang JJ, Ye F, Chen SL. Individual variability in the disposition of and response to clopidogrel: pharmacogenomics and beyond. Pharmacol Ther. 2011;129:267–89. https://doi.org/10.1016/j.pharmthera.2010.10.001.
Cleator JH, Duvernay MT, Holinstat M, Colowick NE, Hudson WJ, Song Y, et al. Racial differences in resistance to P2Y12 receptor antagonists in type 2 diabetic subjects. J Pharmacol Exp Ther. 2014;351:33–43. https://doi.org/10.1124/jpet.114.215616.
Oksjoki R, Kovanen PT, Mäyränpää MI, Laine P, Blom AM, Meri S, et al. Complement regulation in human atherosclerotic coronary lesions. Immunohistochemical evidence that C4b-binding protein negatively regulates the classical complement pathway, and that C5b–9 is formed via the alternative complement pathway. Atherosclerosis. 2007;192:40–8. https://doi.org/10.1016/j.atherosclerosis.2006.06.013.
Xie HG, Ji JZ, Huang BB, Tai T, Mi QY, Xie WJ. Human serum C4b-binding protein alpha (C4BPα) as a biomarker to predict cloidogrel resistance and its clinical applications (in Chinese). In: Patent No. ZL201811515436.9. China National Intellectual Property Administration (CNIPA). 2019. URL: http://epub.cnipa.gov.cn/. [Accessed 3 Aug 2023]. If interested, just inputting the application number of the patent (201811515436.9) into the search frame on that web page and clicking the search icon.
Serebruany VL. The “clopidogrel resistance” trap. Am J Cardiol. 2007;100:1044–6. https://doi.org/10.1016/j.amjcard.2007.04.050.
Wang D, Yang XH, Zhang JD, Li RB, Jia M, Cui XR. Compared efficacy of clopidogrel and ticagrelor in treating acute coronary syndrome: a meta-analysis. BMC Cardiovasc Disord. 2018;18:217. https://doi.org/10.1186/s12872-018-0948-4.
Aswathy SPV, Chandra KR, Jyothikrishna P, Arun KP. Dosage optimization of clopidogrel via a precision medicine approach: the way forward. Pharmacogenomics. 2022;23:195–206. https://doi.org/10.2217/pgs-2020-0198.
Alkattan A, Alkhalifah A, Alsalameen E, Alghanim F, Radwan N. Polymorphisms of genes related to phase II metabolism and resistance to clopidogrel. Pharmacogenomics. 2022;23:61–79. https://doi.org/10.2217/pgs-2021-0092.
Neubauer H, Kaiser AFC, Endres HG, Krüger JC, Engelhardt A, Lask S, et al. Tailored antiplatelet therapy can overcome clopidogrel and aspirin resistance: the BOchum CLopidogrel and Aspirin Plan (BOCLA-Plan) to improve antiplatelet therapy. BMC Med. 2011;9:3. https://doi.org/10.1186/1741-7015-9-3.
Shafiha R, Bahcivanci B, Gkoutos GV, Acharjee A. Machine learning-based identification of potentially novel non-alcoholic fatty liver disease biomarkers. Biomedicines. 2021;9:1636. https://doi.org/10.3390/biomedicines9111636.
Luo X, Liu Y, Wang R, Hu H, Zeng R, Chen H. A high-quality secretome of A549 cells aided the discovery of C4b-binding protein as a novel serum biomarker for non-small cell lung cancer. J Proteom. 2011;74:528–38. https://doi.org/10.1016/j.jprot.2011.01.011.
Sogawa K, Takano S, Iida F, Satoh M, Tsuchida S, Kawashima Y, et al. Identification of a novel serum biomarker for pancreatic cancer, C4b-binding protein alpha-chain (C4BPA) by quantitative proteomic analysis using tandem mass tags. Br J Cancer. 2016;115:949–56. https://doi.org/10.1038/bjc.2016.295.
Sogawa K, Yamanaka S, Takano S, Sasaki K, Miyahara Y, Furukawa K, et al. Fucosylated C4b-binding protein alpha-chain, a novel serum biomarker that predicts lymph node metastasis in pancreatic ductal adenocarcinoma. Oncol Lett. 2021;21:127. https://doi.org/10.3892/ol.2020.12388.
Mikami M, Tanabe K, Matsuo K, Miyazaki Y, Miyazawa M, Hayashi M, et al. Fully-sialylated alpha-chain of complement 4-binding protein: diagnostic utility for ovarian clear cell carcinoma. Gynecol Oncol. 2015;139:520–8. https://doi.org/10.1016/j.ygyno.2015.10.012.
Repetto O, Caggiari L, De ZM, Elia C, Mussolin L, Buffardi S, et al. Quantitative plasma proteomics to identify candidate biomarkers of relapse in pediatric/adolescent Hodgkin lymphoma. Int J Mol Sci. 2022;23:9911. https://doi.org/10.3390/ijms23179911.
Du Y, Xin H, Cao X, Liu Z, He Y, Zhang B, et al. Association between plasma exosomes S100A9/C4BPA and latent tuberculosis infection treatment: proteomic analysis based on a randomized controlled study. Front Microbiol. 2022;13: 934716. https://doi.org/10.3389/fmicb.2022.934716.
Wang Y, Ma Y, Yang C, Huang X, Yang K, Lan F, et al. Potential biomarkers of spinal dural arteriovenous fistula: C4BPA and C1QA. J Neuroinflammation. 2022;19:165. https://doi.org/10.1186/s12974-022-02522-x.
Castiblanco-Valencia MM, Fraga TR, Pagotto AH, Serrano SM, Abreu PA, Barbosa AS, et al. Plasmin cleaves fibrinogen and the human complement proteins C3b and C5 in the presence of Leptospira interrogans proteins: a new role of LigA and LigB in invasion and complement immune evasion. Immunobiology. 2016;221:679–89. https://doi.org/10.1016/j.imbio.2016.01.001.
Potempa M, Potempa J, Okroj M, Popadiak K, Eick S, Nguyen KA, et al. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J Immunol. 2008;181:5537–44. https://doi.org/10.4049/jimmunol.181.8.5537.
Perico L, Morigi M, Pezzotta A, Locatelli M, Imberti B, Corna D, et al. SARS-CoV-2 spike protein induces lung endothelial cell dysfunction and thrombo-inflammation depending on the C3a/C3a receptor signaling. Sci Rep. 2023;13:11392. https://doi.org/10.1038/s41598-023-38382-5.
Beirag N, Varghese PM, Neto MM, Al Aiyan A, Khan HA, Qablan M, et al. Complement activation-independent attenuation of SARS-CoV-2 infection by C1q and C4b-binding protein. Viruses. 2023;15:1269. https://doi.org/10.3390/v15061269.
Pius-Sadowska E, Niedzwiedz A, Kulig P, Baumert B, Sobus A, Roginska D, et al. CXCL8, CCL2, and CMV seropositivity as new prognostic factors for a severe COVID-19 course. Int J Mol Sci. 2022;23:11338. https://doi.org/10.3390/ijms231911338.
di Flora DC, Dionizio A, Pereira HABS, Garbieri TF, Grizzo LT, Dionisio TJ, et al. Analysis of plasma proteins involved in inflammation, immune response/complement system, and blood coagulation upon admission of COVID-19 patients to hospital may help to predict the prognosis of the disease. Cells. 2023;12:1601. https://doi.org/10.3390/cells12121601.
Li Y, Wang H. Complement as new immunotherapy target: past, present, and future. In: Feng X, Xie HG, Malhotra A, Yang CF, editors. Biologics and biosimilars: drug discovery and clinical applications. 1st ed. Boca Raton (FL, USA) and Abingdon (Oxon, UK): Taylor & Francis Group, LLC, CRC Press; 2022. p. 341–69.
Acknowledgments
We appreciate Mr. Michael B. Xie, at the University of Massachusetts Amherst (UMass), Amherst, MA, USA, for his critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the grants for the National Natural Science Foundation of China (82073941), the Research Project of the Social Development of the Province of Jiangsu (BE2021603), the Research Project of the Development of Health Science and Technology of the City of Nanjing (ZKX20035), and a launching grant of Nanjing First Hospital (31010300010339), China (all grants to Hong-Guang Xie). In addition, Hong-Guang Xie is the recipient of the Distinguished Medical Experts of the Province of Jiangsu, China.
Conflicts of Interest
PoonBio Co. Ltd. (Nanjing Puling Shengwu), Nanjing, Jiangsu Province, China is currently a holder of the patent (Patent No. ZL201811515436.9; see [85]), which was transferred from its former owner (Nanjing First Hospital), Nanjing, Jiangsu, China, where Hong-Guang Xie, Ting Tai, Jin-Zi Ji, and Qiong-Yu Mi were inventors of that patent.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
The data that support the findings of this study are available on request from the corresponding author. However, for the data of others, data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Code Availability
Not applicable.
Author Contributions
H-GX had the idea for the article, and drafted, revised, and finalized the manuscript; L-PJ prepared all the figures; and all co-authors critically read and revised the work.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xie, HG., Jiang, LP., Tai, T. et al. The Complement System and C4b-Binding Protein: A Focus on the Promise of C4BPα as a Biomarker to Predict Clopidogrel Resistance. Mol Diagn Ther 28, 189–199 (2024). https://doi.org/10.1007/s40291-023-00691-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00691-w