
Abstract
The emergence of the versatile gene-editing technology using programmable sequence-specific endonuclease system (CRISPR-Cas9) has instigated a major upheaval in biomedical research. In a brief span of time, CRISPR/Cas has been adopted by research labs around the globe because of its potential for significant progress and applicability in terms of efficiency, versatility and simplicity. It is a breakthrough technique for systematic genetic engineering, genome labelling, epigenetic and transcriptional modulation, and multiplexed gene editing, amongst others. This review provides an illustrative overview of the current research trends using CRISPR/Cas technology. We highlight the latest developments in CRISPR/Cas technique including CRISPR imaging, discovery of novel CRISPR systems, and applications in altering the genome, epigenome or RNA in different organisms. Finally, we address the potential challenges of this technique for its future use.
Graphic Abstract

Development of new CRISPR/Cas systems
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In the past decade, the exploitation of the CRISPR/Cas technique has swiftly eclipsed the ZNF and TALEN gene-editing tools. |
CRSIPR/Cas may result in transformative therapies based on the creation of permanent and inheritable changes in the human genome. |
The varying functionality of CRISPR systems have allowed researchers to pursue the therapeutic potential of molecular genetics in relation to disease progression. |
1 Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein (Cas) system, found in 90% of archaea and 40% of bacteria, forms part of the bacterial adaptive immunity against extraneous genetic elements, such as bacteriophages, plasmids and RNA in some cases [1, 2]. This microbial-based RNA-guided endonuclease system has been utilized to engineer biological systems of diverse species, such as zebrafish [3, 4], Drosophila [5, 6], Bombyx mori [7], Caenorhabditis elegans [8], rat [9], mouse [10] and humans [11].
CRISPRs were initially described by a team of Japanese scientists in 1987 as stretches of direct repeats in Escherichia coli [12], and later also described in different bacteria [13]. In the mid-2000s, microbiologists determined that CRISPR/Cas is a pre-existing bacterial defence system that uses antisense RNA as the memory signature of a previous intrusion [14]. This was experimentally confirmed in 2007 using lytic phages and streptococcus thermophilus (lactic acid bacterium) [15]. A significant insight was provided in 2012 from the discovery that Streptococcus pyogenes and S. thermophilus Cas9‐crRNA complexes can act as RNA-guided endonucleases in vitro [16]. These findings, along with previous observations, suggested that the complex of Cas9‐crRNA can be utilized as a targeted genome-engineering technology for making precise double‐stranded breaks. Moreover, newer studies continually aim at optimizing and modifying the CRISPR/Cas system to achieve different outcomes. Over the past three decades, CRISPR has progressed from “curious sequences of unknown biological function” to a global genome-editing tool of choice (Fig. 1).

Timeline of events in the development of CRISPR/Cas technology
Functional CRISPR-Cas loci encompass an operon of cas genes and two discrete RNA components, a fixed tracrRNA (trans-activating crRNA) and a programmable crRNA (CRISPR RNA). The mechanism of action of CRISPR-Cas involves three important steps:
-
(i)
Adaptation: The Cas protein cleaves the invading phage DNA into smaller fragments that get integrated as spacers in the CRISPR array.
-
(ii)
Biogenesis: Subsequently, the CRISPR array is transcribed to generate short RNA sequences (crRNAs), each composed of an invader targeting spacer portion and a direct repeat portion.
-
(iii)
Interference: The crRNA guides the Cas endonuclease to specifically target invading foreign nucleic acids for degradation at sites complementary to the crRNA spacer sequence [17], thereby allowing the bacterial cell to remember, recognize and clear infections.
The protospacer adjacent motif (PAM) is a short sequence distal to the crRNA-targeted sequence in the invading DNA, which is recognized by CRISPR/Cas complex during target DNA binding. The PAM sequence plays a crucial role in the interference and adaptation stages and can alter the affinity between target DNA and Cas protein. It also serves to discriminate between self and non-self target sequences [18].
2 Classification of CRISPR Systems
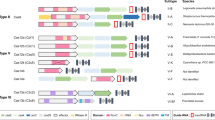
CRISPR systems are majorly divided in two broad classes, Class 1 and Class 2, on the basis of the diversity of Cas proteins. Class 1 systems are usually multi-subunit protein complexes that require a set of Cas proteins for RNA-guided DNA recognition and cleavage, while Class 2 CRISPR systems are less complex, and use only a single Cas protein (Cas9 or Cpf1) for crRNA-guided targeting [19]. This property of Class 2 CRISPR systems has proven to be tremendously useful for genetic manipulation. Class 1 systems include type I, type III, and type IV systems, while Class 2 systems are additionally divided into type II and type V systems (Fig. 2). More specifically, the two main forms of CRISPR systems have recently been categorized into six types (types I–VI) and 33 subtypes (Table 1) [20].

Classification of CRISPR systems (Makarova, K.S, 2020)
2.1 Cas1 and Cas2
Cas1 and Cas2 are the two universally conserved proteins in the CRISPR-based immune system of prokaryotes. Cas1 and Cas2 function in a complex. Two Cas2 copies combine with four Cas1 copies to form a DNA-capturing complex, which scans the cell for invasion of bacteriophage DNA. This complex also identifies the sites of integration in the CRISPR array [21, 22].
2.2 Cas3
Cas3 is a crucial element of the CRISPR-Cas adaptive immune system with translocase and nuclease activity that destroys DNA by restricting it to smaller fragments, neutralizing intruding DNA as part of mechanisms termed “CRISPR interference” [23]. Cas3 allows the identification of non-coding genetic elements and can potentially remove large segments of DNA from a target site in the human genome. This can be helpful in expelling ectopic viruses such as hepatitis B, herpes simplex, Epstein-Barr, etc. [24].
2.3 Cas4
Cas4 is a family of CRISPR adaptation proteins that associate directly with the Cas1-Cas2 complex to select, cut and store DNA [25]. Cas4 proteins (Cas4 and Csa1) play important roles in: (i) identifying the 5′ PAM and 3′ nucleotide motif of protospacers and (ii) defining both the spacer orientation and its length [26]. Cas4 proteins also ensure the insertion of only PAM-bearing sequences into the bacterium’s DNA. This warrants the addition of only usable spacers to the CRISPR array.
2.4 Cas5 and Cas6
Cas5 caps the end of the complex by binding to the CRISPR RNA (crRNA), thereby stabilizing the pre-crRNA. Cas6 is an endoribonuclease required to produce crRNAs in CRISPR-Cas I and III systems for invader defence [27]. Cas5 and Cas6 work in a complex for optimal stability and crRNA processing into discrete crRNA units [28].
2.5 Cas7
The Cas7 proteins form the cascade backbone by attaching multimerically (in a series of six proteins) to mature crRNA. They harbour non-canonical RNA recognition motifs (RRM) and help in nucleation of the surveillance complex [29].
2.6 Cas8
Cas8 proteins offer a security check of the PAM sequence to identify the bacteriophage DNA. It also aids in unwinding of the target DNA and subsequent recruitment of Cas3 for degradation. Furthermore, it interacts with Cas5-Cas7-crRNA complex to stimulate binding with PAM-bearing substrates [30].
3 Cas9 Enzyme: Multifunctional DNA Endonuclease
Crystal structure analysis has revealed the presence of two functional domains in the Cas9 protein:
-
The NUC (nuclease) lobe, which consists of HNH (analogous to phage T4 endonuclease VII), RuvC (analogous to the E. coli RuvC domain, which resolves Holliday junctions), and PAM-interacting (PI) domains.
-
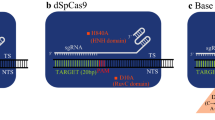
The Cas9-specific REC (recognition) lobe composed of a REC1 and a REC2 domain, and a long helix for interacting with repeat:anti-repeat duplex [31] (Fig. 3a).
Fig. 3 
a Activation of Cas9 protein by guide RNA binding (Adapted from [110]). b Diagram illustrating different types of engineered Cas9 variants

The HNH and RuvC domains of Cas9 utilize a single or two metal mechanisms to cleave the complementary and non-complementary strands, respectively, of the target DNA. The PAM specificity is offered by the PI domain, which is also essential for PAM recognition [31]. The sgRNA is made by fusing the tracrRNA and crRNA to enable the Cas9-mediated cleavage of dsDNA. This has made the CRISPR/Cas9 a handy gene-engineering tool owing to the choice of guide RNA design for identification of the sequence preceding PAM. The tracrRNA consists of three stem loops (loops 1–3) and a 14-nt anti-repeat region, whereas the crRNA is comprised of a 20-nt gRNA and a 12-nt repeat region. This is how the sgRNA bridges the target sequence and the Cas9 enzyme.
The crystal structure of CRISPR/Cas9 reveals the existence of a T-shaped conformation, made up of three stem loops with a linker between stem loops 1 and 2, the heteroduplex of target:gRNA, a tetraloop, and the repeat:anti-repeat duplex. The process starts with the formation of two important duplexes:
-
The target:gRNA heteroduplex through Watson–Crick base pairing.
-
The repeat:anti-repeat duplex.
Simultaneously, the tracrRNA bases use Watson–Crick base pairing to generate stem loops 1, 2, and 3. The NUC and REC lobes of the Cas9 protein recognize the stem-loop1, the target sequence:gRNA, and the repeat:anti-repeat duplex, whereas the NUC lobe specifically recognises the stem loops 2 and 3 and the linker [31]. The stem loops 2 and 3 affect the activity and stability of the CRISPR/Cas9 structure while stem loop 1 is crucial for the development of the functional sgRNA:Cas9 complex. These interactions lead to the formation of a Cas9-sgRNA binary complex. Concurrently, the complex is able to recognize the target sequence complementary to the gRNA. The PI domain helps in the formation of an R-loop configuration, which is followed by the cleavage of the complementary and non-complementary strands by the Cas9-sgRNA-target DNA ternary complex.
The progress and advances of the CRISPR/Cas9 method have chiefly been made around modifications of Cas9 and sgRNA to attain precise editing. Significantly, various versions of Cas9 have been functional in gene editing of both transgenic animals and mammalian cells through modifications of NUC and REV lobes (Fig. 3b).
3.1 Dead Cas9 (dCas9)/Nuclease-Null Deactivated Cas9
Nuclease-null deactivated Cas9 (dCas9), also known as CRISPR interference (CRISPRi), contains a catalytically inactive dCas9 with dead RuvC and HNH domains [32]. It can direct the transcriptional modification of target genes without altering the DNA as a result of the obstruction formed by the sgRNA-Cas9 complex. This scheme offers two discrete functions, dCas9 fusion-mediated activation (CRISPRa) and dCas9 fusion-mediated inhibition (CRISPRi).
3.2 Cas9 Nickase (Cas9n)
Cas9n is generated by incorporation of a loss-of-function mutation in the endonuclease cleavage domains, for example an H847A mutation or a D10A mutation eliminates HNH function or abolishes the nuclease activity of the RuvC domain, respectively. Therefore, combining two separate sgRNAs with Cas9n can result in a staggered DSB to activate the DNA-repair process. This can enhance the specificity and efficiency of the CRISPR/Cas9 system [33].
3.3 Light-Activated Cas9
Recent studies have shown a light-activated Cas9 to be a promising tool for temporal and spatial genome editing [34]. This system consists of two fusion proteins:
-
A light-sensitive cryptochrome 2 (CRY2), which functions as an activator probe in combination with a transcriptional activator domain, and
-
A cryptochrome-interacting basic-helix-loop-helix 1 (CIb1).
The heterodimerization of CIb1 and CRY2 can lead to the recruitment of modified Cas9 to the target sequence for activation of gene transcription upon stimulation of blue light [34, 35].
3.4 Other Modifications of Cas9
-
The dCas9 and CIb1 of light-activated Cas9 can be fused to aim the target site with sgRNAs as the genome anchor probe.
-
The K163 residue of Cas 9, located near gRNA binding sites, is highly conserved and can be used in controlling Cas9-gRNA interactions.
-
Also, the K886 residue of Cas9 could be used to change the structure of Cas9 so as to expose the lysine residue during the process of gRNA binding. The presence of multiple lysines (K) could inhibit the CRISPR/Cas9 system by acting as an important caging site. This can be used for positioning the non-target DNA strand [36].
-
Moreover, by integrating technologies such as flow cytometry [37] and fluorescence microscopy [38], numerous tags and reporters, such as puromycin and GFP, have been attached to Cas9 to attain selection and screening of stable genome-engineered cell lines [37].
4 Comparison of CRISPR/Cas9 with Other Genome-Editing Technologies
Genome-editing technologies, including DNA recombinase-dependent gene replacement, UV and chemical-induced mutagenesis, self-splicing introns, zinc-finger nucleases (ZFNs), and transcriptional activator-like effector nuclease (TALEN) systems, have overwhelmingly contributed to significant and therapeutic developments in biomedical science [39, 40]. However, complications of protein design, synthesis and validation continued to remain a barrier for the widespread adoption of these techniques. Cas9, in comparison, offers numerous potential advantages over ZFNs and TALENs, which are listed below.
4.1 Ease of Customization
The retargeting of TALEN for a new sequence of DNA necessitates the construction of a new pair of TALEN genes, which requires considerably longer hands-on time to generate two new TALENs. However, Cas9 can simply be retargeted to newer DNA sequences by merely procuring a set of two oligos encoding the 20-nt guide sequence [41].
4.2 Pattern of Cleavage
The cleavage of TALENs is non-specific in the 12–24 bp linker flanked by two TALEN monomer-binding regions, whereas Cas9 is highly specific and cuts 3 bp 5′ of the PAM. SpCas9 (Cas9 from WT S. pyogenes) cuts bluntly between the 17th and 18th bases in the target sequence.
4.3 Mechanism of Action
TALEN and ZFNs use an approach that involves tethering the catalytic domain of FokI endonuclease for inducing targeted DNA double-stranded breaks at specific gene loci. On the other hand, Cas9 nuclease is guided by small RNAs via Watson–Crick base pairing with target DNA [11].
4.4 Applicability
The large size of TALENs may hamper the recombination process at a cellular level, and, therefore, tends to be less attractive for therapeutic and biotechnological applications. Also, for efficient functionality, TALENs’ targets need thymidine residues, which further limits their application [42]. Custom ZFNs are extremely difficult to engineer as ZFNs show a necessary interaction with zinc fingers. This limits their application within a biotechnological context, particularly in view of the need for nucleotide sequence specificity. In contrast, Cas9 is well suited to target multiple genomic loci simultaneously in a number of cell types and organisms.
The success of CRISPR-Cas9 genome editing is further evidenced by the substantial growth in scientific publications and federal research grants related to CRISPR as compared to ZNF and TALEN. As seen in Table 2, the National Institutes of Health (NIH) support for CRISPR-related work escalated from US$5 million in 2011 to US$1.8 billion in 2019. Likewise, the number of CRISPR-related research publications show a 60-fold increase from 2011 to 2019 (Table 2). As can also be seen graphically, the steep increase of CRISPR methodology has swiftly eclipsed the ZNF and TALEN gene-editing tools.
5 Experimental Design
This section provides a general guide/experimental workflow for completion of a successful CRISPR experiment along with important considerations regarding the specific CRISPR components, delivery technique and analysis method.
5.1 Target Selection for sgRNA
The 20-nt guide sequence in the sgRNA determines the specificity of the Cas9 endonuclease. There are two core considerations in the selection of the guide sequence for gene targeting:
-
(i)
The 5′-PAM sequence for Streptococcus pyogenes Cas9 [43]: The 5′-NGG PAM is not a part of the 20-nt sgRNA but immediately follows the target DNA locus. The target sequence must precede (i.e. be 5′) a 5′-NGG PAM. The sgRNA base pairs with the complementary strand to facilitate cleavage of Cas9 at ∼ 3 bp upstream of the PAM.
-
(ii)
The minimization of off-target activity [44], i.e. the gRNA sequence, should strongly match the target sequence.
Many online CRISPR design tools have been generated that search a genomic sequence of interest for identification of appropriate target sites. The CRISPR Design Tool facilitates the target selection by providing the sequences for all primers and oligos essential for:
-
(i)
Preparation of the sgRNA constructs,
-
(ii)
Analysing efficiency of target modification, and
-
(iii)
Evaluating cleavage at possible off-target sites.
Although rare, some sgRNAs may not function for reasons as yet unknown; hence, it is safer to design two or more sgRNAs for each locus. Also, when the promoter for CRISPR plasmid is the human U6 RNA polymerase III, a guanine (G) residue is placed at the 5′ end of the sgRNA since the promoter favours a G as the first nucleotide to start sgRNA expression.
5.2 Construction and Delivery of sgRNA
sgRNAs can be delivered either as expression plasmids or as PCR amplicons consisting of an expression cassette, depending on the desired application. Indeed, construction of an expression plasmid for sgRNA is quick and easy, requiring a single step of cloning with just a pair of partly complementary oligonucleotides. This follows the annealing and ligation of the guide sequence encoding oligonucleotide pairs into an appropriate plasmid encompassing both the Cas9 enzyme and the rest of the sgRNA as an invariant scaffold immediately preceding the oligonucleotide cloning site. On the other hand, PCR-driven sgRNA delivery adds the custom sgRNA sequence on the reverse PCR primer that amplifies a U6 promoter template. The resultant amplicon is co-transfected along with a Cas9 expression plasmid pSpCas9. This technique is ideal for quick screening of multiple candidate sgRNAs. As this simple method precludes the requirement for plasmid-based cloning, it is best suited for co-transfecting a huge number of sgRNAs for making large knockout libraries. Also, compared to the ∼ 20-bp-long oligonucleotides essential for plasmid-based sgRNA delivery, the sgRNA-encoding primers are more than 100 bp in length. Besides plasmid-based and PCR delivery methods, sgRNAs and Cas9 can be incorporated into cells as RNA and mRNA, respectively.
5.3 Design of the Template Single-Stranded DNA Oligonucleotide (ssODN)
The double-stranded break caused by Cas9 enzyme stimulates at least two diverse DNA repair processes, including non-homologous end-joining (NHEJ) repair and homology-directed repair (HDR).The NHEJ repair system re-joins both the ends of the DSB, thereby inducing unpredictable deletions, insertions or substitutions with around 50% efficiency, whereas the HDR pathway results in specific mutations, deletions or insertions by using homologous donor DNA sequences with a lower frequency (Fig. 4). The CRISPR/Cas9 method uses a small single-stranded DNA oligonucleotide (ssODN) as a repair template in comparison with the conventional large homologous arms used in HDR. The ssODN comprises a complementary sequence of the sense or antisense target region and encompasses at least 40-nt in the 3′ and 5′ directions [33]. Also, the phases of the cell cycle govern the choice of pathway, for example HDR is restricted to S and G2 phases while NHEJ prefers G1, S and G2 phases [45].

Working of the CRISPR/Cas9 gene-editing system
5.4 Efficiency Verification of the CRISPR/Cas9 System
There are a few bioinformatic tools that can be utilized to predict the effectiveness of the designed gRNA. Since the mutations or indels (insertions and deletions) initiated by the Cas9 cleavage can be produced via the DNA repair mechanism, quite a few technologies are available to predict the competence of the CRISPR/Cas9 method, for example the deep-sequencing [46] and the SURVEYOR assays [47]. The deep-sequencing assay uses the Hiseq 2500 and Illumina Miseq machines to crosscheck the indels in the 100- to 200-bp size range against the reference sequence. On the other hand, the SURVEYOR procedure uses the SURVEYOR nuclease T7E1 to cleave the reannealed heteroduplexes in the 200- to 400-bp target region, leaving the homoduplexes intact. The cleavage efficiency of Cas9 is determined by the strength of the gel bands in gel electrophoresis.
6 Development of New CRISPR Systems: Beyond Cas9
CRISPR systems have witnessed dramatic progress as cutting-edge biotechnology tools enabling new lines of biological inquiry. The natural variety in CRISPR/Cas structures has contributed to the discovery of other Cas effectors with the ability to extend the capacities of CRISPR-based developments. Additional subtypes of CRISPR/Cas systems have been established by analysing the microbial variety for signatures of CRISPR-Cas systems.
6.1 SaCas9
Owing to the difficulty in delivering the large-sized SpCas9, smaller Cas9 orthologs have been identified that function effectively in mammalian cells while upholding a wide target range. SaCas9 (PAM 5′-NNGRRT) from Staphylococcus aureus is 1 kb smaller in size than SpCas9 and has shown high efficiency in human cells [49]. Because of its smaller size, SaCas9 has enabled the use of a single-AAV vector for delivery of the enzyme with guide RNA [48]. SaCas9 has further emerged as the first in vivo genome-engineering tool in humans [49].
6.2 Cas12
Cas12 are members of the Class2 system that have recently gained much traction as efficient biological gene-editing tools. The first Cas12 enzyme identified was Cas12a (previously known as Cpf1) [50]. It requires a T-rich PAM sequence [51]. Cas12a-mediated genome editing presents enormous advantages over Cas9:
-
(i)
It has a higher specificity and less off-target activity, making it more important for therapeutic applications [52, 53].
-
(ii)
It has inherent RNAase activity, through which it can process its own pre-crRNA to mature crRNA [54].
-
(iii)
It does not require tracrRNA and therefore offers a simplified design of guide RNA [51].
-
(iv)
It generates 5′ overhangs rather than blunt double-stranded breaks of Cas9 [51]. This is beneficial for incorporation of newer sequences [55].
-
(v)
It is best suited for multiplex genome editing.
-
(vi)
It is suitable for viral packaging owing to its smaller size.
Cas12b (previously known as C2c1) are dual RNA-guided nucleases that are similar to Cas12a but need a tracrRNA [56]. Other Cas12 members have also been recognized (designated types V-A to V-I). They include Cas12c [56], Cas12d (CasY), Cas12e (CasX), Cas12f (Cas14), Cas12g (smallest of the Cas12 family), Cas12h and Cas12i [57, 58]. Recently, two Cas12e orthologs (DpbCasX and PlmCasX) have been in use for targeted gene knockout in human cells [59].
6.3 Cas13
Cas13 is a subset of large highly conserved proteins with the higher eukaryotic-prokaryotic nuclease (HEPN) domain [60]. They are single effector crRNA-guided RNAses operating at the RNA level instead of DNA [61]. They show a collateral effect, i.e. they cleave the non-target bystander RNA at specific sites. This optimally suits them for targeting bacterial and viral infections. All members of the Cas13 family have been modified for selective RNA repression in mammalian cells [61, 62].
6.4 Cas14
Cas14 is a family of extremely compact RNA-guided nucleases (Cas14a-Cas14h) that cleave the ssDNA without the requirement of any restrictive sequence [58]. Cas14 proteins trigger non-specific DNA cleavage that enables high-fidelity SNP genotyping (Cas14 DETECTR). This can be potentially used for detection of ssDNA viruses of compelling therapeutic, socioeconomic and ecological importance that target hosts in all three realms of life [63]. CRSIPR/Cas14 can thus be highly useful in the field of CRISPR-based molecular diagnostics.
6.5 Cascade
Cas proteins of the Type I CRISPR/Cas system interact to form an immunosurveillance effector complex called Cascade (CRISPR-associated complex for anti-viral defence). It consists of five Cas proteins and assembles on DNA in a site-specific manner. The Cascade complex uses Cas3 for DNA cleavage. Cascade-mediated gene silencing has been used to simultaneously silence multiple gene targets with lesser off-target effects and greater interference [64].
6.6 Cast
Recently, CRISPR/Cas has been harnessed to guide transposons to specific sites in the genome giving rise to a new technique (CRISPR/Cas + Transposon = CAST) [65]. CAST (CRISPR-associated transposase) has overcome the biggest hurdle to the use of transposons as precise gene-engineering tools. CAST uses Cas12k, which further associates with tniQ59 and tnsB/C. For this purpose, several complexes consisting of Cas6, Cas7, Cas8, tniQ and tns A/B/C have also been tested [66]. In eukaryotic cells, the CAST technique can be engineered to enable selective sequence insertion of 10 kb or more. This would increase the frequencies of integrations and expand the range of target sites.
7 Applications of the CRISPR/Cas Technique
Some examples of potential and current uses of CRISPR-Cas are summarized here.
7.1 Basic Research
CRISPR/Cas gene editing has added new possibilities and adaptability to basic research. Disease modelling has served as an effective tool for understanding the clinical condition of the disease and designing appropriate therapies. CRISPR-Cas has rendered disease-model development more reliable, less labour-intensive, and far more cost effective. The CRISPR/Cas platform has outperformed the RecA-based recombineering approach in gene knockouts. This has considerable application to redirect the metabolic flux to desirable pathways, and also to identify key functional genes. Visualizing cells has become more accurate, so researchers may add a fluorescent protein tag more precisely to a gene of interest [67]. CRISPR has also emerged as an imaging method for visualizing chromatin in living cells, for example dCas9, when fused to a fluorophore such as green fluorescent protein (GFP), enables the identification and study of chromatin by binding to a particular target sequence on a living cell’s genome.
CRISPR/Cas has shown considerable application in regulating gene expression and modifying epigenetic states, in addition to enabling alteration in mammalian genomes. Advancements to the CRISPR/Cas9 method have made this new feature possible without adding DSBs [68], thus avoiding undesirable mutations in the candidate genes. The fusing of a nuclease-deficient Cas9 (dCas9) with the viral transcriptional activation domain VP64 has enabled the induction of the expression of a broad variety of genes in their native chromosomal scenario [69]. With CRISPR, gain-of-function (GOF) and loss-of-function (LOF) mutations could be rapidly induced in the oncogenes, tumour suppressor genes or several other important players of the tumour-progression process [70]. The CRISPR/Cas method is also an effective means of introducing chromosomal translocations that imitate cancers, like acute myeloid leukaemia, lung cancer or Ewing’s sarcoma [71].
7.2 Human Health and Medicine
The potential of the CRISPR/Cas gene-editing platform to cure, treat or prevent disease or medical conditions can produce substantial cuts in direct or indirect economic costs and reduce the burden resulting from debilitation, pain and death. Below are a few notable applications, although the list is not exhaustive.
7.2.1 Diabetes
According to the International Diabetes Federation 2019, diabetes, a profligate disease of impaired glucose metabolism, affects approximately 463 million people worldwide, which means that around one in every five persons is diabetic. CRISPR/Cas9 has been exploited for precise evaluation of diabetes pathogenesis. CRISPR/Cas has enabled the replacement of insulin-producing cells in pre-clinical animal models of Type 1 diabetes. It is advantageous over previous methods as it uses the individual’s own cells, thereby reducing the chance of transplant rejection and eliminating the inadequate availability of donors [72]. Chung et al. have recently used the CRISPRi technique to silence the expression of Fabp4 to reduce lipid storage in adipocytes. This has successfully reduced the body weight of obese mice by about 20%. It has also diminished the indicators of type 2 diabetes such as insulin resistance, inflammation, high glucose levels and hepatic steatosis [73].
7.2.2 Malaria
Malaria is one of the most fatal and widespread diseases in the world. A number of CRISPR-based methods are being considered with the aim of minimizing or eradicating malaria by effective elimination, alteration or reduction of the primary vector, the Anopheles mosquito. These include infertility of the Anopheles mosquito [74], gene engineering for preferentially producing male offspring, or the creation of a malaria-resistant Anopheles mosquito [75]. For instance, CRISPR/Cas9 has enabled the inactivation of the fibrinogen-related protein 1 (FREP1) gene in Anopheles gambiae. FREP1 knockout mutants have shown effective suppression of infection with both rodent and human malaria parasites at the sporozoite and oocyst stages [76]. With the aim of reducing or eradicating the Anopheles mosquito in sub-Saharan Africa, the Bill and Melinda Gates Foundation, a non-profit research consortium, has granted approximately US$75 million toward decreasing the count of female mosquitoes in closely linked Anopheles species. Similar strategies are being explored to reduce the spread of other mosquito-borne viral diseases such as St. Louis encephalitis, West Nile, Zika, yellow fever and dengue fever [77].
7.2.3 Sickle Cell Disease
Sickle cell disease (SCD) is a hereditary life-threatening disease affecting more than 30 million people worldwide. The only curative therapy for sickle cell disease is allogeneic hematopoietic stem cell transplantation (HSCT), which is limited to donor availability. However, in some pre-clinical trials, CRISPR/Cas gene-editing technology has resulted in a novel restorative therapy for SCD by autologous, genome-modified HSPC transplantation in CD34 + hematopoietic stem and progenitor cells (HSPCs), which leads to continuous generation of fetal haemoglobin (HbF) with functional RBCs [78]. Another study has optimized the CRISPR/Cas system for treating SCD and other single gene disorders by clinical translation of hematopoietic stem and progenitor cell genome-correction strategy [79]. Also, CRISPR-Cas strategy has been utilised for the treatment of SCD and homozygous β-thalassemia by modification of bone marrow HSPCs in another pre-clinical trial [80].
7.2.4 Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD), one of the most common human hereditary disorders, affects nearly 1:5000 newborn males. The molecular basis of this severe X-linked disease is a mutation in the dystrophin gene that results in progressive muscle wasting. While no effective treatments exist for this disorder, CRISPR/Cas9 has emerged as a promising tool to correct the genetic basis of this disease. The unique multiplex gene-editing competences of the CRISPR/Cas system have been used to target the mutational hotspot (exon 45-55) in the dystrophin gene to restore its expression [81]. A related approach utilizes single and dual AAV vectors to specifically edit the mutation in dystrophic mdx4cv mice using CRISPR/Cas9. Muscle-restricted Cas9 expression in myogenic cells has enabled multi-exon deletion, direct editing of the mutation, or full correction of the gene through homologous recombination with treated muscles expressing up to 70% of the dystrophin gene [82]. Studies have also developed a representative model of DMD by targeting the point mutation in exon 23 of the mdx mouse using the CRISPR/Cas system to generate a premature stop codon [83, 84]. Also, genetic correction of patient-derived induced pluripotent stem cells (iPSCs) using CRISPR/Cas has shown much promise for DMD gene therapy [85].
7.2.5 Cystic Fibrosis
Cystic fibrosis (CF), the most common autosomal multi-organ disorder, is associated with mutations within the cystic fibrosis transmembrane conductance regulator (CFTR) gene encoding for the chloride ion channel. Several supportive therapies such as inhaled antimicrobials, mucolytics, nutritional support and systemic anti-inflammatories have been used for improvement in life expectancy over time, but curative therapies are not available. However, the evolution of CRISPR-nucleases has augmented the progress of gene correction for cystic fibrosis. A complete restoration of CFTR protein functionality has been achieved in cultured intestinal stem cells of paediatric CF patients using the CRISPR/Cas technique [86]. Similarly, rectification of CFTR mutation has been done in iPSCs by reprogramming somatic skin fibroblasts of CF patients into an embryonic stem cell state using the CFTR/Cas9 gRNA vector [87]. Recently, the CRISPR/Cas system has been used to permanently edit at least two types of the mutations that result in CF in airway epithelial cells and intestinal organoids derived from CF patients [88]. The technique repairs the intronic splicing defects in a gene by producing isolated indels within intron and is termed “SpliceFix” as it fixes the gene and restores the mechanism of protein production simultaneously.
7.2.6 Persistent Viral Infection
Many viral pathogens like hepatitis viruses, HIV, papillomaviruses, herpesviruses, etc., develop persistent infection by incorporating their DNA into the human genome or keeping it episomally within the host cells. The CRISPR/Cas technique has been successfully used to minimize or eradicate such recurrent viral infections in animal models, thereby providing new hope for cures for chronic and latent viral infections. HIV-1 infection has been effectively prevented in TZM-bI cells by targeting its long terminal repeat (LTR) sequences using a gRNA-Cas9 construct [89]. This construct has also been useful in inactivating HIV gene expression in infected T cells and microglial cells. Similarly, from multiple other cell lines, LTR, gag and env genes of HIV have been targeted to remove HIV proviral DNA [90]. Another study in a transgenic mouse model using lentiviral vector delivery of CRISPR/Cas has been successful in eliminating the proviral HIV DNA from infected human peripheral blood mononuclear cells [91]. Likewise, CRISPR/Cas has been utilized for clearance of herpes simplex virus 1 (HSV-1) infection by targeting 12 crucial genes to reduce its replication in Vero cells [92]. Another study has used the CRISPR/Cas system to limit HSV-1 infection by suppressing its replication in human oligodendroglioma cells [93]. CRISPR/Cas is also being targeted at other herpes viruses in vitro including human cytomegalovirus (CMV), which causes severe illness when acquired by people with compromised immune systems, and Epstein-Barr virus (EBV), which leads to certain nasopharyngeal cancers and lymphomas [92].
7.3 Industrial Biotechnology
CRISPR/Cas has had a pervasive impact on industries and companies that depend on bacteria, yeast and fungi. CRISPR-Cas has not only widened the range of microorganisms used for industrial production [94], but also made virus-resistant industrially relevant strains. This has potentially enhanced the generation of chemicals used in manufacturing biofuels and engineering probiotics [1]. The first step towards producing long-chain hydrocarbons in yeast has been the production of a yeast strain that produces useful polymers and lipids [95]. This has led to advances in developing new precursors for specialized polymers, biofuels, fragrances and adhesives. In this way, non-renewable raw materials generated in petroleum-processing plants could be substituted with cheaper raw materials via a safer and more effective bio-manufacturing procedure.
7.4 Agriculture
Besides interest in its prospects for clinical and biomedical research, CRISPR/Cas technology has contributed substantially to international agriculture. CRISPR/Cas has made it possible to incorporate or remove gene sequences with much greater precision than the conventional methods of plant genetic engineering and livestock breeding. This has provided the opportunity for making changes in major food crops by supplementing the current plant DNA sequences with required ones, or by enabling differential gene expression.
CRISPR/Cas has opened up a radically novel way of developing new plant varieties, for example production of a wheat strain resistant to powdery mildew [96] and alteration of a plethora of agricultural products, including oranges, tomatoes, potatoes, soybeans, rice and sorghum [97]. Recent CRISPR-enabled livestock research includes chickens that only deliver female-egg-laying chicks, more tender and beefier Brazilian cattle, cattle that only produce males for better fodder-to-meat output, virus-resistant pigs, and hornless dairy cattle, a development that may lead to financial benefits, improved health for agricultural workers, and advancements in animal welfare [98]. CRISPR/Cas has recently been used for the cultivation of genetically modified mushrooms immune to browning and a specialized variety of corn with rare starch (“waxy” corn) characteristics [99].
7.5 Management and Conservation of Ecosystem
The CRISPR genome manipulation method has been proposed as a possible management tool for coping with the problems presented by plant pests (e.g. Palmer amaranth) and invasive species (e.g. zebra mussels, Japanese beetles and spotted knapweed) [100]. It may also be used to skew an invading population’s gender ratio to males, and thereby promote a population collapse. For example, a sex-determining gene drive for resistant foreign organisms has been recommended as an effective method to conserve island biodiversity. CRISPR/Cas can also reconstruct extinct animals like the passenger pigeon and woolly mammoth by modifying the contemporary animal’s DNA to contain the missing traits like enhancing hair growth and subcutaneous fat [101].
8 Ethical challenges
While the use of the CRISPR/Cas technique in the delicate realm of gene-editing technologies has improved convenience and performance, it entails a range of ethical and biosafety issues, as discussed below.
8.1 Risk of Off-Targets
The primary concern among them is its safety, owing to the risk of off-target effects (edits in the wrong place). The off-target concerns are especially important if they turn out to be heritable in humans [102, 103].
8.2 Ecological Disequilibrium
Recently, the CRISPR/Cas9-based mutagenic chain reaction (MCR) has fostered a novel means of producing homozygous autocatalytic loss-of-function mutations in Drosophila [5]. Nonetheless, the possible serious implications on the ecological balance due to a complete shift in the species [104] has raised several concerns against the use of this technique. This can be even more detrimental if the negative trait is passed to related organisms across political borders. This demands security precautions to deter the dissemination of organisms that can inflict ecological harm or impact human health.
8.3 Germline Genome Editing
Gene alteration of human germ cells employing the CRISPR/Cas to generate “engineered babies” [105] has triggered debates and ethical concerns among researchers, clinicians and the public. Increasingly contentious among scientists is embryo modification or manipulation in human fetuses. In fact, a few countries have even limited use of CRISPR/Cas technique, while others have prohibited its use in humans altogether. In response to this, The International Summit on Human Gene Editing, convened in December 2015, supported basic research and studies using human embryos that would not be considered for pregnancy establishment. It was also agreed upon with credentials that the therapeutic use of human germline editing should be revisited periodically [106, 107]. Its second summit was held in November 2018 in the midst of the announcement of the birth of the first genetically engineered baby. Even though this research was strongly condemned, the forum vetoed an embargo on germline editing and instead permitted a “translational approach to germline gene editing [108].
8.4 Commercialization of Products
The effectiveness of the CRISPR/Cas method in achieving correct genetic alterations makes it more difficult to recognise a genetically engineered organism until it is outside the lab and to put it on the market. It needs proper approval from the regulatory bodies, such as the US Food and Drug Administration [109]. The patenting of the product is another problem as there are a variety of commercial interests involved.
References
Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–41.
Sorek R, Lawrence CM, Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem. 2013;82:237–66.
Jao LE, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci USA. 2013;110:13904–9.
Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ. Genome editing with RNA-guided Cas9 nuclease in Zebrafish embryos. Cell Res. 2013;23:465–72.
Gratz SJ, Rubinstein CD, Harrison MM, Wildonger J, O’Connor-Giles KM (2015) CRISPR-Cas9 genome editing in Drosophila. Curr Protoc Mol Biol. 2015:31.2.1–31.2.20.
Bassett AR, Kong L, Liu JL. A genome-wide CRISPR Library For High-Throughput Genetic Screening In Drosophila Cells. J Genet Genom. 2015;42:301–9.
Ma S, Chang J, Wang X, Liu Y, Zhang J, Lu W, Gao J, Shi R, Zhao P, Xia Q. CRISPR/Cas9 mediated multiplex genome editing and heritable mutagenesis of BmKu70 in Bombyx mori. Sci Rep. 2014;4:1–6.
Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–34.
Li D, Qiu Z, Shao Y, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681–3.
Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–62.
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–8.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakatura A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–33.
Mojica FJM, Díez-Villaseñor C, Soria E, Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol. 2000;36:244–6.
Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:1–26.
Barrangou Rodolphe, Fremaux Christophe, Deveau Hélène, Richards Melissa, Boyaval Patrick, Moineau Sylvain, Romero Dennis A, Horvath Philippe. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007. https://doi.org/10.1126/science.1136466.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA—guided. Science. 2012;337:816–22.
Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78.
Westra ER, Semenova E, Datsenko KA, Jackson RN, Wiedenheft B, Severinov K, Brouns SJJ. Type I-E CRISPR-Cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013. https://doi.org/10.1371/journal.pgen.1003742.
Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–36.
Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83.
Nuñez JK, Kranzusch PJ, Noeske J, Wright AV, Davies CW, Doudna JA. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat Struct Mol Biol. 2014;21:528–34.
Xiao Y, Ng S, Hyun Nam K, Ke A. How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature. 2017;550:137–41.
He L, St. John James M, Radovcic M, Ivancic-bace I, Bolt EL. Cas3 protein—a review of a multi-tasking machine. Genes (Basel) 2020;11(2):208.
Dolan AE, Hou Z, Xiao Y, Gramelspacher MJ, Heo J, Howden SE, Freddolino PL, Ke A, Zhang Y. Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using type I CRISPR-Cas. Mol Cell. 2019;74:936–50.
Lee H, Dhingra Y, Sashital DG (2019) The Cas4-Cas1-Cas2 complex mediates precise prespacer processing during CRISPR adaptation. eLife 8:1–26.
Zhang Z, Pan S, Liu T, Li Y, Peng N. Cas4 nucleases can effect specific integration of CRISPR spacers. J Bacteriol. 2019. https://doi.org/10.1128/JB.00747-18.
Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–96.
Brendel J, Stoll B, Lange SJ, et al. A complex of cas proteins 5, 6, and 7 is required for the biogenesis and stability of clustered regularly interspaced short palindromic repeats (CRISPR)-derived RNAs (crRNAs) in haloferax volcanii. J Biol Chem. 2014;289:7164–77.
Hrle A, Su AAH, Ebert J, Benda C, Randau L, Conti E. Structure and RNA-binding properties of the type III-A CRISPR-associated protein Csm3. RNA Biol. 2013;10:1670–8.
Kalwani P, Rath D, Ballal A. Novel molecular aspects of the CRISPR backbone protein “Cas7” from cyanobacteria. Biochem J. 2020;477:971–83.
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–49.
Brezgin S, Kostyusheva A, Kostyushev D, Chulanov V. Dead cas systems: types, principles, and applications. Int J Mol Sci. 2019;20:1–26.
Ran FA, Hsu PD, Lin CY, et al. Erratum: double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity (Cell (2013) 154 (1380-1389)). Cell. 2013;155:479–80.
Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198–200.
Nihongaki Y, Kawano F, Nakajima T, Sato M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol. 2015;33:755–60.
Hemphill J, Borchardt EK, Brown K, Asokan A, Deiters A. Optical control of CRISPR/Cas9 gene editing. J Am Chem Soc. 2015;137:5642–5.
Rojas-Fernandez A, Herhaus L, Macartney T, Lachaud C, Hay RT, Sapkota GP. Rapid generation of endogenously driven transcriptional reporters in cells through CRISPR/Cas9. Sci Rep. 2015;5:1–6.
Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep. 2015;5:1–6.
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–51.
Bedell VM, Wang Y, Campbell JM, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–8.
Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405.
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ. TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:117–22.
Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6.
Lin S, Staahl BT, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3:e04766.
Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, Sorek R. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–10.
Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33:102–6.
Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–91.
Wang Y, Wang B, Xie H, et al. Efficient Human genome editing using SaCas9 ribonucleoprotein complexes. Biotechnol J. 2019;14:1–8.
Schunder E, Rydzewski K, Grunow R, Heuner K. First indication for a functional CRISPR/Cas system in Francisella tularensis. Int J Med Microbiol. 2013;303:51–60.
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell. 2015;163:759–71.
Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, McCaw ZR, Aryee MJ, Joung JK. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–74.
Kim HK, Song M, Lee J, et al. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat Methods. 2017;14:153–9.
Fonfara I, Richter H, BratoviÄ M, le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517–21.
Moreno-Mateos MA, Fernandez JP, Rouet R, Vejnar CE, Lane MA, Mis E, Khokha MK, Doudna JA, Giraldez AJ. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat Commun. 2017;8:1–9.
Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–97.
Burstein D, Harrington LB, Strutt SC, Probst AJ, Anantharaman K, Thomas BC, Doudna JA, Banfield JF. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–41.
Harrington LB, Harrington LB, Burstein D, et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;4294:1–8.
Liu JJ, Orlova N, Oakes BL, et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature. 2019;566:218–23.
Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, Scott DA. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol Cell. 2018;70:327–39.
Smargon AA, Cox DBT, Pyzocha NK, et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell. 2017;65:618–30.
Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018;173:665–76.
Aquino-Jarquin G. CRISPR-Cas14 is now part of the artillery for gene editing and molecular diagnostic. Nanomed Nanotechnol Biol Med. 2019;18:428–31.
Rath D, Amlinger L, Hoekzema M, Devulapally PR. Efficient programmable gene silencing by Cascade. Cascade. 2015;43:237–46.
Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, Zhang F. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365(6448):48–53.
Klompe SE, Vo PLH, Halpin-healy TS, Sternberg SH. Article Transposon-encoded CRISPR—Cas systems. Nature. https://doi.org/10.1038/s41586-019-1323-z.
Chen B, Gilbert LA, Cimini BA, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–91.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83.
Gilbert LA, Larson MH, Morsut L, et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442.
Sánchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387–95.
Choi PS, Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun. 2014;5:1–6.
Gerace D, Martiniello-Wilks R, Nassif NT, Lal S, Steptoe R, Simpson AM. CRISPR-targeted genome editing of mesenchymal stem cell-derived therapies for type 1 diabetes: a path to clinical success? Stem Cell Res Ther. 2017;8:1–10.
Chung JY, Ul Ain Q, Song Y, Yong SB, Kim YH. Targeted delivery of CRISPR interference system against Fabp4 to white adipocytes ameliorates obesity, inflammation, hepatic steatosis, and insulin resistance. Genome Res. 2019;29:1442–52.
Hammond A, Galizi R, Kyrou K, et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol. 2016;34:78–83.
Alphey L. Can CRISPR-Cas9 gene drives curb malaria? Nat Biotechnol. 2016;34:149–50.
Dong Y, Simões ML, Marois E, Dimopoulos G. CRISPR/Cas9-mediated gene knockout of Anopheles gambiae FREP1 suppresses malaria parasite infection. PLoS Pathog. 2018;14:1–16.
Adelman ZN, Tu Z. Control of mosquito-borne infectious diseases: sex and gene drive. Trends Parasitol. 2016;32:219–29.
Yu Vionnie W C, PhD Yi Liu, PhD Matthew Curran, Pu Zhang MD, Jennifer Snead PhD, Schmedt Christian, Yi Yang PhD, Lin Victor Guosheng, Tschantz William R, PhD Lisa Quinn, Russ Carsten, Clarkson Scott, Janiak Amy, Morag Stewart PhD, Yanick Mulumba SS. CRISPR/Cas9 gene-edited hematopoietic stem cell therapy for sickle cell disease. Blood. 2017;130:535.
Park So Hyun, Lee Ciaran M, Deshmukh Harshavardhan, Gang Bao P. Therapeutic Crispr/Cas9 genome editing for treating sickle cell disease. Blood. 2016;128:4703.
Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, Kan YW. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci USA. 2016;113:10661–5.
Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause duchenne muscular dystrophy. Nat Commun. 2015;6:1–13.
Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR, Chamberlain JS. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:1–9.
Long C, Long C, Amoasii L, et al (2015) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 5725.
Tabebordbar M, Zhu K, Cheng JKW, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science (New York, NY). 2015;5177:1–9.
Li HL, Fujimoto N, Sasakawa N, et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015;4:143–54.
Schwank G, Koo BK, Sasselli V, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–8.
Crane AM, Kramer P, Bui JH, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015;4:569–77.
Maule G, Casini A, Montagna C, Ramalho AS, de Boeck K, Debyser Z, Carlon MS, Petris G, Cereseto A. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat Commun. 2019. https://doi.org/10.1038/s41467-019-11454-9.
Hu W, Kaminski R, Yang F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci USA. 2014;111:11461–6.
Wang G, Zhao N, Berkhout B, Das AT. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018;244:321–32.
Bella R, Kaminski R, Mancuso P, et al. Removal of HIV DNA by CRISPR from patient blood engrafts in humanized mice. Mol Ther Nucleic Acids. 2018;12:275–82.
van Diemen FR, Kruse EM, Hooykaas MJG, Bruggeling CE, Schürch AC, van Ham PM, Imhof SM, Nijhuis M, Wiertz EJHJ, Lebbink RJ. CRISPR/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog. 2016;12:1–29.
Roehm PC, Shekarabi M, Wollebo HS, Bellizzi A, He L, Salkind J, Khalili K. Inhibition of HSV-1 replication by gene editing strategy. Sci Rep. 2016;6:1–11.
Donohoue PD, Barrangou R, May AP. Advances in industrial biotechnology using CRISPR-Cas systems. Trends Biotechnol. 2018;36:134–46.
Schwartz CM, Hussain MS, Blenner M, Wheeldon I. Synthetic RNA polymerase III promoters facilitate high-efficiency CRISPR-Cas9-mediated genome editing in yarrowia lipolytica. ACS Syn Biol. 2016;5:356–9.
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu JL. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol. 2014;32:947–51.
Montenegro M (2016) CRISPR is coming to agriculture—with big implications for food, farmers, consumers and nature. Ensia 1–15.
Carlson DF, Lancto CA, Zang B, Kim ES, Walton M, Oldeschulte D, Seabury C, Sonstegard TS, Fahrenkrug SC. Production of hornless dairy cattle from genome-edited cell lines. Nat Biotechnol. 2016;34:479–81.
Waltz E. Gene-edited CRISPR mushroom escapes US regulation. Nature. 2016;532:293.
Esvelt KM, Smidler AL, Catteruccia F, Church GM (2014) Concerning RNA-guided gene drives for the alteration of wild populations. eLife 3:1–21.
Reardon S. The crispr zoo. Nature. 2016;531:160–3.
Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, Joung JK, Peterson RT, Yeh JRJ. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS ONE. 2013;8:1–9.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. XOne-step generation of mice carrying reporter and conditional alleles by CRISPR/cas-mediated genome engineering. Cell. 2013;154:1370.
Bohannon J. Biologists devise invasion plan for mutations. Science. 2014;347(6228):1300.
Liang P, Xu Y, Zhang X, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6:363–72.
On human gene editing: international summit statement. The National Academies of Sciences. 2015. http://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=12032015a. Accessed 3 Dec 2015.
International Summit on Human Gene Editing. The National Academies of Sciences, Engineering, and Medicine. http://nationalacademies.org/gene-editing/Gene-Edit-Summit/. Accessed Dec 2015.
On Human Genome Editing II, Statement by the Organizing Committee of the Second International Summit on Human Genome Editing. In: The National Academies of Sciences, Engineering, and Medicine. 2018. http://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=11282018b. Accessed 28 Nov 2018.
Mo O. CRISPR-Cas9 human genome editing: challenges, ethical concerns and implications. J Clin Res Bioethics. 2015;06:5–7.
Garrity C. CRISPR mechanism. Medford: Tufts University; 2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Financial support in favour of Asiya Batool from the DBT-RA Program in Biotechnology and Life Sciences is gratefully acknowledged.
Conflicts of interest/competing interests
The authors confirm that they do not have any financial or non-financial competing interest in the publication of this paper.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Rights and permissions
About this article

Cite this article
Batool, A., Malik, F. & Andrabi, K.I. Expansion of the CRISPR/Cas Genome-Sculpting Toolbox: Innovations, Applications and Challenges. Mol Diagn Ther 25, 41–57 (2021). https://doi.org/10.1007/s40291-020-00500-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-020-00500-8