
Abstract
The widespread use of drugs for unapproved purposes remains common in children, primarily attributable to practical, ethical, and financial constraints associated with pediatric drug research. Pharmacometrics, the scientific discipline that involves the application of mathematical models to understand and quantify drug effects, holds promise in advancing pediatric pharmacotherapy by expediting drug development, extending applications, and personalizing dosing. In this review, we delineate the principles of pharmacometrics, and explore its clinical applications and prospects. The fundamental aspect of any pharmacometric analysis lies in the selection of appropriate methods for quantifying pharmacokinetics and pharmacodynamics. Population pharmacokinetic modeling is a data-driven method (‘top-down’ approach) to approximate population-level pharmacokinetic parameters, while identifying factors contributing to inter-individual variability. Model-informed precision dosing is increasingly used to leverage population pharmacokinetic models and patient data, to formulate individualized dosing recommendations. Physiologically based pharmacokinetic models integrate physicochemical drug properties with biological parameters (‘bottom-up approach’), and is particularly valuable in situations with limited clinical data, such as early drug development, assessing drug–drug interactions, or adapting dosing for patients with specific comorbidities. The effective implementation of these complex models hinges on strong collaboration between clinicians and pharmacometricians, given the pivotal role of data availability. Promising advancements aimed at improving data availability encompass innovative techniques such as opportunistic sampling, minimally invasive sampling approaches, microdialysis, and in vitro investigations. Additionally, ongoing research efforts to enhance measurement instruments for evaluating pharmacodynamics responses, including biomarkers and clinical scoring systems, are expected to significantly bolster our capacity to understand drug effects in children.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pharmacometrics uses mathematical models to understand how drugs work. It is a valuable discipline to help fill unmet research needs on drugs in children. |
The gathering of high-quality data is crucial for constructing models that provide insights into the exposure actions of drugs in children. |
Population-pharmacokinetic models create customized dosing recommendations for a large group of children with specific diseases, using data from studies with samples of a limited number of children. |
Physiologically based pharmacokinetic models predict how drugs spread in the body using information about physiology and the physicochemical characteristics of the drug. |
Pharmacometric models can be complex, and many pediatricians may not be familiar with them. Therefore, it is crucial to foster collaborations between pediatricians and pharmacometrics to bring this knowledge to children. |
1 Introduction
Most pediatricians frequently prescribe medications to children and adolescents, but only a minority participate actively in drug development and research, even though there are clear unmet needs in the field of pediatric pharmacology [1]. In recent years, there has been an increasing recognition of the pressing demand for safe and effective drugs specifically targeted to pediatric populations. This consensus is shared by professional societies, regulatory bodies, and the general public [2,3,4]. In response to this imperative, regulatory authorities introduced various incentives and mandates to promote pediatric drug research. For example, the European Medicines Agency has required the submission of a Pediatric Investigation Plan for all new drug submissions since 2007 [5]. Furthermore, the Best Pharmaceuticals for Children Act was enacted by the US Congress and the US Food and Drug Administration in 2002 to catalyze research on new medications. On the contrary, the Pediatric Research Equity Act incentivizes research on existing medicines [6, 7], which is of particular importance, as a significant proportion of licensed drugs administered to children still lack specific dosages, indications, or age-related labeling [8,9,10,11,12]. Pediatric drug studies, marked by their time-consuming and resource-intensive nature, often face premature discontinuation because of challenges in patient recruitment [13, 14]. Pharmacometrics, a discipline focused on mathematically modeling how drugs are processed by the body (pharmacokinetics) and their effects on the body (pharmacodynamics), has emerged as a pivotal discipline to address some of the limitations to advance pediatric drug research. This discipline significantly contributes to pediatric drug development, personalized pharmacotherapy, and drug repurposing. Nevertheless, clinicians frequently lack familiarity with the methodology inherent in pharmacometric studies, a proficiency crucial for leveraging its advantages in clinical practice. In this review, we first succinctly review developmental alterations influencing pharmacokinetics and pharmacodynamics in children. Subsequently, we outline methods for the quantification and analysis of pharmacokinetic (PK) and pharmacodynamic (PD) responses. Following this, we discuss the clinical applications of pharmacometric study designs. Finally, we contemplate prospective developments within the field.
2 Pediatric Drug Responses: Age and Maturational Alterations in Pharmacokinetics and Pharmacodynamics
Developmental pharmacology investigates changes in therapeutic and adverse drug reactions during infancy and childhood. Table 1 outlines the developmental aspects of PK processes, and readers interested in more in-depth insights are encouraged to explore previously published comprehensive reviews on this topic [15,16,17,18].
Drug absorption involves movement from the administration site to the bloodstream, critical for all routes of administration except an intravascular injection. Bioavailability, a parameter most pertinent to drug absorption, denotes the proportion of a drug that reaches the systemic circulation, which is inherently 100% after intravascular administration [19]. Drugs distribute rapidly to highly perfused organs and later to tissues with slower perfusion rates. The extent of distribution is influenced by physicochemical properties of the drug and biological factors. Egress from the intravascular space involves passive diffusion, carrier-mediated, or active transport processes [20]. Volume of distribution describes the relationship between the amount of drug administered and the measured concentration. It is an apparent volume, as the volume of distribution can surpass any physiologic volume required to contain all the drug within the body at the measured concentration. Metabolism modifies a drug into a polar hydrophilic molecule, promoting drug elimination, a process crucial for lipophilic drugs [21]. Elimination (clearance), which begins as soon as the drug is absorbed, involves drug excretion (i.e., removal from the body), with renal and hepatic routes primarily involved.
Research on maturational changes in pharmacodynamics is limited compared to pharmacokinetics. Nevertheless, structural modifications and alterations in signal transduction pathways during infancy and childhood affect the potency, denoting organ or tissue sensitivity, and the efficacy, signifying the maximum response, of a drug [29]. Substantial disparities in drug responses between pediatric and adult populations are documented, such as fluoroquinolone-induced tendon rupture being exceedingly rare in children but observed more frequently in the elderly [30]. Moreover, children exhibit a higher tolerance for digoxin, but may paradoxically display heightened susceptibility to the immunosuppressive effects of dexamethasone and cyclosporine [29, 31]. Additionally, the pediatric population exhibits different PD responses compared with adults to benzodiazepines, antipsychotics, and antidepressants [32, 33]. Children may manifest paradoxical responses to benzodiazepines, showcasing heightened agitation and cognitive effects contrary to the anticipated sedating effects observed in adults. With antipsychotics, pediatric patients are more susceptible to metabolic side effects, such as weight gain, dyslipidemia, and insulin resistance, along with a greater likelihood of hormonal disturbances such as hyperprolactinemia. Additionally, the use of antidepressants in children involves an elevated risk of suicidality, especially during the initial phases of treatment. These diverse PD responses underscore the imperative for tailored and vigilant prescribing practices when administering these medications to children.
3 Quantifying Pharmacokinetics
Selecting an appropriate specimen for a drug concentration analysis is crucial (Table 2) [34]. For instance, considering the ease of collection, dried blood spots or saliva may be preferable for children, offering a practical alternative to venipuncture-based samples such as plasma. However, it is imperative to acknowledge that the choice of specimen method may impact the measured drug concentrations. In this context, it is essential to recognize that precision, gauging the consistency and reproducibility of repeated measurements, and accuracy, assessing the proximity of measurements to the true value, play pivotal roles in ensuring the reliability of PK data. Furthermore, microdialysis has gained popularity to obtain samples to analyze tissue concentrations of drugs, for example, in the brain and in the muscles, bones, and brain [35, 36]. This technique involves the insertion of fine probes into tissues or extracellular spaces, allowing the continuous sampling of interstitial fluid, which proved real-time data on local drug concentrations at the target site [37].
Standardized operating procedures are essential for all sample collections to mitigate pre-analytical factors, such as patient and sample misidentification. Potential contamination during a sample collection, such as through lines also used for medication administration, should be avoided. The selection of the appropriate collection medium is as crucial, as exposure to light, heat, or substances within collection media may contribute to drug degradation. Rigorous documentation of sampling and drug administration times is paramount.
Chromatographic analysis methods, in particular liquid chromatography/mass spectrometry and high-performance liquid chromatography, have become indispensable for measuring drug concentrations [43, 44]. Liquid chromatography separates drug compounds based on their structural properties, while mass spectrometry identifies and quantifies them based on their mass-to-charge ratio. Liquid chromatography/mass spectrometry is ideal for detecting drugs in complex biological samples with high precision and accuracy [44]. It is particularly valuable in measuring low concentrations of drugs and metabolites. In contrast, high-performance liquid chromatography utilizes high-pressure pumps to move a liquid solvent containing the drug through a chromatographic column. The separated components are then detected, using a spectrophotometric detector, often based on ultraviolet light. Compared with liquid chromatography/mass spectrometry, high-performance liquid chromatography is a slightly less sensitive, but efficient, simpler, and less expensive method [43]. Meanwhile, immunoassays are also capable of quantifying drug concentrations, and are the method most often available through clinical laboratories because of their efficiency and costs, but decreased selectivity, sensitivity, and specificity compared with chromatographic methods remain limitations [45, 46].
4 Measuring Pharmacodynamic Responses
Evaluating pharmacodynamic responses introduces more challenges than investigating pharmacokinetic responses. While a conventional therapeutic window spanning from minimally effective to toxic concentrations exists, these precise values are often elusive [47]. For certain drugs such as antibiotics and anti-cancer medications, defined thresholds, such as the minimal inhibitory concentration, provide in vitro response benchmarks. However, diverse measurement instruments are essential to quantify PD responses for other drugs, necessitating adherence to criteria related to relevance, reliability, reproducibility, responsiveness, and appropriateness concerning the child’s age and developmental stage [48]. In chronic conditions, adjusting measurement instruments becomes imperative to accommodate the expected spontaneous evolution influencing the PD response over time. Further details and examples of common PD metrics are provided in Table 3 [49].
Clinical scoring systems, which are structured tools employed to quantify health conditions, demand validation to precisely gauge meaningful changes in disease severity following drug treatments [50]. Exemplarily, the Acute Otitis Media Severity of Symptom Scale score was designed to measure the therapy response in children receiving treatment for acute otitis media, and included crying, ear tugging, eating behavior, and fever [51].
Biomarkers, objective indicators of normal biological, pathogenic, or pharmacologic processes, prove indispensable when quantifying clinical responses is challenging, or as a proxy for events in the future [48]. As an illustration, creatinine is a widely available biomarker for assessing the glomerular filtration rate and monitoring renal toxicities of drugs. Yet, increments in creatinine do not necessarily represent renal failure. This is most exemplary in patients taking trimethoprim, in whom creatinine may underestimate the glomerular filtration rate, as trimethoprim inhibits the tubular secretion of creatinine [52]. Therefore, multiple biomarkers or other instruments may be used to measure pharmacodynamics. Recently, Walsh et al. investigated the PD responses of salbutamol in children experiencing asthma attacks, by integrating the Pediatric Asthma Severity Scale score and the biomarkers lactate, pH, and glucose [53]. Using this approach, the authors established infusion rates that achieve maximal clinical effects while minimizing toxicities. In conclusion, the process of selecting and validating biomarkers as indicators of PD response or proxies for later PD outcomes presents multiple challenges. These challenges necessitate careful consideration to ensure the accuracy and relevance of biomarker measurements in clinical and research contexts.
5 Population Pharmacokinetic Modeling
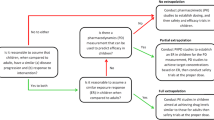
In traditional PK studies (Fig. 1a), concentration–time profiles are individually constructed for each participant. Subsequently, classical non-compartmental analysis methods are employed to compute descriptive statistics of exposure metrics such as maximum concentration, area under the curve, and half-life [54]. These studies necessitate nearly identical sampling schedules for each subject and are limited in their capacity to analyze covariates explaining inter-individual variability [54, 55]. In contrast, the utilization of mathematically advanced, non-linear mixed-effects models has gained popularity in pediatric population PK (popPK) studies, as illustrated in Fig. 1b. The term ‘mixed effects’ encompasses a combination of fixed parameters, variables that describe the behavior of a typical individual in the population under study, and random-effects parameters that can fluctuate across individuals and within an individual over time. In these studies, drug concentrations from multiple individuals are aggregated into a single dataset. Non-linear mixed-effects regression methods are then applied to analyze both central tendencies for the population and variations between individuals and timepoints [54]. This approach allows the model to include both predictable sources of variability (e.g., weight, renal function) and unpredictable sources (e.g., measurement error) to describe the pharmacokinetics of a drug. Furthermore, the design allows flexibility in sampling schedules and sparse sampling, which is a major advantage in children compared to traditional PK studies.

a Traditional or non-compartmental pharmacokinetic (PK) study. Individual PK profiles are analyzed per participant, which requires an almost identical sampling schedule for every subject. Pharmacokinetic parameters, such as maximum concentration (Cmax), area under the curve (AUC), or half-life (t1/2) determined per subject, after which population values are calculated using descriptive statistics. The therapeutic window is defined as the area between the minimum effective and minimum toxic concentration. b The structural model provides a mathematical representation of the typical PK profile for the studied population. This model is enhanced by the covariate model, which incorporates subject-specific characteristics, or covariates, to explain variations in drug pharmacokinetics within a population. Covariates can include demographic factors such as age, sex, and weight; physiological parameters such as renal and liver function; genetic factors, such as polymorphisms in drug-metabolizing enzymes or transporters; and other clinical variables, including concurrent medications. For example, in this context, Subject 1 and Subject 2 illustrate the PK pattern for the covariate weight, where Subject 2 is described as a “high weight” individual compared to Subject 1. This contrast demonstrates the fixed effect of a covariate in pharmacokinetics relative to the “typical subject”. The statistical model addresses residual or unexplained variability, representing the proportion of the data not captured by the structural model and covariates. This component of the model helps account for differences in PK profiles that are not explained by the identified factors
Developing popPK models, as depicted in Fig. 1b, involves three interconnected steps. First, a structural model is fitted to the population data to delineate the central tendency of the concentration–time profile for the entire population. This structural model can be characterized as either a one-compartment or multiple-compartment model, with the central compartment being an abstract representation of plasma and tissues directly impacted by drug distribution [56]. Multiple-compartment models introduce peripheral compartments, where drug distribution occurs at a slower rate. Additionally, distinct absorption kinetics are incorporated for drugs not administered intravascularly. Second, a statistical model is constructed to compute the variability between subjects, across timepoints, and the unexplained variability around the structural model. Last, the analysis of covariates is undertaken to identify predictors contributing to the variation around the structural model. The process of selecting the best-fitting model for describing the population data is arduous and consists of goodness-of-fit plots and mathematical diagnostics.
A covariate analysis plays a pivotal role in facilitating dosing adjustments within specific patient subsets. For instance, renal function significantly impacts the clearance of medications eliminated by the kidneys. To comprehensively assess the influence of renal function on drug clearance, an ideal popPK study would encompass a pediatric population with a spectrum of renal function estimates, which may result from maturation, disease-related factors, or a combination of both. In such a scenario, a dosing nomogram could be developed to offer tailored guidance for individuals with normal, impaired, or enhanced renal function. The covariate model can encompass various clinical parameters, biomarkers, or co-medications. However, the selection of potential covariates is primarily contingent on scientific plausibility, statistical significance, and clinical relevance. It is important to note that the number of predictors is limited by the data that are available for analysis as employing an excessive number of predictors may compromise the precision of predictions [58].
Ideally, popPK models should undergo an evaluation in an external population to broaden their applicability beyond the model development cohort. Additionally, prescribers should remain cognizant of the specific population for which a drug was originally labeled, as the sources of PK variability may not become apparent until the drug is administered to a more diverse patient group. When a unique population was not included in the initial study, it becomes imperative to conduct supplementary popPK studies. This necessity is particularly pronounced in neonates, given their distinctive and evolving physiology. For instance, propofol dosing in neonates traditionally considered body weight and gestational age. However, a popPK model revealed that clearance in neonates is more accurately predicted by separating gestational and postnatal maturation, with postnatal maturation taking precedence after birth [57]. Consequently, reduced doses of propofol are recommended for premature infants during their postnatal maturation phase. In contrast, for other drugs in infants, such as propranolol, weight or size may be the primary covariate predicting pharmacokinetics [58].
Critical illness is a condition warranting specific popPK studies, as this condition potentially alters all PK processes. For instance, a popPK model examining phenobarbital in neonates and infants undergoing extracorporeal membrane oxygenation unveiled significantly increased clearance when continuous venovenous hemodiafiltration was employed, necessitating higher phenobarbital doses for these children [59]. Similarly, other popPK studies have identified subsets of critically ill children with augmented renal clearance [60]. This recognition informs dosing decisions, suggesting that intermittent administration of renally eliminated antibiotics might suffice for critically ill children with acute renal failure, while higher doses, prolonged infusions, or continuous infusions may be more appropriate for those with augmented renal clearance [61,62,63]. In childhood obesity, the volume of distribution for lipophilic drugs such as fentanyl undergoes changes, leading to unpredictability in serum steady-state concentrations. A popPK study conducted in obese children identified a model-based continuous infusion strategy, aimed at optimizing the probability of reaching target serum fentanyl concentrations in this population [64].
6 Physiologically Based Pharmacokinetic Modeling
Physiologically based PK (PBPK) models predict how drugs distribute in various tissues by incorporating the drug’s physicochemical properties and biological parameters into mathematical functions. In contrast to a data-driven popPK model, a PBPK model is predominantly a knowledge-based method. Physiologically based PK models comprise three primary components: model structure, organism parameters, and drug parameters [65]. A typical PBPK model features compartments representing organs or tissues interconnected through the circulatory system, as depicted in Fig. 2a [66]. Organism parameters encompass anatomical and physiological characteristics; these are typically available in literature resources. Each compartment is defined by specific attributes such as volume, tissue composition, and perfusion blood flow rates [67]. Enzymatic processes occur within relevant intracellular compartments, with connections to the extracellular space facilitated by passive diffusion or active drug transport. Blood flow governs mass transfer between compartments, and its rate varies for each organ [68, 69]. Maturation of physiological processes or changes in pathophysiological trends, such as the glomerular filtration rate, can be captured in mathematical functions. These functions can subsequently aid in exploring or predicting the pharmacokinetics of other compounds following a similar elimination pathway [70, 71].

a Overview of a physiologically based pharmacokinetic (PBPK) model structure, adapted from [65]. Each block represents a compartment, either organs or tissues connected through the blood circulation. C represents the rest of the organs and tissues that composed the model structure. b Building blocks of a pediatric PBPK model. An intravenous (IV) PBPK model of healthy adults issued as a basis, information about absorption kinetics needs to be added to an oral (PO) PBPK model. The adult PBPK model needs to be adjusted for ontogeny data and pharmacokinetic data in healthy children to transform into PBPK models for healthy children. Subsequently, data on pathophysiological changes are needed to develop a PBPK model for ill children. Finally, pharmacokinetic data in ill children are required for model validation in ill children. PK pharmacokinetics
Finally, drug parameters encompass substance-specific characteristics describing the interaction between the drug and the system. These parameters comprise physicochemical properties including molecular weight, lipophilicity, solubility, the fraction of unbound drug, and tissue-plasma partitioning. All these factors hold significant importance in predicting membrane permeability, availability for gastrointestinal absorption, transmembrane transport, and partitioning within tissues, organs, and plasma [67]. Clearly, the biological system in a specific subgroup of children with a given trait (e.g., chronic kidney disease, cystic fibrosis) differs from healthy children. As a result, both organ and drug parameters must be integrated into highly specific models that mathematically represent the drug’s interaction with the system. To achieve this, there are commercially available validated tools, such as PK-Sim, MoBI®, and Simcyp® [72, 73]. Those platforms also have the capability to account for the maturation of physiological processes and trends in pathophysiological processes, enabling the modeling of drug exposure in populations of children with specific comorbidities.
Figure 2b simplifies and illustrates the development of a pediatric PBPK model. It starts with a validated PBPK model in healthy adults as a foundation, with subsequent adaptations of organ parameters to match the anatomical and physiological parameters of healthy children. This includes incorporating ontogeny functions related to drug metabolism and elimination. Ultimately, by adjusting the model to account for pathophysiological alterations in healthy children, drug exposure can be estimated for the population of children with a specific disease [74]. The evaluation of the model is critical to ensure its accuracy, often accomplished by comparing PBPK-based simulated data to in vivo observations for a predictive performance analysis. These simulations are considered acceptable when they fall within a two-fold prediction error or, for more stringent criteria, within a 70–130% prediction range.
Physiologically based models prove particularly valuable when limited clinical data are available, such as in early-phase drug development or when adapting existing drugs for new indications. Enhanced understanding of maturation and pathophysiology can improve our comprehension of PK alterations related to diseases in children. Physiological functions within PBPK models can be adjusted to account for disease states, enabling predictions of drug disposition under these circumstances [75, 76]. Exemplarily, a pediatric PBPK model estimated the impact of reduced cardiac output on propofol clearance, which is age dependent and proportionally greater in adults. Consequently, age-specific dose reductions for propofol were recommended in low cardiac output states. Additionally, a PBPK study in children with obesity revealed that relative to children with a healthy weight, those with obesity experienced reduced weight-normalized clearance for clindamycin and trimethoprim/sulfamethoxazole [77]. Nonetheless, simulations still supported the recommended weight-based dosing in children with obesity. Last, other PBPK models supported the use of adult metformin doses in older children and adolescents with obesity, offering valuable insights into potential drug labeling for this unique population [78, 79].
Physiologically based PK models offer significant potential to enhance drug therapies for children across various clinical scenarios. For example, PBPK models can address knowledge gaps concerning drug distribution in anatomical sites that are challenging to sample [80]. The adequate penetration of antibiotics into the cerebrospinal fluid is crucial for treating central nervous system infections such as meningitis and encephalitis. A generic PBPK model was developed to predict the passive drug transfer in the cerebrospinal fluid in children, both with and without meningitis. This model incorporated age-appropriate parameters and underwent validation for drugs such as paracetamol, ibuprofen, flurbiprofen, naproxen, and meropenem [81]. It serves as a valuable template for models focused on cerebrospinal fluid penetration. Furthermore, an adult PBPK model was scaled down for pediatrics, accounting for different elimination routes, and successfully predicted the disposition of drugs such as meropenem, colistin, and sulbactam in the blood, lung, skin, and heart tissues [82]. Physiologically based modeling can also estimate drug penetration in human milk, contributing to the understanding of drug safety during lactation. A workflow concerning lactation-related exposure to lamotrigine illustrates its feasibility, including incorporation of the variability in milk concentrations [83].
Finally, PBPK modeling proves invaluable in adapting dosing recommendations to account for drug–drug interactions. For example, rifampicin, a potent cytochrome P450 (CYP) inducer, can reduce the effective concentrations of many drugs, including antiretroviral drugs such as lopinavir and ritonavir, potentially compromising HIV control. A PBPK model simulated lopinavir/ritonavir exposure in children between 2 months and 8 years of age across different rifampicin doses, suggesting a super-boosted lopinavir/ritonavir regimen that could achieve adequate drug concentrations when co-administered with rifampicin [84]. Similarly, phenobarbital, another potent enzyme inducer, was found to have a greater impact on the pharmacokinetics of tacrolimus than in adults, as estimated by a pediatric PBPK model [85]. Moreover, in the absence of PK data for tadalafil in children aged under 2 years with pulmonary arterial hypertension, a recent pediatric PBPK model was developed, incorporating CYP3A-mediated intrinsic clearance and its ontogeny (CYP3A4 and CYP3A7) [86]. This model accounted for the common co-administration of tadalafil with bioseston, a CYP3A4 inducer, and recommended dosing adjustments for children taking both drugs.
Integration of PD responses into PBPK models is contingent upon the availability of quantitative mechanic insights into the drug under investigation. For drugs that exert their effect via binding to a receptor, efficacy hinges on the availability of receptors for binding, as the classic receptor occupancy theory posits that drug effects are proportionate to the fraction of receptors occupied [87]. Hence, acquiring information pertaining to the maturation of drug receptors and signaling processes during infancy and childhood becomes imperative for predicting PK/PD outcomes. Verscheijden et al. undertook a modeling effort to elucidate the PK/PD responses to morphine in both children and adults, recognizing substantial differences in therapeutic responses even when serum concentrations remained comparable [88]. They sourced data regarding intracellular morphine transport through in vitro studies and leveraged the existing literature to glean insights into blood–brain permeability over the course of lifetime. Remarkably, in their final model, disparities in PD responses among neonates were not attributable to variations in morphine disposition in brain tissues, but rather to differences in the brain’s susceptibility to morphine.
7 Therapeutic Drug Monitoring and Model-Informed Precision Dosing
Therapeutic drug monitoring (TDM) is a clinical practice employed to measure and analyze drug concentrations, aiming to optimize dosages, ensure therapeutic efficacy, and monitor for drug toxicities [89]. In conventional TDM, drug concentrations in blood (either serum or plasma) guide individual patient dosing [90]. Specific criteria must be met to consider TDM for a drug [91]. First, a therapeutic window must be established with a target for effectiveness and/or toxicity. Therapeutic drug monitoring proves futile in the absence of a predictable relationship between drug concentrations and outcomes, and for drugs whose drug responses are obvious clinically. Second, a validated assay must exist to quantify drug concentrations in a timeframe that is relevant to the patient. For drugs administered long term on an outpatient basis, it may be reasonable to get results of TDM a day or two later. However, long turnaround times can render TDM ineffective in acute care settings. Third, a significant inter-individual variation in PK responses is essential. Last, the measured drug concentration should reflect the free (i.e., unbound) drug, critical for a pharmacological effect. Clinical assays frequently measure total drug concentrations, and the variability in protein binding, both intra-individually and inter-individually, can present challenges in establishing a therapeutic window. Additionally, therapeutic windows are commonly defined at a population level, neglecting patient-specific factors affecting treatment outcomes or toxicity at any dose. Relying on a single drug concentration measurement (e.g., maximum or minimum concentration) often restricts clinicians to fractional dose adjustments, limiting the precision of personalized dosing based on only TDM. Fortunately, more sophisticated tools in the form of model-informed precision dosing (MPID) have become increasingly available to guide individual dosing [92]. Model-informed precision dosing leverages drug behavior knowledge from popPK models along with patient data (i.e., drug concentrations, relevant covariate data) to estimate individual PK parameters more accurately [93, 94]. In brief, MIPD utilizes data from a popPK model, known as the Bayesian prior, to describe the typical PK parameters (and their variability) in a population in the absence of any patient-specific information. When patient information becomes available (e.g., weight, renal function, measured drug concentrations), mathematical algorithms are then updated to determine how this new individual’s data fit within the pre-existing population data, calculating what is referred to as a (Bayesian) posterior probability and generating predictions of the individual’s pharmacokinetics. From this, individualized target-oriented dosing recommendations can be made. While most MIPD software programs can generate dosing recommendations simply based on covariate information (e.g., weight, renal function), incorporation of measured drug concentrations greatly improves the estimation of patient-specific PK/PD parameters and the precision of dosing guidance [95]. Model-informed precision dosing has proven efficacy in promoting PK/PD target attainment for individual patients for vancomycin and aminoglycosides, among other drugs [96,97,98]. There are several commercial software programs available to assist clinicians in MIPD implementation [99, 100]. Furthermore, MIPD allows the use of multiple models at the same time by either applying model selection or model averaging techniques, which allows clinicians to tailor the predictions to their specific patient populations without having to develop their own model [101, 102].
8 Future Perspectives
Despite advancements in pediatric pharmacology, significant knowledge gaps persist for many drugs in children. Pharmacometrics emerges as a promising discipline with the potential to enhance the safety and efficacy of pediatric pharmacotherapy. It accomplishes this by expediting drug development, extending drug applications to new indications, and enabling personalized care through MIPD [103,104,105]. In this review, we outlined diverse pharmacometric study designs and underscored their potential to expand our understanding of pharmacotherapy for children. Population PK models elucidate predictors for PK responses, facilitating optimized dosing within specific subpopulations. Pediatric PBPK models integrate the dynamic physiological and biochemical changes of childhood and disease into mathematical functions, predicting drug disposition across tissues and PD responses if quantitative mechanistic data are available [106, 107]. These models prove instrumental in managing drug–drug interactions, adapting dosing regimens to various clinical scenarios, and assessing different drug formulations tailored to the pediatric population [108,109,110]. However, overcoming barriers is crucial to fully integrate pharmacometrics into routine clinical practice. First and foremost, increasing the data available for analysis is paramount to advance reliable models. Opportunistic and non-invasive sampling techniques, along with microdialysis, are promising avenues for expanding data availability for popPK models [111]. Microdosing studies provide insights into absorption and drug disposition. Moreover, routinely collected clinical data can potentially be transformed into PD endpoints, offering insights into variations in therapy responses. Furthermore, opportunistic sampling in children involves collecting clinical specimens during routine medical procedures [112, 113]. This method maximizes data acquisition without additional discomfort or invasive procedures and minimizes the burden on young patients while enhancing the efficiency of research endeavors. Ongoing research on the validation of biomarkers for PD is pertinent for understanding therapy response differences [114]. Biobanks play a vital role by increasing data for PBPK models through collecting body samples for in vitro studies and furthering our knowledge of developmental pathophysiology. Leveraging machine learning and artificial intelligence can efficiently manage the growing volume of data needed for pharmacometrics [115]. These tools enable the rapid identification of complex patterns within large diverse datasets, facilitating the design of more tailored studies for children, including improved covariate modeling, but are not yet routinely used in pediatric drug development or clinical practice and were therefore not within the scope of this review. In the same sense, quantitative systems pharmacology models, which aim to model the interaction between the drug and the biological system in which it operates on a fundamental level, are not yet used at the same frequency as the techniques described in this review. Quantitative systems pharmacology models are, however, readily applied in the drug discovery space and might in future contribute to pediatric drug development [116]. Developing pediatric pharmacometric models is an arduous task, underscoring the importance of intensive collaborations between pharmacometricians and clinicians. Such collaborations are essential for data collection and the translation of advanced modeling techniques into clinical practice.
9 Conclusions
Pharmacometrics, employing mathematical models to understand pharmacokinetics and pharmacodynamics, emerges as a pivotal discipling in pediatric drug development, facilitating drug repurposing endeavors, and enhancing personalized pharmacotherapy. Selecting an appropriate specimen, adherence to standardized operating procedures, consideration of pre-analytical factors, and utilization of a validated measurement assay are essential for accurately and precisely measuring drug concentrations. Instruments for measuring pharmacodynamics should demonstrate validity, precision, suitability for the age and developmental stage, responsiveness to changes over time, and consideration of the natural progression of diseases. Non-linear mixed-effect popPK models analyze inter-individual variability and predictors for PK responses in a population. Ideally, these models should be validated in external populations. Physiologically based PK modeling is a knowledge-based approach that predicts drug distribution in tissues by incorporating physicochemical characteristics and biological parameters. This is a particularly helpful approach in situations where there are very limited in vivo data available. Therapeutic drug monitoring optimizes subsequent dosages using drug concentrations, while MIPD employs popPK models and individual patient data for a more sophisticated and personalized approach to guide drug dosing and enhance therapeutic efficacy. Collaborations between clinicians and pharmacometricians are indispensable, not only for robust data collection to inform models, but also for the translation and integration of these models in clinical practice.
References
Subramanian D, Cruz CV, Garcia-Bournissen F. Systematic review of early phase pediatric clinical pharmacology trials. J Pediatr Pharmacol Ther. 2022;27:609–17.
Joseph PD, Craig JC, Caldwell PHY. Clinical trials in children. Br J Clin Pharmacol. 2015;79:357–69.
Schupmann W, Li X, Wendler D. Acceptable risks in pediatric research: views of the US public. Pediatrics. 2021;149:1–10.
Burckart GJ, Kim C. The revolution in pediatric drug development and drug use: therapeutic orphans no more. J Pediatr Pharmacol Ther. 2020;25:565–73.
Bourgeois FT, Kesselheim AS. Promoting pediatric drug research and labeling: outcomes of legislation. N Engl J Med. 2022;381(9):875–81.
Baum VC, Bax R, Heon D, Yang Z, Sakiyama M. Pediatric drug regulation: international perspectives. Paediatr Anaesth. 2019;29:572–82.
Moore-Hepburn C, Rieder M. Paediatric pharmacotherapy and drug regulation: moving past the therapeutic orphan. Br J Clin Pharmacol. 2022;88:4250–7.
Sharif R, Aamir M, Shakeel F, Faisal S, Khan JA. Pharmacoepidemiological assessment of off-label drug use in pediatric ambulatory departments at four tertiary care hospitals in Pakistan. Trop J Pharm Res. 2020;19:2219–25.
Allen HC, Garbe MC, Lees J, Aziz N, Miller JL, Johnson P, et al. Off-label medication use in children, more common than we think: a systematic review of the literature. J Okla State Med Assoc. 2019;111:776–83.
Hoon D, Taylor MT, Kapadia P, Gerhard T, Strom BL, Horton DB. Trends in off-label drug use in ambulatory settings: 2006–2015. Pediatrics. 2019;144:1–19.
Tukayo BLA, Sunderland B, Parsons R, Czarniak P. High prevalence of off-label and unlicensed paediatric prescribing in a hospital in Indonesia during the period Aug–Oct 2014. PLoS ONE. 2020;15:1–13.
Yackey K, Stukus K, Cohen D, Kline D, Zhao S, Stanley R. Off-label medication prescribing patterns in pediatrics: an update. Hosp Pediatr. 2019;9:186–93.
Hwang TJ, Orenstein L, Kesselheim AS, Bourgeois FT. Completion rate and reporting of mandatory pediatric postmarketing studies under the US Pediatric Research Equity Act. JAMA Pediatr. 2019;173:68–74.
Srivastava A, Bourgeois FT. Evaluation of publication of pediatric drug trials. JAMA Netw Open. 2021;4:14–7.
van den Anker J, Reed MD, Allegaert K, Kearns GL. Developmental changes in pharmacokinetics and pharmacodynamics. J Clin Pharmacol. 2018;58:S10-25.
Smits A, Annaert P, Cavallaro G, De Cock PAJG, de Wildt SN, Kindblom JM, et al. Current knowledge, challenges and innovations in developmental pharmacology: a combined conect4children Expert Group and European Society for Developmental, Perinatal and Paediatric Pharmacology White Paper. Br J Clin Pharmacol. 2022;88:4965–84.
Van Groen BD, Nicolaï J, Kuik AC, Van Cruchten S, Van Peer E, Smits A, et al. Ontogeny of hepatic transporters and drug-metabolizing enzymes in humans and in nonclinical species. Pharmacol Rev. 2021;73:597–678.
Lu H, Rosenbaum S. Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther. 2014;19:262–76.
Nicolas JM, Bouzom F, Hugues C, Ungell AL. Oral drug absorption in pediatrics: the intestinal wall, its developmental changes and current tools for predictions. Biopharm Drug Dispos. 2017;38:209–30.
Wanat K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol Biol Rep. 2020;47:3221–31.
Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx. 2005;2(4):554–71. https://doi.org/10.1602/neurorx.2.4.554.
Debotton N, Dahan A. A mechanistic approach to understanding oral drug absorption in pediatrics: an overview of fundamentals. Drug Discov Today. 2014;19:1322–36.
Batchelor HK, Marriott JF. Paediatric pharmacokinetics: key considerations. Br J Clin Pharmacol. 2015;79:395–404.
Bansal N, Momin S, Bansal R, Gurram Venkata SKR, Ruser L, Yusuf K. Pharmacokinetics of drugs: newborn perspective. Pediatr Med. 2024;7:19.
Kiss M, Mbasu R, Nicolaï J, Barnouin K, Kotian A, Mooij MG, et al. Ontogeny of small intestinal drug transporters and metabolizing enzymes based on targeted quantitative proteomics. Drug Metab Dispos. 2021;49:1038–46.
Baptista JP. Augmented renal clearance. In: Antibiotic pharmacokinetic/pharmacodynamic considerations in the critically ill. Editors: Udy A, Roberts J, Lipman J. Adis, Singapore. https://doi.org/10.1007/978-981-10-5336-8_7
Quigley R. Developmental changes in renal function. Curr Opin Pediatr. 2012;24:184–90.
Gattineni J, Baum M. Developmental changes in renal tubular transport: an overview. Pediatr Nephrol. 2015;30:2085–98.
Mulla H. Understanding developmental pharmacodynamics: importance for drug development and clinical practice. Pediatr Drugs. 2010;12:223–33.
Ross RK, Kinlaw AC, Herzog MM, Funk MJ, Gerber JS. Fluoroquinolone antibiotics and tendon injury in adolescents. Pediatrics. 2021;147: e2020033316.
Pacifici GM. Clinical Pharmacology of Digoxin in Infants and Children. Clin Med. 2021;3(2):1036.
Moon YE. Paradoxical reaction to midazolam in children. Korean J Anesthesiol. 2013;65:2–3.
Liu XI, Schuette P, Burckart GJ, Green DJ, La J, Burnham JM, et al. A comparison of pediatric and adult safety studies for antipsychotic and antidepressant drugs submitted to the United States Food and Drug Administration. J Pediatr. 2019;208:236-242.e3.
Plebani M. Errors in clinical laboratories or errors in laboratory medicine? Clin Chem Lab Med. 2006;44:750–9.
Himebauch AS, Sankar WN, Flynn JM, Sisko MT, Moorthy GS, Gerber JS, et al. Skeletal muscle and plasma concentrations of cefazolin during complex paediatric spinal surgery. Br J Anaesth. 2016;117:87–94.
Dalmage MR, Nwankwo A, Sur H, Nduom E, Jackson S. A scoping review of pediatric microdialysis: a missed opportunity for microdialysis in the pediatric neuro-oncology setting. Neurooncol Adv. 2022;4:1–8.
Ketharanathan N, Yamamoto Y, Rohlwink UK, Wildschut ED, Mathôt RAA, De Lange ECM, et al. Combining brain microdialysis and translational pharmacokinetic modeling to predict drug concentrations in pediatric severe traumatic brain injury: the next step toward evidence-based pharmacotherapy? J Neurotrauma. 2019;36:111–7.
Dilo A, Daali Y, Desmeules J, Chalandon Y, Uppugunduri CRS, Ansari M. Comparing dried blood spots and plasma concentrations for busulfan therapeutic drug monitoring in children. Ther Drug Monit. 2020;42:111–7.
Klak A, Pauwels S, Vermeersch P. Preanalytical considerations in therapeutic drug monitoring of immunosuppressants with dried blood spots. Diagnosis. 2019;6:57–68.
Doriety LJ, Farrington EA. Urine drug screening: what pediatric clinicians need to know to optimize patient care. J Pediatr Health Care. 2021;35:449–55.
Hutchinson L, Sinclair M, Reid B, Burnett K, Callan B. A descriptive systematic review of salivary therapeutic drug monitoring in neonates and infants. Br J Clin Pharmacol. 2018;84:1089–108.
Kuwayama K, Miyaguchi H, Iwata YT, Kanamori T, Tsujikawa K, Yamamuro T, et al. Time-course measurements of drug concentrations in hair and toenails after single administrations of pharmaceutical products. Drug Test Anal. 2017;9:571–7.
Sabir AM, Moloy M, Bhasin PS. Hplc method development and validation: a review. Int Res J Pharm. 2016;4:39–46.
Decosterd LA, Widmer N, André P, Aouri M, Buclin T. The emerging role of multiplex tandem mass spectrometry analysis for therapeutic drug monitoring and personalized medicine. TrAC. 2016;84:5–13.
Dasgupta A. Limitations of immunoassays used for therapeutic drug monitoring of immunosuppressants. In: Personalized immunosuppression in transplantation role of biomarker monitoring and therapeutic drug monitoring. Editors: Oellerich M, Dasgupta A. Amsterdam: Elsevier Inc.;2016.
Aucella F, Lauriola V, Vecchione G, Tiscia GL, Grandone E. Liquid chromatography-tandem mass spectrometry method as the golden standard for therapeutic drug monitoring in renal transplant. J Pharm Biomed Anal. 2013;86:123–6.
Cooney L, Loke YK, Golder S, Kirkham J, Jorgensen A, Sinha I, et al. Overview of systematic reviews of therapeutic ranges: methodologies and recommendations for practice. BMC Med Res Methodol. 2017;17:1–9.
Kelly LE, Sinha Y, Barker CIS, Standing JF, Offringa M. Useful pharmacodynamic endpoints in children: selection, measurement, and next steps. Pediatr Res. 2018;83:1095–103.
Timsit JF, de Kraker MEA, Sommer H, Weiss E, Bettiol E, Wolkewitz M, et al. Appropriate endpoints for evaluation of new antibiotic therapies for severe infections: a perspective from COMBACTE’s STAT-Net. Intensive Care Med. 2017;43:1002–12.
Shaikh N, Hoberman A, Paradise JL, Rockette HE, Kurs-Lasky M, Colborn DK, et al. Responsiveness and construct validity of a symptom scale for acute otitis media. Pediatr Infect Dis J. 2009;28:9–12.
Blussé Van Oud-Alblas HJ, Brill MJE, Peeters MYM, Tibboel D, Danhof M, Knibbe CAJ. Population pharmacokinetic-pharmacodynamic model of propofol in adolescents undergoing scoliosis surgery with intraoperative wake-up test: a study using Bispectral index and composite auditory evoked potentials as pharmacodynamic endpoints. BMC Anesthesiol. 2019;19:1–12.
Sierra CM, Tran Y, Oana L, Bahjri K. Renal impairment associated with trimethoprim-sulfamethoxazole use in the pediatric population. J Pediatr Pharmacol Ther. 2022;27:663–8.
Walsh S, Pan S, Sheng Y, Kloprogge F, Standing JF, Anderson BJ, et al. Optimising intravenous salbutamol in children: a phase 2 study. Arch Dis Child. 2023;108:316–22.
De Cock RFW, Piana C, Krekels EHJ, Danhof M, Allegaert K, Knibbe CAJ. The role of population PK-PD modelling in paediatric clinical research. Eur J Clin Pharmacol. 2011;67(Suppl. 1):5–16.
Barker CIS, Standing JF, Kelly LE, Hanly Faught L, Needham AC, Rieder MJ, et al. Pharmacokinetic studies in children: recommendations for practice and research. Arch Dis Child. 2018;103:695–702.
Chen B, Abuassba AOM. Compartmental models with application to pharmacokinetics. Proc Comput Sci. 2021;187:60–70.
Sandra L, Smits A, Allegaert K, Nicolaï J, Annaert P, Bouillon T. Population pharmacokinetics of propofol in neonates and infants: gestational and postnatal age to determine clearance maturation. Br J Clin Pharmacol. 2021;87:2089–97.
Del Frari L, Léauté-Labrèze C, Guibaud L, Barbarot S, Lacour JP, Chaumont C, et al. Propranolol pharmacokinetics in infants treated for infantile hemangiomas requiring systemic therapy: modeling and dosing regimen recommendations. Pharmacol Res Perspect. 2018;6: e00399.
Thibault C, Massey SL, Abend NS, Naim MY, Zoraian A, Zuppa AF. Population pharmacokinetics of phenobarbital in neonates and infants on extracorporeal membrane oxygenation and the influence of concomitant renal replacement therapy. J Clin Pharmacol. 2021;61:378–87.
Rhoney DH, Metzger SA, Nelson NR. Scoping review of augmented renal clearance in critically ill pediatric patients. Pharmacotherapy. 2021;41:851–63.
Zylbersztajn B, Parker S, Navea D, Izquierdo G, Ortiz P, Torres JP, et al. Population pharmacokinetics of vancomycin and meropenem in pediatric extracorporeal membrane oxygenation support. Front Pharmacol. 2021;12: 709332.
Rapp M, Urien S, Foissac F, Béranger A, Bouazza N, Benaboud S, et al. Population pharmacokinetics of meropenem in critically ill children with different renal functions. Eur J Clin Pharmacol. 2020;76:61–71.
Li S, Shu C, Wu S, Xu H, Wang Y. Population pharmacokinetics and dose optimization of ganciclovir in critically ill children. Front Pharmacol. 2020;11: 614164.
Maharaj AR, Wu H, Zimmerman KO, Speicher DG, Sullivan JE, Watt K, et al. Dosing of continuous fentanyl infusions in obese children: a population pharmacokinetic analysis. J Clin Pharmacol. 2020;60:636–47.
Jones HM, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometr Syst Pharmacol. 2013;2:1–12.
Dahl SG, Aarons L, Gundert-Remy U, Karlsson MO, Schneider Y-J, Steimer J-L, et al. Incorporating physiological and biochemical mechanisms into pharmacokinetic–pharmacodynamic models: a conceptual framework. Basic Clin Pharmacol Toxicol. 2010;106:2–12.
Kuepfer L, Niederalt C, Wendl T, Schlender J, Willmann S, Lippert J, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometr Syst Pharmacol. 2016;5:516–31.
Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, et al. Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood–brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther. 2005;313:1254–62.
Hoehme S, Brulport M, Bauer A, Bedawy E, Schormann W, Hermes M, et al. Prediction and validation of cell alignment along microvessels as order principle to restore tissue architecture in liver regeneration. Proc Natl Acad Sci. 2010;107:10371–6.
De Cock RFW, Allegaert K, Brussee JM, Sherwin CMT, Mulla H, de Hoog M, et al. Simultaneous pharmacokinetic modeling of gentamicin, tobramycin and vancomycin clearance from neonates to adults: towards a semi-physiological function for maturation in glomerular filtration. Pharm Res. 2014;31:2643–54.
O’Hanlon CJ, Holford N, Sumpter A, Al-Sallami HS. Consistent methods for fat-free mass, creatinine clearance, and glomerular filtration rate to describe renal function from neonates to adults. CPT Pharmacometr Syst Pharmacol. 2023;12:401–12.
Simcyp®. Available from www.simcyp.com. Accessed 8 May 2024.
Open Systems Pharmacology. Available from http://www.open-systems-pharmacology.org/. Accessed 8 May 2024.
Balbas-Martinez V, Michelet R, Edginton AN, Meesters K, Trocóniz IF, Vermeulen A. Physiologically-based pharmacokinetic model for ciprofloxacin in children with complicated urinary tract infection. Eur J Pharm Sci. 2019;128:171–9.
Edginton AN, Willmann S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet. 2008;47:743–52.
Watt KM, Cohen-Wolkowiez M, Barrett JS, Sevestre M, Zhao P, Brouwer KLR, et al. Physiologically-based pharmacokinetic approach to determine dosing on extracorporeal life support: fluconazole in children on ECMO. CPT Pharmacometr Syst Pharmacol. 2018;7:629–37.
Allegaert K, Abbasi MY, Michelet R, Olafuyi O. The impact of low cardiac output on propofol pharmacokinetics across age groups: an investigation using physiologically based pharmacokinetic modelling. Pharmaceutics. 2022;14:1957.
Gerhart JG, Carreño FO, Edginton AN, Sinha J, Perrin EM, Kumar KR, et al. Development and evaluation of a virtual population of children with obesity for physiologically based pharmacokinetic modeling. Clin Pharmacokinet. 2022;61:307–20.
Ford JL, Gerhart JG, Edginton AN, Yanovski JA, Hon YY, Gonzalez D. Physiologically based pharmacokinetic modeling of metformin in children and adolescents with obesity. J Clin Pharmacol. 2022;62:960–9.
Johnson TN, Small BG, Rowland YK. Increasing application of pediatric physiologically based pharmacokinetic models across academic and industry organizations. CPT Pharmacometr Syst Pharmacol. 2022;11:373–83.
Verscheijden LFM, Koenderink JB, de Wildt SN, Russel FGM. Development of a physiologically-based pharmacokinetic pediatric brain model for prediction of cerebrospinal fluid drug concentrations and the influence of meningitis. PLoS Comput Biol. 2019;15: e1007117.
Zhu S, Zhang J, Lv Z, Zhu P, Oo C, Yu M, et al. Prediction of tissue exposures of meropenem, colistin, and sulbactam in pediatrics using physiologically based pharmacokinetic modeling. Clin Pharmacokinet. 2022;61:1427–41.
Yeung CHT, Ito S, Autmizguine J, Edginton AN. Incorporating breastfeeding-related variability with physiologically based pharmacokinetic modeling to predict infant exposure to maternal medication through breast milk: a workflow applied to lamotrigine. AAPS J. 2021;23:70.
Salerno SN, Capparelli EV, McIlleron H, Gerhart JG, Dumond JB, Kashuba ADM, et al. Leveraging physiologically based pharmacokinetic modeling to optimize dosing for lopinavir/ritonavir with rifampin in pediatric patients. Pharmacotherapy. 2023;3(7):638–49.
Zhao X, Lu X, Zuo M, Wang N, Zhang Y, Chen J, et al. Drug-drug interaction comparison between tacrolimus and phenobarbital in different formulations for paediatrics and adults. Xenobiotica. 2021;51:877–84.
Verscheijden LFM, van der Zanden TM, van Bussel LPM, de Hoop-Sommen M, Russel FGM, Johnson TN, et al. Chloroquine dosing recommendations for pediatric COVID-19 supported by modeling and simulation. Clin Pharmacol Ther. 2020;108:248–52.
Felmlee MA, Morris ME, Mager DE. Mechanism-based pharmacodynamic modeling. Methods Mol Biol. 2012;929:583–600.
Verscheijden LFM, Litjens CHC, Koenderink JB, Mathijssen RHJ, Verbeek MM, de Wildt SN, et al. Physiologically based pharmacokinetic/pharmacodynamic model for the prediction of morphine brain disposition and analgesia in adults and children. PLoS Comput Biol. 2021;17:1–21.
Abdulla A, Edwina EE, Flint RB, Allegaert K, Wildschut ED, Koch BCP, et al. Model-informed precision dosing of antibiotics in pediatric patients: a narrative review. Front Pediatr. 2021;9: 624639.
Downes KJ, Goldman JL. Too much of a good thing: defining antimicrobial therapeutic targets to minimize toxicity. Clin Pharmacol Ther. 2021;109:905–17.
Roberts JA, Norris R, Paterson DL, Martin JH. Therapeutic drug monitoring of antimicrobials. Br J Clin Pharmacol. 2012;73:27–36.
Holford N, Ma G, Metz D. TDM is dead. Long live TCI! Br J Clin Pharmacol. 2022;88:1406–13.
Kantasiripitak W, Wicha SG, Thomas D, Hoffman I, Ferrante M, Vermeire S, et al. A model-based tool for guiding infliximab induction dosing to maximise long-term deep remission in children with inflammatory bowel diseases. J Crohns Colitis. 2023;17:896–908.
Darwich AS, Polasek TM, Aronson JK, Ogungbenro K, Wright DFB, Achour B, et al. Model-informed precision dosing: background, requirements, validation, implementation, and forward trajectory of individualizing drug therapy. Annu Rev Pharmacol Toxicol. 2021;61:225–45.
Maier C, Hartung N, Kloft C, Huisinga W, de Wiljes J. Reinforcement learning and Bayesian data assimilation for model-informed precision dosing in oncology. CPT Pharmacometr Syst Pharmacol. 2021;10:241–54.
Frymoyer A, Stockmann C, Hersh AL, Goswami S, Keizer RJ. Individualized rmpiric vancomycin dosing in neonates using a model-based approach. J Pediatr Infect Dis Soc. 2019;8:97–104.
Allegaert K, Flint R, Smits A. Pharmacokinetic modelling and Bayesian estimation-assisted decision tools to optimize vancomycin dosage in neonates: only one piece of the puzzle. Expert Opin Drug Metab Toxicol. 2019;15:735–49.
Barras MA, Serisier D, Hennig S, Jess K, Norris RLG. Bayesian estimation of tobramycin exposure in patients with cystic fibrosis. Antimicrob Agents Chemother. 2016;60:6698–702.
Kumar AA, Burgard M, Stacey S, Sandaradura I, Lai T, Coorey C, et al. An evaluation of the user-friendliness of Bayesian forecasting programs in a clinical setting. Br J Clin Pharmacol. 2019;85:2436–41.
Kantasiripitak W, Van Daele R, Gijsen M, Ferrante M, Spriet I, Dreesen E. Software tools for model-informed precision dosing: how well do they satisfy the needs? Front Pharmacol. 2020;11:620.
Kantasiripitak W, Outtier A, Wicha SG, Kensert A, Wang Z, Sabino J, et al. Multi-model averaging improves the performance of model-guided infliximab dosing in patients with inflammatory bowel diseases. CPT Pharmacometr Syst Pharmacol. 2022;11:1045–59.
Uster DW, Stocker SL, Carland JE, Brett J, Marriott DJE, Day RO, et al. A model averaging/selection approach improves the predictive performance of model-informed precision dosing: vancomycin as a case study. Clin Pharmacol Ther. 2021;109:175–83.
Bi Y, Liu J, Li L, Yu J, Bhattaram A, Bewernitz M, et al. Role of model-informed drug development in pediatric drug development, regulatory evaluation, and labeling. J Clin Pharmacol. 2019. https://doi.org/10.1002/jcph.1478.
Kluwe F, Michelet R, Mueller-Schoell A, Maier C, Klopp-Schulze L, van Dyk M, et al. Perspectives on model-informed precision dosing in the digital health era: challenges, opportunities, and recommendations. Clin Pharmacol Ther. 2021;109:29–36.
Yellepeddi V, Rower J, Liu X, Kumar S, Rashid J, Sherwin CMT. State-of-the-art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin Pharmacokinet. 2019;58:1–13.
Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6:140–8.
Johnson TN, Rostami-Hodjegan A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr Anaesth. 2011;21:291–301.
Kovarik JM, Hartmann S, Bartlett M, Riviere G, Neddermann D. Oral-intravenous crossover study of fingolimod. Biopharm Drug Dispos. 2007;28:97–104.
Schaller S, Willmann S, Lippert J, Schaupp L, Pieber TR, Schuppert A, et al. A generic integrated physiologically based whole-body model of the glucose insulin-glucagon regulatory system. CPT Pharmacometr Syst Pharmacol. 2013;2: e65.
Willmann S, Thelen K, Becker C, Dressman JB, Lippert J. Mechanism-based prediction of particle size-dependent dissolution and absorption: cilostazol pharmacokinetics in dogs. Eur J Pharm Biopharm. 2010;76:83–94.
Hammarlund-Udenaes M. Microdialysis as an important technique in systems pharmacology: a historical and methodological review. AAPS J. 2017;19:1294–303.
Girdwood ST, Kaplan J, Vinks AA. Methodologic progress note: opportunistic sampling for pharmacology studies in hospitalized children. J Hosp Med. 2020;16:35–7.
Laughon MM, Benjamin DK, Capparelli EV, Kearns GL, Berezny K, Paul IM, et al. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol. 2011;4:643–52.
Shores DR, Everett AD. Children as biomarker orphans: progress in the field of pediatric biomarkers. J Pediatr. 2018;193:14-20.e31.
McComb M, Bies R, Ramanathan M. Machine learning in pharmacometrics: opportunities and challenges. Br J Clin Pharmacol. 2022;88:1482–99.
Bradshaw EL, Spilker ME, Zang R, Bansal L, He H, Jones RDO, et al. Applications of quantitative systems pharmacology in model-informed drug discovery: perspective on impact and opportunities. CPT Pharmacometr Syst Pharmacol. 2019;8:777–91.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Kevin J. Downes is supported by The Eunice Kennedy Shriver National Institute of Child Health and Human Development (K23HD091365). Kevin Meesters is the recipient of the 2023 Bertram Hoffmeister Postdoctoral Fellowship Award at BC Children’s Hospital Research Institute. The other authors received no additional funding.
Conflicts of Interest/Competing Interests
Violeta Balbas-Martinez is an employee and shareholder of Eli-Lilly and Company. Kevin Meesters, Violeta Balbas-Martinez, Karel Allegaert, Kevin J. Downes, and Robin Michelet have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Authors’ Contributions
KM, VB-M, KJD, RM, and KA conceptualized this article, performed the extensive literature review, contributed to the writing of the manuscript, and critically reviewed the manuscript. All authors read and approved the final manuscript as submitted.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meesters, K., Balbas-Martinez, V., Allegaert, K. et al. Personalized Dosing of Medicines for Children: A Primer on Pediatric Pharmacometrics for Clinicians. Pediatr Drugs 26, 365–379 (2024). https://doi.org/10.1007/s40272-024-00633-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-024-00633-x