
Abstract
As part of its Single Technology Appraisal process, the National Institute for Health and Care Excellence (NICE) invited the manufacturer (Pfizer) of tofacitinib (TOF; Xeljanz®) to submit evidence of the drug’s clinical and cost-effectiveness in the treatment of rheumatoid arthritis (RA) after the failure of conventional disease-modifying antirheumatic drugs (cDMARDs). The School of Health and Related Research Technology Appraisal Group at the University of Sheffield was commissioned to act as the independent Evidence Review Group (ERG). The ERG produced a detailed review of the evidence for the clinical and cost-effectiveness of the technology, based upon the company’s submission to NICE. The clinical effectiveness evidence in the company’s submission for TOF is based predominantly on four randomised controlled trials (RCTs) comparing the efficacy of TOF against placebo. Three RCTs investigated TOF in combination with methotrexate (MTX), and one RCT investigated TOF monotherapy. All four RCTs compared TOF with placebo plus cDMARDs, one RCT also included adalimumab as a comparator. The study population in the four RCTs comprised patients who were MTX inadequate responders or cDMARD inadequate responders (cDMARD-IR). The company performed network meta-analyses (NMA) to assess the relative efficacy of TOF compared with biologic DMARDs (bDMARDs) in patients who were cDMARD-IR or bDMARD-IR with moderate-to-severe RA for European League Against Rheumatism (EULAR) response and change in the Health Assessment Questionnaire Disability Index at 6 months. The company’s NMA concluded that TOF had comparable efficacy to bDMARDs currently recommended by NICE. The company submitted a de novo model that assessed the cost-effectiveness of TOF versus its comparators in six different populations: (1) cDMARD-IR with severe RA; (2) cDMARD-IR with severe RA for whom MTX is contraindicated or not tolerated; (3) bDMARD-IR; (4) bDMARD-IR for whom rituximab (RTX) is contraindicated or not tolerated; (5) bDMARD-IR for whom MTX is contraindicated or not tolerated; and, (6) cDMARD-IR with moderate RA. According to the company’s economic analyses, in cDMARD-IR with severe RA, TOF plus MTX dominates or extendedly dominates most comparators, whilst TOF monotherapy is slightly less effective and less expensive than its comparators, with the cost saved per quality-adjusted life year (QALY) lost always higher than £50,000. In bDMARD-IR with severe RA, RTX plus MTX dominated TOF plus MTX, but in patients for whom RTX was not an option, TOF plus MTX dominated all comparators included in the analysis (four comparators recommended by NICE were not included). In cDMARD-IR with moderate RA, the cost per QALY for TOF in combination with MTX or as monotherapy compared with a sequence of cDMARDs was estimated to be greater than £50,000/QALY. The ERG identified a number of limitations in the company’s analyses, including use of a fixed-effects model in the NMA and the use of treatment sequences in the cost-effectiveness model which did not reflect NICE recommendations. These limitations were addressed partly by the company during the clarification round and partly by the ERG. The exploratory analyses undertaken by the ERG resulted in similar conclusions: (1) TOF plus MTX was dominated by RTX plus MTX; (2) TOF in combination with MTX or as monotherapy dominates or extendedly dominates some of its comparators in cDMARD-IR and bDMARD-IR with severe RA for whom RTX plus MTX was not an option; and (3) in cDMARD-IR with moderate RA, the cost per QALY of TOF in combination with MTX or as a monotherapy versus cDMARDs was in excess of £47,000. The NICE Appraisal Committee consequently recommended TOF plus MTX as an option for patients whose disease has responded inadequately to intensive therapy with a combination of cDMARDs only if (1) disease is severe [a Disease Activity Score (DAS28) of more than 5.1] and (2) the company provides TOF with the discount agreed in the Patient Access Scheme (PAS). TOF plus MTX is also recommended as an option for adults whose disease has responded inadequately to, or who cannot have, other DMARDs, including at least one bDMARD, only if (1) disease is severe, (2) they cannot have RTX, and (3) the company provides TOF with the discount agreed in the PAS. For patients who are intolerant of MTX, or where MTX is contraindicated, TOF monotherapy is recommended where TOF plus MTX would be recommended.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tofacitinib (TOF) plus methotrexate (MTX) has shown similar clinical efficacy and comparable costs to other recommended biologic disease-modifying antirheumatic drugs (bDMARDs) plus MTX in patients with rheumatoid arthritis (RA). |
In patients who are inadequate responders to bDMARDs (bDMARD-IR) and who are eligible for rituximab (RTX) in combination with MTX, RTX plus MTX is of similar clinical efficacy to TOF plus MTX, but has a significantly lower cost. Therefore, RTX plus MTX should be preferred to TOF plus MTX. |
In patients who have had an inadequate response to conventional disease-modifying antirheumatic drugs for whom MTX is contraindicated or not tolerated, TOF monotherapy has a similar efficacy and comparable costs to bDMARD monotherapies recommended by NICE in this population. There is no available evidence to compare the effectiveness of TOF monotherapy to that of recommended bDMARD monotherapies in bDMARD-IR. |
The relative simplicity of the decision when bDMARDs were the main comparator provides supportive evidence that fast-track appraisals, which have been proposed by NICE where efficacy and costs are comparable, can be delivered. |
1 Introduction
The National Institute for Health and Care Excellence (NICE) is an independent organisation responsible for providing national guidance on promoting good health and preventing and treating ill health in priority areas with significant impact. Health technologies must be shown to be clinically effective and to represent a cost-effective use of National Health Service (NHS) resources in order for NICE to recommend their use within the NHS in England. The NICE Single Technology Appraisal (STA) process usually covers new single health technologies within a single indication, soon after their UK market authorisation [1]. Within the STA process, the company provides NICE with a written submission, alongside a mathematical model that summarises the company’s estimates of the clinical and cost-effectiveness of the technology. This submission is reviewed by an external organisation independent of NICE [the Evidence Review Group (ERG)], which consults with clinical specialists and produces a report. After consideration of the company’s submission (CS), the ERG report and testimony from experts and other stakeholders, the NICE Appraisal Committee (AC) formulates preliminary guidance, the Appraisal Consultation Document (ACD), which indicates the initial decision of the AC regarding the recommendation (or not) of the technology. Stakeholders are then invited to comment on the submitted evidence and the ACD, after which a further ACD may be produced or a Final Appraisal Determination (FAD) issued, which is open to appeal. An ACD is not produced when the technology is recommended within its full marketing authorisation; in this case, a FAD is produced directly.
This paper presents a summary of the ERG report [2] for the STA of tofacitinib (TOF; Xeljanz®) in combination with methotrexate (MTX) for treating rheumatoid arthritis (RA) for patients whose disease has responded inadequately to at least one conventional disease-modifying antirheumatic drug (cDMARD) or biologic DMARD (bDMARD), and TOF as monotherapy in the case of intolerance to MTX or when treatment with MTX is inappropriate. The paper includes a summary of the subsequent development of the NICE guidance for the use of this technology in England. Full details of all relevant appraisal documents (including the appraisal scope, ERG report, company and consultee submissions, FAD and comments from consultees) can be found on the NICE website [3].
2 The Decision Problem
RA is a chronic inflammatory disease characterised by progressive, irreversible, joint damage; impaired joint function; and pain and tenderness caused by swelling of the synovial lining of joints [4]. The condition is associated with increasing disability and reduced quality of life [4]. The primary symptoms are pain; morning stiffness; swelling; tenderness; loss of movement; redness of the peripheral joints; and fatigue [5, 6]. RA is associated with substantial costs both directly (associated with drug acquisition and hospitalisation) and indirectly due to reduced productivity [7]. RA has long been reported as being associated with increased mortality [8, 9], particularly due to cardiovascular events [10]. NICE estimates that there are 400,000 patients in the UK with RA [11], based on a prevalence of 0.8% reported by Symmons et al. [12]. The incidence of RA is greater in females (3.6 per 100,000 per year) than in males (1.5 per 100,000 per year) [13]. For both genders, the peak age of incidence in the UK is in the eighth decade of life, but all ages can develop the disease [13].
Two classifications have dominated the measurement of improvement in RA symptoms: American College of Rheumatology (ACR) responses [14] and European League Against Rheumatism (EULAR) responses [15]. ACR response has been widely adopted in randomised controlled trials (RCTs) although studies have shown that the value of the measure can vary between studies due to the timing of the response [16]. In the UK, monitoring the progression of RA is often undertaken using the Disease Activity Score of 28 joints (DAS28). The DAS28 can be used to classify both the disease activity of the patient and the level of improvement estimated within the patient. The EULAR response criteria use the individual change in DAS28 and the absolute DAS28 score to classify a EULAR response as good, moderate or none [15]. EULAR response has been reported less frequently in RCTs than ACR responses [17]. However, EULAR response is much more closely aligned to the treatment continuation rules stipulated by NICE, which require either a moderate or good EULAR response or a DAS28 improvement of more than 1.2 to continue treatment with bDMARDs.
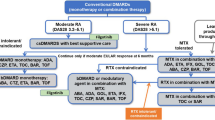
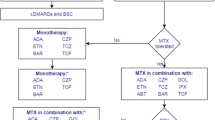
2.1 Current Treatment
For patients with newly diagnosed RA, NICE recommends considering a combination of cDMARDs, including MTX and at least one other cDMARD plus short-term glucocorticoids, as first-line treatment, ideally beginning within 3 months of the onset of persistent symptoms [11]. NICE currently recommends the use of the bDMARDs baricitinib (BARI), abatacept (ABA), adalimumab (ADA), certolizumab pegol (CZP), etanercept (ETA), golimumab (GOL), infliximab (IFX), and tocilizumab (TCZ), each in combination with MTX for patients who have severe active RA (defined as a DAS28 score greater than 5.1) after the failure to respond to cDMARD treatment. For patients who meet these criteria but for whom MTX is contraindicated or has been withdrawn, NICE recommends the use of ADA, CZP, ETA and TCZ as monotherapy [18]. Most of these bDMARDs (all except BARI, ABA and TCZ) are tumour necrosis factor alpha inhibitors (TNFi). After the failure of the first TNFi, NICE recommends rituximab (RTX) in combination with MTX for the treatment of severe active RA [19]. If RTX is contraindicated or withdrawn because of an adverse event (AE), NICE recommends ABA, ADA, BARI, CZP, ETA, GOL, IFX or TCZ in combination with MTX [18, 20, 21]. If MTX is contraindicated or withdrawn because of an AE, NICE recommends ADA or ETA [22] as monotherapy. NICE also recommends TCZ in combination with MTX as a third-line biologic after inadequate response to RTX in combination with MTX [20].
Treatment continuation criteria vary across Technology Assessments (TAs): TA375 [19] states that for patients to continue treatment with their first bDMARD, they must achieve and maintain at least a moderate EULAR response. For RTX, TA195 [23] states that treatment should be continued only if there is an improvement in the DAS28 score of at least 1.2 points at initiation of treatment and whilst this response is maintained. If the relevant continuation criterion is not met, then the treatment should be stopped and the next treatment in the sequence initiated.
3 The Independent Evidence Review Group (ERG) Review
In accordance with the process for STAs, the ERG and NICE had the opportunity to seek clarification on specific points in the CS [24], in response to which the company provided additional information. The ERG also modified the company’s decision analytic model to produce an ERG base case and to assess the impact of alternative parameter values and assumptions on the model results. The evidence presented in the CS and the ERG’s review of that evidence is summarised here.
3.1 Clinical Evidence Provided by the Company
Evidence was presented in the CS for the efficacy of TOF in combination with MTX and other cDMARDs or as monotherapy in the treatment of moderate to severe RA in patients after the failure of disease-modifying antirheumatic drugs (DMARDs). This evidence was based primarily on four RCTs (ORAL Standard [25], ORAL Scan [26] and ORAL Sync [27] for TOF plus MTX; ORAL Solo for TOF monotherapy [28]). MTX plus placebo was the comparator in ORAL Scan and ORAL Sync; placebo without MTX was the comparator in ORAL Solo; and an active treatment (ADA) and placebo were the comparators in ORAL Standard. In addition to treatment groups receiving the licensed dose of TOF at 5 mg twice daily, all four RCTs also included treatment groups receiving TOF 10 mg twice daily (a currently unlicensed dose). Preliminary results were also provided for a recently completed, head-to-head trial (ORAL Strategy) of TOF plus MTX versus ADA plus MTX versus TOF monotherapy.
The population in ORAL Standard and ORAL Scan related to adults with active moderate-to-severe RA who were cDMARD experienced and MTX inadequate responders (MTX-IRs). The population in ORAL Sync and ORAL Solo was adults with active moderate-to-severe RA who were DMARD inadequate responders (DMARD-IRs). ORAL Solo had a 24-week randomised period, ORAL Standard and ORAL Sync had a 52-week randomised period, and ORAL Scan had a 104-week randomised period. In ORAL Standard, ORAL Scan and ORAL Sync, patients receiving placebo advanced to TOF 5 mg at month 3 if trial response criteria were not met (defined as a 20% reduction in the number of tender and swollen joints). A co-primary outcome for ORAL Standard, ORAL Scan and ORAL Sync was the proportion of patients achieving an ACR20 response at 6 months. A co-primary outcome for ORAL Solo was the proportion of patients achieving an ACR20 response at 3 months. The primary endpoint for the ORAL Strategy trial was ACR50 response at 6 months. Other relevant endpoints in the included trials included the proportion of patients achieving low disease activity at 3 and 6 months, and the proportion of patients achieving disease remission at 3 months using the DAS28 outcome.
Using the co-primary outcome of ACR20, the three trials of TOF plus MTX showed TOF 5 mg twice daily plus MTX to be statistically superior to placebo plus cDMARD (p ≤ 0.001) at 6 months. Other statistically significant results (p ≤ 0.001) were demonstrated across these trials versus placebo for ACR50, ACR70, and treatment response using EULAR criteria and Health Assessment Questionnaire Disability Index (HAQ-DI) at both 3 and 6 months, with the following exceptions:
-
1.
The proportion achieving disease remission using DAS28 erythrocyte sedimentation rate (ESR) in ORAL Scan at 6 months when using the step-down statistical approach
-
2.
The change in baseline HAQ-DI in ORAL Scan at 6 months when using the step-down statistical approach.
ACR20 was significant for TOF monotherapy versus placebo at 3 months in one trial (ORAL Solo), but not significant for the primary endpoint of the proportion achieving remission using DAS28ESR at 3 months. As all patients crossed over from placebo to receive TOF at 3 months in ORAL Solo, there are no placebo-controlled results at 6 months for the other relevant endpoints. The ORAL Strategy trial showed TOF combination therapy with MTX to be non-inferior to ADA plus MTX (< 0.0001), but TOF monotherapy was not found to be non-inferior to both TOF plus MTX and ADA plus MTX for the primary endpoint of ACR50 at 6 months (p = 0.0512).
A revised summary of safety data for TOF provided by the company following an ERG request showed that the highest incidence rates of AEs were for serious infection events and herpes zoster. Additional data provided by the company indicated bronchitis, pneumonia and all cardiac disorders occurred most commonly in the TOF treatment arms.
Network meta-analyses (NMA) were performed to assess the relative efficacy of TOF compared with the comparators in patients who were inadequate responders to cDMARDs (cDMARD-IR) or to bDMARDs (bDMARD-IR) with moderate-to-severe RA for EULAR response and change in the HAQ-DI at 6 months. For the base case NMA cDMARD-IR population, the odds of achieving a EULAR response were all statistically higher for TOF in combination with MTX (TOF plus cDMARD) compared to cDMARD at 6 months. No statistically significant differences were found for TOF plus cDMARD versus bDMARDs plus cDMARD, except for TOC plus cDMARD, which was statistically superior in attaining at least a good EULAR response.
Whilst the odds of all EULAR responses were higher in TOF monotherapy compared to cDMARD, only the effect for a good response was statistically significant. No statistically significant differences were found in TOF versus bDMARDs. Both TOF plus cDMARD and TOF monotherapy were associated with significant reduction in HAQ-DI compared with cDMARD at 6 months.
For the base case NMA bDMARD-IR population, the odds of all EULAR responses were all statistically higher in TOF plus cDMARD compared with cDMARD at 6 months. No statistically significant differences were found for TOF plus cDMARD versus ABA plus cDMARD. TOF plus cDMARD was statistically superior compared to GOL plus cDMARD in attaining both at least a moderate and a good EULAR response, but statistically inferior versus RTX plus cDMARD, TOF plus cDMARD, non-TNFi plus cDMARD and TNFi plus cDMARD. TOF in combination with cDMARD was associated with a significant reduction in HAQ-DI compared with cDMARD at 6 months.
3.1.1 Critique of the Clinical Evidence and Interpretation
The eligibility criteria applied in the selection of evidence for the clinical effectiveness review were considered by the ERG to be reasonable and generally consistent with the decision problem as outlined in the final NICE scope.
Whilst the ERG considered the company’s literature searches to be sufficient and comprehensive to retrieve relevant and up-to-date data for clinical effectiveness, the searches for safety data were not sufficient to identify all up-to-date relevant AE data for TOF. The CS reported that a “data cut” was imposed on the safety data such that only trials included in the published pooled analysis [29], which included trial data up to March 2015, were included in the safety overview. In a response to the ERG’s request for clarification, the company confirmed that a separate search for AEs was not undertaken and that data on AEs were identified as part of a broader search of efficacy, safety and health-related quality of life.
The ERG noted that pooling safety data across all trials and providing incidence rates may be inappropriate to fully document the potentially different safety profiles of TOF combination therapy with MTX versus TOF monotherapy. The 2017 European Public Assessment Report highlighted “a higher incidence of adverse events for the combination of Xeljanz with MTX, compared with Xeljanz as monotherapy” and that “combination of tofacitinib with methotrexate increased the risk of ALT [alanine transaminase] elevation compared with tofacitinib monotherapy” [30].
The quality of the included RCTs for TOF was assessed using well established and recognised criteria. Data for extra-articular manifestations of disease were not included in the CS. Mortality data were presented for “death within 30 days of last dose of study drug” in the pooled safety analysis.
NMA were performed separately for both the cDMARD-IR and bDMARD-IR populations using a Bayesian approach for EULAR response at month 6 and change from baseline HAQ-DI score at month 6. Trials in the analysis of the cDMARD-IR population were largely the same as those in the NMA undertaken by the independent Assessment Group in NICE TA375 [19]. A similar comparison could not be made for the bDMARD-IR population, as this was outside of the scope of TA375.
The ERG believes that the results presented in NMA of clinical effectiveness should be treated with caution, as the ordered categorical EULAR data were dichotomised in the cDMARD-IR population, which ignores the natural ordering and correlations between the EULAR response categories. A fixed-effects model was used in all the analyses in the bDMARD-IR population, and EULAR response (moderate response and good response) in the cDMARD-IR population. Heterogeneity is expected, and this approach underestimates uncertainty in the treatment effects. For TOF trials with early escape, the results from non-responder imputation (NRI) without advancement penalty (NRI only applied for the placebo arm, not the TOF arm) were used in the base case NMA. This imputation approach potentially overestimates the relative treatment effect of TOF in these trials. Depending on the NRI approach applied to the TOF trials with early escape, the conclusion for the efficacy ranking of TOF among the bDMARDs varies markedly.
3.2 Cost-Effectiveness Evidence Provided by the Company
The company supplied a de novo discrete event simulation model constructed in Microsoft Excel® that was largely based on the model developed by the assessment group in TA375 [19]. The model simulates patients’ disease progressions as they go through the sequences of treatments being compared. For each line of treatment, patients may achieve good, moderate or no EULAR response, which is assessed at 6 months after treatment initiation. The EULAR response rates for TOF as a monotherapy or in combination with MTX are estimated using a regression model calculated based on TOF trial data. The EULAR response rates for the comparator treatments are calculated by applying odds ratios (ORs) based on the company’s NMA to TOF response rates. Patients who achieve moderate or good EULAR response are assumed to have an improvement in HAQ-DI score and remain on treatment until loss of efficacy (as assessed by a clinician), AE occurrence or death. Time to treatment discontinuation for responders is estimated using survival curves fitted to TOF trial data using patient characteristics as predictive covariates. Patients who fail to achieve a moderate or good EULAR response at 6 months discontinue treatment and move on to the next treatment in the sequence. HAQ-DI is assumed to remain constant whilst on bDMARDs or TOF, whilst for patients on cDMARDs and palliative care, HAQ-DI progression is assumed to be non-linear based on latent HAQ-DI trajectory classes [31]. Patients are assumed to experience an increase in HAQ-DI equal to the value of the decrease in HAQ-DI achieved on treatment initiation, with the increase occurring over the 6 months before treatment discontinuation, when the next treatment in the sequence is employed. The mortality rate is assumed to be affected by the patient’s HAQ-DI score at baseline, but not by HAQ-DI progression. The model estimates the costs and quality-adjusted life years (QALYs) over a lifetime horizon from the perspective of the NHS and Personal Social Services. Health-related quality of life was modelled using values from the EuroQol 5 Dimensions (EQ-5D) questionnaire. Changes in EQ-5D are estimated based on a mapping algorithm from HAQ-DI scores and patient characteristics. Hospitalisation costs and resource use estimates were based on HAQ-DI score bands as in NICE TA375 [19], with unit costs taken from the British National Formulary (BNF) [32] and NHS Reference Costs for 2015/2016 [33]. A Patient Access Scheme (PAS) for TOF had been agreed to with the Department of Health.
The analyses presented in the CS relate to six different populations of RA patients: (1) cDMARD-IR with severe RA who can tolerate MTX; (2) cDMARD-IR with severe RA for whom MTX is contraindicated or not tolerated; (3) bDMARD-IR for whom RTX is an option; (4) patients who are bDMARD-IR and RTX ineligible; (5) bDMARD-IR for whom MTX is contraindicated or not tolerated; and (6) patients with moderate RA who are cDMARD-IR. Severe RA was defined as a DAS28 of > 5.1, whilst moderate RA was defined as a DAS28 of > 3.2 and ≤ 5.1. Baseline characteristics of patients are based on the relevant TOF studies.
In the analyses presented by the company for cDMARD-IR with severe RA who could tolerate MTX, TOF plus MTX dominated or extendedly dominated the majority of bDMARD comparators; the incremental cost-effectiveness ratios (ICERs) of the remaining comparators were higher than £80,000 per QALY gained. In cDMARD-IR with severe RA for whom MTX was contraindicated or not tolerated, TOF is less effective and less expensive than the recommended bDMARDs (ETA, ADA and TCZ), but the cost saved per QALY lost (southwest quadrant) is higher than £50,000. In bDMARD-IR with severe RA for whom RTX was an option, RTX plus MTX dominated TOF plus MTX. In bDMARD-IR with severe RA for whom RTX is not an option, TOF plus MTX dominated all the comparators included in the analysis (although four recommended comparators were not included). In bDMARD-IR with severe RA for whom MTX was contraindicated or not tolerated, the ICER for TOF compared with TCZ was estimated to be £25,932 per QALY gained. However, TCZ monotherapy is not recommended by NICE in this population and none of the comparators recommended by NICE were included in the analysis. In cDMARD-IR with moderate RA, the ICER for TOF plus MTX compared with a sequence of cDMARD treatments was estimated to be £51,693 per QALY gained and the ICER for TOF monotherapy compared with a different sequence of cDMARDs was estimated to be £51,370 per QALY. All of the analyses presented excluded the commercial-in-confidence PASs in place for TCZ and ABA, as requested by NICE.
The company presented additional analyses during the clarification round amending the NMA and incorporating the following corrections requested by the ERG: (1) amended sequences in line with TA375; (2) used non-linear latent class HAQ-DI trajectories for palliative care; (3) amended changes in HAQ-DI scores upon moderate or good EULAR response; (4) used age at onset instead of age as predictor of class membership for the latent class mixture model; and (5) activated the flag that establishes a patient as a bDMARD-IR after going through their first bDMARD or Janus kinase inhibitor. The analyses undertaken with the revised model resulted in slightly different ICERs, but did not modify the conclusions of the analyses included in the CS.
3.2.1 Critique of the Cost-Effectiveness Evidence and Interpretation
The company’s original economic analysis contained several issues, the most important being that the sequences used in the model did not appropriately reflect NICE recommendations and the model assumed a constant worsening of HAQ-DI instead of using the non-linear HAQ-DI trajectories observed by Norton et al. [31]. The ERG communicated these shortcomings during the clarification round, and the company presented new analyses after addressing these issues. The ERG believes that the company’s revised analyses included a number of limitations. First, relevant comparators recommended by NICE were missing from the company’s analyses: in the analysis for bDMARD-IR MTX-intolerant patients with severe RA, all relevant comparators (ADA, ETA and CZP as monotherapies) were missing, and in the analysis for bDMARD-IR with severe RA who were RTX-ineligible, four comparators (ADA, ETA, IFX and CZP with concomitant MTX) were missing. The company justified these omissions citing the lack of evidence for the missing comparators in the relevant populations. Second, the company’s NMA suffered from a series of limitations, as described in Sect. 3.1.1. For example, the treatment effect was estimated by applying an NRI only in the placebo arm (estimate 1), instead of applying NRI in both arms (estimate 2). The ERG believes that the true treatment effect lies between these two estimates and that this uncertainty should have been explored in sensitivity analyses. Third, the company assumed TOF as monotherapy to have equal efficacy to TOF plus MTX in terms of moderate and good EULAR response rates. However, in the ORAL Strategy trial [34], TOF monotherapy was not found to be non-inferior to TOF plus MTX and the results of the NMA show that TOF monotherapy results in slightly lower rates of response compared with TOF plus MTX. Fourth, the company assumed sulfasalazine to have the same efficacy as placebo in the analysis for the cDMARD-IR MTX-intolerant population. The ERG believes this leads to an underestimation of the efficacy of sulfasalazine. Finally, the company rounded modified HAQ-DI values to the nearest valid HAQ-DI score rather than allowing the valid HAQ-DI score to be sampled based on the continuous HAQ-DI value. The ERG notes that this approach might lead to inaccurate estimations of HAQ-DI scores, as values might be rounded up more often than they are rounded down or vice versa.
3.3 Additional Work Undertaken by the ERG
The ERG undertook additional analyses after applying two changes to the company’s model: (1) calculating the ORs for all treatments including monotherapies compared to TOF plus MTX instead of assuming TOF monotherapy to have the same efficacy as TOF plus MTX; and (2) sampling a valid HAQ-DI score based on the modified HAQ-DI score, as in TA375 [19]. The ERG undertook two sets of analyses for each population: one based on the company’s NMA and the other based on the NMA requested by the ERG (referred to as clarification NMA) addressing the issues described in Sect. 3.1.1. and applying NRI in both arms. The results presented here do not include the confidential PASs in place for TCZ and ABA.
For cDMARD-IR with severe RA who can tolerate MTX, based on the company’s NMA, TOF plus MTX dominated all bDMARD comparators except ETA biosimilar plus MTX. Based on the clarification NMA, TOF plus MTX dominated ADA plus MTX, but was extendedly dominated in the full incremental analysis. For cDMARD-IR with severe RA for whom MTX was contraindicated or not tolerated, TOF and TCZ monotherapy extendedly dominated ADA and ETA biosimilar regardless of the NMA used. The ICER of TCZ compared with TOF was £51,488 and £50,430 per QALY gained using the company’s NMA and using the clarification NMA, having removed the constraint that TOF monotherapy had the same efficacy as TOF plus MTX, respectively.
In the bDMARD-IR with severe RA for whom RTX was an option, RTX plus MTX dominated TOF plus MTX regardless of the NMA used. Replacing TCZ plus MTX with TOF plus MTX after RTX plus MTX was estimated to result in ICERs of £67,852 and £90,846 per QALY lost using the company’s and the clarification NMA, respectively. In the bDMARD-IR with severe RA for whom RTX was not an option, TOF plus MTX dominated GOL plus MTX regardless of the NMA used, and dominated ABA plus MTX also when using the company’s NMA. The ICER of ETA biosimilar and TCZ in combination with MTX compared with TOF plus MTX was higher than £30,000 per QALY gained regardless of the NMA used. Finally, in patients with moderate RA who are cDMARD-IR, the ICER of TOF plus MTX compared with MTX was £47,594 and £50,708 per QALY gained using the company’s and the clarification NMA, respectively.
3.4 Conclusions of the ERG Report
The systematic review of clinical effectiveness showed TOF plus MTX to be superior to placebo plus cDMARD in the target population across a number of relevant primary endpoints. The company’s NMA of clinical effectiveness showed that TOF plus cDMARD was superior to cDMARD and comparable to bDMARDs. Evidence to support the clinical effectiveness of TOF monotherapy in those who cannot tolerate MTX is less robust. The company presented results of analyses based on a de novo economic model. According to the company’s analyses, TOF plus MTX dominates some of its comparators in cDMARD-IR and bDMARD-IR patients with severe RA who can tolerate MTX and for whom RTX is not an option. TOF monotherapy also dominates some of its comparators in cDMARD-IR with severe RA for whom MTX is contraindicated or not tolerated (no evidence exists for the relevant comparators in bDMARD-IR with severe RA who are MTX intolerant). In the cDMARD-IR population with moderate RA, the ICERs of TOF plus MTX versus MTX and TOF monotherapy versus cDMARDs are in excess of £47,000 per QALY gained. TOF plus MTX was dominated by RTX plus MTX. The ERG identified a number of limitations in the company’s analyses. The company addressed some of these limitations during the clarification round, but the following issues remained: relevant comparators recommended by NICE were not included in the analyses; the NMA is subject to potential limitations; the company assumed equal efficacy for TOF as monotherapy and for TOF in combination with MTX; and the company rounded modified HAQ-DI values to the nearest valid HAQ-DI score rather than allowing the valid HAQ-DI score to be sampled based on the continuous HAQ-DI value. The ERG undertook exploratory analyses alleviating these issues except for the first and providing results based on two alternative NMAs. The results of the exploratory analyses carried out by the ERG were slightly different to those presented by the company, but did not significantly impact the conclusions.
4 Key Methodological Issues
The main limitation of the amended economic analysis is the exclusion of relevant comparators from two of the six populations because of the lack of available evidence. The ERG believes that the analyses would have been improved by making assumptions on the efficacy of these comparators based on their relative efficacy in other populations and exploring the uncertainty in sensitivity analyses. The ERG notes that the conclusions of the company’s analyses tally with the expectations before constructing a mathematical model, given the comparable efficacy and costs of the intervention to that of the comparators. The relative simplicity of this decision provides supportive evidence that abbreviated appraisals, which have been proposed by NICE [35], can be delivered under conditions such as those in the TOF STA.
5 National Institute for Health and Care Excellence Guidance
In October 2017, on the basis of the evidence available (including verbal testimony of invited clinical experts and patient representatives), the NICE AC produced guidance that TOF in combination with MTX was recommended as an option for patients with severe RA whose disease has responded inadequately to intensive therapy with a combination of cDMARDs and also for patients with severe RA who have responded inadequately to, or who cannot have, other DMARDs, including at least one bDMARD and where treatment with RTX plus MTX was not an option. The AC also produced guidance that TOF monotherapy was recommended under the same criteria as TOF plus MTX, where RTX plus MTX was contraindicated or not tolerated. All recommendations were conditional on the company providing TOF with the agreed PAS.
5.1 Consideration of Clinical and Cost-Effectiveness Issues Included in the Final Appraisal Determination
This section summarises the key issues considered by the AC. The full list of the issues considered by the AC can be found in the FAD [3].
5.1.1 Current Clinical Management
The AC considered the current clinical management of severe active RA following inadequate response to a TNFi in England and noted that the NICE guidance recommends BARI, CZP, ADA, ETA, IFX, ABA, TCZ and GOL (each with MTX) as options when RTX (plus MTX) is contraindicated or not tolerated, and ADA and ETA monotherapy as alternative options if RTX therapy cannot be given because MTX is contraindicated or not tolerated. The AC heard from clinical experts that responses to bDMARDs differ between patients and therefore it is important to have a range of options for bDMARD treatments. The AC was aware that the marketing authorisation covers the use of TOF in moderate to severe disease, but that TA375 [19] recommends that treatment with a bDMARD should only be started when disease is severe, that is a disease activity (DAS28) score of more than 5.1.
5.1.2 Uncertainties in the Clinical Evidence
The AC considered the problems highlighted by the ERG with the methods used in the company’s NMA. These problems included different models for EULAR response in the two populations; a random effects model for DMARD-IRs and a fixed-effects model for patients whose disease responded to bDMARDs; a uniform prior in the random effects model; the use of estimate 1 in their base case; and the method of linking ETA to the network. Also, studies reporting EULAR responses were synthesised with converted EULAR response outcomes from studies that only reported ACR responses. At the clarification stage, the company corrected the errors in their NMA. The committee was satisfied that the corrected NMA was suitable for decision-making and showed that TOF works as well as bDMARDs.
The AC heard from clinical experts that there is a need for new treatment options, particularly when there is an inadequate response to cDMARDs or bDMARDs. They also noted that there are subtly different AEs across the different classes of drugs for RA, but the AEs associated with Janus kinase inhibitors are unlikely to influence their desire to prescribe the drug. Both the clinical and patient experts highlighted that TOF is given orally, which has benefits for both patients and the health system. The patient experts emphasised that this is an important factor for patients who have difficulty injecting themselves because of the disease affecting their hands.
5.1.3 Uncertainties in the Economic Modelling
The AC concluded that the ERG’s amended model was adequate for its decision-making. For bDMARD-IR who could receive RTX plus MTX, the AC noted the uncertainty on the incremental cost-effectiveness of the elongated sequence where TOF plus MTX was provided after RTX plus MTX. The AC concluded that TOF was not a cost-effective use of NHS resources in this population. The AC recognised the considerable uncertainty about the effectiveness of TOF monotherapy in bDMARD-IR, but noted that in the appraisal of BARI [36], the AC concluded that BARI monotherapy has similar clinical effectiveness to that of BARI in combination with cDMARDs. The AC concluded that its recommendations for TOF plus MTX should also apply to TOF monotherapy for bDMARD-IR with severe RA for whom MTX was contraindicated or not tolerated.
6 Conclusions
The evidence suggests that TOF plus MTX or as monotherapy has a similar efficacy for treating severe active RA following inadequate response to DMARDs to that of bDMARDs already recommended by NICE. Therefore, TOF plus MTX or as monotherapy was considered by NICE to be a cost-effective use of NHS resources for patients for whom RTX or MTX are contraindicated or not tolerated. However, the cost of RTX treatment is significantly lower than that of TOF with comparable efficacy so TOF was not considered by NICE to be a cost-effective use of NHS resources when RTX and MTX is a treatment option for a patient.
References
National Institute for Health and Care Excellence. Guide to the methods of technology appraisal. 2013. https://www.nice.org.uk/article/pmg9/. Accessed 26 Jan 2018.
Uttley L, et al. Evidence review group report: tofacitinib for treating moderate to severe active rheumatoid arthritis after the failure of disease-modifying anti-rheumatoid drugs: a Single Technology Appraisal. 2017. https://www.nice.org.uk/guidance/ta480/documents/committee-papers. Accessed 26 Feb 2018.
National Institute for Health and Care Excellence. Rheumatoid arthritis—tofacitinib [ID526]. NICE Guidance, UK 2017. https://www.nice.org.uk/guidance/indevelopment/gid-tag438. Accessed 26 Jan 2018.
Scott DL, Steer S. The course of established rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2007;21(5):943–67.
Pincus T, et al. Early radiographic joint space narrowing and erosion and later malalignment in rheumatoid arthritis: a longitudinal analysis. J Rheumatol. 1998;25(4):636–40.
Drossaers-Bakker KW, et al. Radiographic damage of large joints in long-term rheumatoid arthritis and its relation to function. Rheumatology. 2000;39(9):998–1003.
Allaire S, et al. Current risk factors for work disability associated with rheumatoid arthritis: recent data from a US national cohort. Arthritis Care Res. 2009;61(3):321–8.
Naz SM, Symmons DPM. Mortality in established rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2007;21(5):871–83.
Dadoun S, et al. Mortality in rheumatoid arthritis over the last fifty years: systematic review and meta-analysis. Jt Bone Spine. 2013;80(1):29–33.
Meune C, et al. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology. 2009;48(10):1309–13.
National Collaborating Centre for Chronic Conditions, Rheumatoid arthritis: national clinical guideline for management and treatment in adults. 2009, Royal College of Physicians (UK).
Symmons D, et al. The prevalence of rheumatoid arthritis in the United Kingdom: new estimates for a new century. Rheumatology. 2002;41(7):793–800.
Symmons DPM, et al. The incidence of rheumatoid arthritis in the United Kingdom: results from the Norfolk Arthritis Register. Rheumatology. 1994;33(8):735–9.
Felson DT, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheumat. 1995;38(6):727–35.
van Gestel AM, et al. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis: comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism criteria. Arthritis Rheumat. 1996;39(1):34–40.
Felson DT. Assessing the efficacy and safety of rheumatic disease treatments: obstacles and proposed solutions. Arthritis Rheumat. 2003;48(7):1781–7.
Stevenson M, et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol Assess. 2016;20(35):1–610.
National Institute for Health and Care Excellence (NICE), Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for rheumatoid arthritis not previously treated with DMARDs or after conventional DMARDs only have failed. 2016.
National Institute for Health and Clinical Excellence (NICE), Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for rheumatoid arthritis not previously treated with DMARDs or after conventional DMARDs only have failed. 2016.
National Institute for Health and Clinical Excellence (NICE), Tocilizumab for the treatment of rheumatoid arthritis. 2012.
National Institute for Health and Clinical Excellence (NICE), Golimumab for the treatment of rheumatoid arthritis after the failure of previous disease-modifying anti-rheumatic drugs. 2011.
National Institute for Health and Clinical Excellence (NICE), Adalimumab, etanercept, infliximab, rituximab and abatacept for the treatment of rheumatoid arthritis after the failure of a TNF inhibitor. 2010, NICE: London.
Wu EQ, et al. Mapping FACT-P and EORTC QLQ-C30 to patient health status measured by EQ-5D in metastatic hormone-refractory prostate cancer patients. Value Health. 2007;10:408–14.
Pfizer I. Tofacitinib citrate for treating moderate to severe rheumatoid arthritis. Company Submission to NICE. 2017.
van Vollenhoven RF, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367(6):508–19.
van der Heijde D, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheumat. 2013;65(3):559–70.
Kremer J, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159(4):253–61.
Fleischmann R, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. 2017;390(10093):457–68.
Cohen SB, et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheumat Dis. 2017.
European Medicines Agency. Assessment report Xeljanz. 2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004214/WC500224913.pdf. Accessed 26 Feb 2018.
Norton S, et al. Health Assessment Questionnaire disability progression in early rheumatoid arthritis: systematic review and analysis of two inception cohorts. Semin Arthritis Rheumat. 2014;44(2):131–44.
British National Formulary, British National Formulary (BNF) 64. 2012.
Department of Health. National Schedule of Reference Costs 2015-16 for NHS Trusts. Total average cost of outpatient attendance for Service Code 410 (Rheumatology). Sheet: Total Outpatient Attendances. 2016. https://www.gov.uk/government/publications/nhs-reference-costs-2015-to-2016. Accessed 26 Feb 2018.
Pfizer I. Protocol A3921187. A phase 3b/4 randomized double blind study of 5 mg of tofacitinib with and without methotrexate in comparison to adalimumab with methotrexate in subjects with moderately to severely active rheumatoid arthritis. Statist Anal Plan. 2014.
National Institute for Health and Care Excellence. Abbreviated technology appraisal process consultation. 2016. https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-technology-appraisal-guidance/abbreviated-technology-appraisal-process-consultation. Accessed 26 Jan 2018.
National Institute for Health and Care Excellence (NICE). Baricitinib for moderate to severe rheumatoid arthritis. 2017. https://www.nice.org.uk/guidance/ta466. Accessed 26 Feb 2018.
Acknowledgements
This summary of the ERG report was compiled after NICE issued the FAD. All authors have commented on the submitted manuscript and have given their approval for the final version to be published. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of NICE or the Department of Health. Any errors are the responsibility of the authors.
Author information
Authors and Affiliations
Contributions
LU and MM-SJ critiqued the clinical effectiveness data reported by the company. IB and MS critiqued the mathematical model provided and the cost-effectiveness analyses submitted by the company. SR critiqued the NMA performed by the company. Ruth Wong critiqued the literature searches undertaken by the company. DLS and AY provided clinical advice to the ERG throughout the project. All authors were involved in drafting and commenting on the final document. LU acts as the guarantor of the manuscript. This summary has not been externally reviewed by PharmacoEconomics.
Corresponding author
Ethics declarations
Funding
This project was funded by the National Institute for Health Research (NIHR) Health Technology Assessment Programme (Project Number 19/04/17). Visit the HTA programme website for further project information (http://www.hta.ac.uk).
Conflict of interest
David Scott has no conflicts of interest relating to tofacitinib. However, his department has received a peer-reviewed grant from Pfizer within the last 12 months to undertake academic research on polypharmacy in arthritis. The department has also received free etanercept from Pfizer to use in an NIHR-funded programme grant in rheumatoid arthritis. David Scott and Matt Stevenson declare that they have written a commentary on janus kinase inhibitors for The Lancet (http://dx.doi.org/10.1016/S0140-6736(17)31659-8). The Lancet editorial was written after the completion of this STA. Lesley Uttley, Iñigo Bermejo, Shijie Ren, Marrissa Martyn-St James, Ruth Wong and Adam Young have no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Uttley, L., Bermejo, I., Ren, S. et al. Tofacitinib for Treating Rheumatoid Arthritis After the Failure of Disease-Modifying Anti-rheumatic Drugs: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. PharmacoEconomics 36, 1063–1072 (2018). https://doi.org/10.1007/s40273-018-0639-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-018-0639-0