
Abstract
Systemic autoinflammatory diseases (SAIDs) are a group of rare genetic and nongenetic immune dysregulatory disorders associated with high morbidity and mortality if left untreated. Therefore, early diagnosis and initiation of targeted treatment is vital in SAID patients to control the disease activity and prevent long-term immune-mediated damage. A specific group of genetically defined SAIDs is associated with increased inflammasome-mediated production of active interleukin (IL)-1. Even though progress in immunobiology and genetics has brought forth diagnostic tools and novel treatments that have been described in the literature extensively, many challenges remain in the clinical setting. Some challenges that health care providers may face on a day-to-day basis include the requirement of a multidisciplinary approach due to the complexity of these diseases, limited evidence-based treatment options, and barriers to access available therapies. Primarily, IL-1 inhibitors anakinra, canakinumab, and rilonacept are used to control the inflammation in these patients, with the goal of achieving sustainable remission. Recently published provisional points to consider from the European Alliance of Associations for Rheumatology (EULAR) and American College of Rheumatology (ACR) provide diagnosis, management, and monitoring recommendations for four IL-1-mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes (CAPS), tumour necrosis factor receptor-associated periodic syndrome (TRAPS), mevalonate kinase deficiency (MKD), and deficiency of the IL-1 receptor antagonist (DIRA). The goal of this paper is to aid health care professionals by providing a practical approach to diagnosis and management of these four IL-1 mediated SAIDs on the basis of the recent EULAR/ACR recommendations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
High index of suspicion is essential for early diagnosis of CAPS, TRAPS, MKD and DIRA. |
To provide a diagnosis for patients with suspected phenotypes, genetic testing using next-generation sequencing (NGS) is necessary. Sanger sequencing of a single gene may be utilized in the presence of family history and/or classic clinical features. |
Early treatment with IL-1 inhibitors is required to reduce morbidity and mortality. |
The lifelong necessity for IL-1 inhibition in monogenic autoinflammatory diseases CAPS, TRAPS, MKD and DIRA mandates ensuring safety monitoring. |
1 Introduction
Systemic autoinflammatory diseases (SAIDs) are a large and heterogeneous group of disorders characterized by dysregulation of the innate immune system. Most SAIDs are associated with mutations in genes involved in the innate immune response. A specific group of genetically defined SAIDs are associated with increased inflammasome-mediated production of active interleukin (IL)-1 that are usually referred as “Hereditary Periodic Fever Syndromes” [1].
The IL-1 mediated autoinflammatory disorders are ultra-rare systemic autoinflammatory diseases with an estimated prevalence of less than one in a million. Accepted strategies to study these disorders, such as placebo-controlled trials are often not feasible due to the rarity, small sample size and severity of these diseases [2, 3]. Therefore, it is challenging to conduct research in various areas such as the development of biomarkers, outcome instruments or new treatment approaches. Extensive knowledge has been gained from functional and/or genetic studies over the past 20 years since the term “autoinflammation” was first described [4], however, many obstacles still remain when applying this knowledge in clinical setting for the benefit of individual patients. Until confirming the diagnosis, these complex patients are often seen by multiple health practitioners and disease flares may result in frequent emergency room visits, and/or hospitalizations. In addition, there are limited number of physicians who specialize in these disorders. All these challenges cause diagnostic delays, high costs to the health care system, and frustration from patients and their families. Early identification of these diseases, initiation of targeted therapy, and development of a unified delivery of multidisciplinary care are vital to prevent diagnostic delays, organ damage, morbidity, and mortality as well as to improve quality of life (QOL) measures [5, 6].
In July 2022, the European Alliance of Associations for Rheumatology (EULAR) and American College of Rheumatology (ACR) published provisional points to consider for the diagnosis, management and monitoring of four IL-1 mediated autoinflammatory diseases, including cryopyrin-associated periodic syndromes (CAPS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), mevalonate kinase deficiency (MKD), and deficiency of the IL-1 receptor antagonist (DIRA) [5]. In this review, we focus on those four IL-1-mediated SAIDs: CAPS, TRAPS, MKD, and DIRA, and provide insights into the general pathophysiological concepts, clinical features, suggested diagnostic workup, and current and future perspectives for therapy on the basis of the EULAR and ACR points to consider. Our goal is to increase awareness and provide health care professionals with a practical approach on the diagnosis and management of these four diseases.
2 Immunopathological Mechanisms of IL-1-Mediated SAIDs
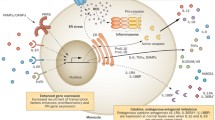
In CAPS, TRAPS, MKD, and DIRA, different pathophysiologic mechanisms cause increased IL-1β (Fig. 1). To date, it is still unclear how increased IL-1 signaling causes different clinical phenotypes. Nevertheless, it is now well established that IL-1-mediated cytokines, and in the context of monogenic autoinflammatory diseases, mainly IL-1β, IL-1α, and IL-1 receptor antagonist (IL-1RA), are tightly regulated inflammatory mediators involved in response to tissue damage, the presence of pathogen- and danger-associated molecular patterns (DAMPs and PAMPs), and cell death pathways [7]. Inflammasomes, intracellular ensembles of multiple proteins, usually remain inactive under normal circumstances, but can be activated in the presence of specific triggers, such as pathogen toxins, intracellular metabolites, or stress, triggering innate inflammatory responses [7, 8].

Pathophysiology of the IL-1-mediated diseases. Pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) stimulate innate immune sensors NLRP3 and pyrin in the cytoplasm. Stimulated NLRP3 and pyrin form ASC and activate the inflammasome complex, which subsequently drives pro-caspase 1 and caspase 1 formation. Caspase 1 mediates cleavage of pro-IL-1β and pro-IL-18 into bioactive forms of proinflammatory cytokines, IL-1β and IL-18, which then activate intranuclear NF-κB for the transcription of proinflammatory cytokines. Gain-of-function mutations in the NLRP3 gene result in CAPS. In healthy cells, IL-1β binding to its cytoplasmic IL-1 receptor is naturally inhibited by IL-1RA. DIRA causes uncontrolled IL-1β binding, resulting in a hyperinflammatory response. RhoA GTP molecules regulate the pyrin inflammasome through serine–threonine kinases. Loss-of-function mutations in the mevalonate kinase gene result in reduced mevalonate kinase activity, inactivation of the RhoA GTPase, and increased pyrin inflammasome formation, which then leads to excessive cytokine production in MKD. In TRAPS, disease-causing variants affect receptor structure and binding to the TNF ligand. Although disease mechanisms remain unclear, intracellular accumulation of the misfolded TNF receptor 1 is thought to cause ER stress, overproduction of ROS, and ultimately activation of different pathological immune responses. ASC apoptosis-associated speck-like protein containing a caspase-recruitment domain, CAPS cryopyrin-associated periodic syndrome, DIRA deficiency of interleukin-1 receptor antagonist, ER endoplasmic reticulum, GSDMD gasdermin D, IL interleukin, IL-1R interleukin-1 receptor, IL-1RA interleukin-1 receptor antagonist, MKD mevalonate kinase deficiency, MVK mevalonate kinase, NF-κB nuclear factor kappa B, NLRP3 nucleotide-binding domain-like receptor 3, ROS reactive oxygen species, TNFR1 tumor necrosis factor receptor 1, TRAPS TNF receptor-associated periodic syndrome
Inflammasomes consist of oligomers of a sensor protein, caspase 1, and an adaptor. The sensor is responsible for recognizing danger signals and gives the inflammasome its specificity. The most common sensors are one of ten different nod-like receptor (NLR)-family proteins and the sensor protein pyrin. After recognition of microbial or endogenous ligands, the sensors oligomerize with an adaptor protein, and subsequently, inactive caspase 1 is converted to its active version forming the inflammasome complex. Activated caspase 1 then cleaves pro-IL-1 and pro-IL-18 to produce biologically active IL-1β and IL-18, which in turn trigger acute inflammation, innate immune cell activation, and classical clinical features seen in these patients, such as fever [9, 10].
2.1 Cryopyrin-Associated Periodic Syndromes (CAPS)
Autosomal dominant (AD) or de novo heterozygous gain-of-function mutations in the NOD-like receptor protein 3 (NLRP3) gene result in persistent intrinsic activation of the NLRP3 inflammasome, or a decreased threshold for its activation, constituting the prototype of inflammasomopathies [11]. NLRP3 protein was shown to contain a pyrin domain in the amino terminus, and it is a critical sensor triggered by diverse molecular and cellular mechanisms, including microbial toxins, ionic influx, and mitochondrial reactive oxygen species (mROS), leading to innate immune system activation [12, 13]. Overactivated NLRP3 in CAPS patients drives consequent NLRP3 inflammasome assembly, caspase 1 activation, and overproduction of active IL-1β and IL-18 [12].
2.2 Tumor Necrosis Factor Receptor-Associated Periodic Syndrome (TRAPS)
TRAPS is caused by autosomal dominant (AD) heterozygous gain-of-function (GOF) mutations on chromosome 12, in the TNFRSF1A gene, which encodes for tumor necrosis factor (TNF) receptor 1 (TNFR1). Although precise TRAPS immunopathological mechanisms have not been completely defined, a few affected immune pathways have been discovered in recent studies. The discoveries are as follows: (1) intracellular accumulation of the misfolded mutant TNF receptor proteins were found to be associated with increased endoplasmic reticulum (ER) stress and unfolded protein response, causing excessive production of mROS, leading to increased cell death and abnormal autophagy [7, 14]; (2) ER stress and mROS have been shown to activate nuclear factor ĸB (NF-κB) and, consequently, transcription of proinflammatory target genes, promoting proinflammatory cytokine production; and (3) heterozygous variants affect the structure of the extracellular domain of the TNFR1 and impair its binding to the TNF ligand and shedding from the cell surface, affecting TNFR1 counter regulatory mechanisms [14,15,16].
2.3 Mevalonate Kinase Deficiency (MKD)
MKD is caused by autosomal recessive loss-of-function (LOF) mutations in the gene of the mevalonate kinase (MVK) enzyme [17], causing decreased or absent MVK enzymatic activity, and affecting a crucial step in the isoprenoid biosynthesis pathway that yields many lipid products that diminish inflammatory responses [7, 18]. Recent human and murine models have shown the role of Rho GTPase in regulating pyrin activity in normal conditions which has helped clarify the mechanism by which loss of MVK leads to autoinflammation [19]. Shortage of nonsterol isoprenoids causes defective protein prenylation and impaired attachment of isoprenoids to GTPases, such as the Rho family of GTPase, thereby resulting in an uncontrolled release of IL-1β from innate immune cells, presumably in a pyrin–inflammasome-dependent pathway [7, 19, 20].
2.4 Deficiency of the IL-1 Receptor Antagonist (DIRA)
IL-1RA competitively binds to the IL-1 receptor (IL-1R), thus preventing IL-1α and IL-1β binding to the IL-1R and acting as a natural inhibitor of IL-1-mediated inflammatory responses [7]. In DIRA, autosomal recessive homozygous LOF mutations in IL1RN, the gene encoding the IL-1RA, lead to unopposed IL-1 signaling and uncontrolled IL-1-mediated hyperinflammation, which is responsible for the disease phenotype [21, 22].
3 Clinical Evaluation and Diagnostic Approach
Early diagnosis is vital in patients with autoinflammatory disorders to control the disease activity and prevent long-term immune-mediated damage by starting targeted treatment in a timely manner. However, diagnostic delays still occur and remain an important issue, often due to limited awareness and limited experience in autoinflammatory disorders [6]. Detailed patient and family history, as well as physical examination should be the first steps in diagnosis because recognition of the signs and symptoms of autoinflammatory disorders warrants further evaluation and genetic testing [23].
Patients with CAPS, TRAPS, MKD, and DIRA present with systemic inflammation; however, clinical signs, symptoms, and disease severity vary based on diagnosis. Each disease has a specific pattern of clinical features that may determine the workup for evaluation and diagnosis. Common and disease-specific signs and symptoms of CAPS, TRAPS, MKD, and DIRA are summarized in Table 1.
3.1 CAPS
CAPS is a spectrum with mild, moderate, and severe forms. Across the spectrum of severity, patients with CAPS share symptoms of urticaria-like rash with neutrophilic infiltrate, ocular manifestations, fevers, and joint pain. The mildest form of CAPS is familial cold autoinflammatory syndrome (FCAS), which is characterized by cold-triggered episodes of neutrophilic urticaria, typically lasting less than 24 h [24,25,26]. Muckle–Wells syndrome (MWS) is the moderate form of CAPS, and patients typically present with progressive sensorineural hearing loss, neutrophilic urticaria, and arthritis [27,28,29]. Neonatal-onset multisystem inflammatory disease (NOMID)/chronic infantile neurologic cutaneous and articular syndrome (CINCA) is the most severe form of CAPS [30]. It is associated with nearly persistent systemic inflammation with neonatal-onset neurologic manifestations (chronic aseptic meningitis, increased intracranial pressure, papilledema, cognitive impairment), severe progressive sensorineural hearing loss, and skeletal manifestations (frontal bossing, distal femur overgrowth) [31]. Age at disease onset varies for each CAPS, which can aid the diagnosis. MWS presents in childhood, while CINCA/NOMID manifests early in life, even in the neonatal period [32]. FCAS may present at any age. Family history is often positive in patients with FCAS and MWS, but often negative in patients with CINCA/NOMID [33].
Markers of systemic inflammation should be monitored during clinical disease flares, even if the the patient is clinically quiescent. These markers include complete blood count (CBC), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and, if available, serum amyloid A (SAA) and S100 [34, 35]. Additionally, organ involvement and damage should be assessed during the diagnostic process (Table 2). Most FCAS and some MWS patients develop typical skin lesions triggered by cold air, but not by contact with an ice cube or cold water. The ice cube test is helpful in differentiating FCAS from cold urticaria [36]. Skin biopsy of urticaria-like rashes reveals a neutrophilic dermatosis that is common in CAPS patients [36]. Comprehensive ophthalmologic and audiologic evaluation is necessary for all patients with suspected CAPS. Although acute conjunctivitis is the most common eye manifestation during disease flares, it can also become chronic. Other eye manifestations include keratitis, episcleritis, and uveitis. Most CINCA/NOMID patients develop papilledema due to chronic elevated intracranial pressure. Papilledema and uveitis can cause progressive vision loss [37,38,39]. Due to ongoing cochlear inflammation, progressive sensorineural hearing loss is a main concern for MWS and CINCA/NOMID phenotypes. Hearing loss can be irreversible if left untreated or if treatment is delayed; however, reversing or stabilizing the progression of hearing loss may be possible by treating patients early in the disease course [40,41,42].
3.2 TRAPS
Typical clinical symptoms of TRAPS include recurrent fever episodes lasting longer than 7 days, sometimes lasting for weeks, along with maculopapular migratory rash, periorbital edema, abdominal pain, chest pain, and/or musculoskeletal complaints. Some patients present with episodes of inflammations, and others present with a continuous disease course [1, 43,44,45]. Periorbital edema is a pathognomonic feature of the disease in the context of the other typical clinical findings, and it may be a sign of a disease flare when combined with myalgia. Other ocular manifestations include recurrent conjunctivitis or anterior uveitis. Skin lesions are typically migratory, painful/tender macular, edematous, or urticarial lesions. Abdominal pain in TRAPS can also mimic an acute abdomen similar to what is observed in patients with familial Mediterranean fever (FMF). Arthralgia during attacks is more common than frank synovitis, but patients can present with nonerosive, asymmetric, and monoarticular arthritis. Neurological manifestations such as headache, aseptic meningitis, optic neuritis, and pleuritis are also seen in TRAPS [44,45,46]. Although presence of family history and febrile episodes lasting longer than 7 days are highly predictive clinical features of TRAPS, patients with low-penetrance TNFRSF1A mutations may have a negative family history and a later clinical onset [47, 48].
Systemic inflammation is present during febrile episodes, demonstrated by elevated acute phase reactants. Depending on the disease severity, inflammatory markers may remain elevated between fever episodes. Systemic amyloid A (AA) amyloidosis may occur especially in the context of mutations in cysteine residues, delayed diagnosis and/or prolonged disease activity; in these cases, monitoring of S100 and serum amyloid A (SAA) can have clinical utility [44, 45, 49]. Lesioned skin biopsies display a mild perivascular lymphocytic infiltrate in the edematous areas of the papillary dermis [49].
3.3 MKD
This disorder is commonly divided into two forms based on MVK activity: hyper immunoglobulinemia D and periodic fever syndrome (HIDS) describes the milder form with reduced MVK enzyme activity, whereas mevalonic aciduria is the severe form with nearly absent MVK enzymatic activity [1, 50].
Patients usually present with MKD features in the first year of life with disease episodes, whereas some patients present with a continuous course through exacerbations. Inflammatory episodes of MKD are generally triggered by immunizations, surgery, stress, or trauma and characterized by fever lasting for 4–6 days. Severe abdominal pain is a common feature and is mostly associated with diarrhea or vomiting. Lymphadenopathy is also frequently observed, with most presentations being bilateral, tender, cervical nodes, though generalized lymphadenopathy may also be present. Most patients present with a variety of mucocutaneus symptoms such as a rash (maculopapular or urticarial-like), aphthous stomatitis and/or pharyngitis. Patients can have arthralgia and nonerosive arthritis. Persistent arthritis has also been reported and recorded in these patients [50,51,52,53]. Patients with mevalonic aciduria, the most severe form of MKD, can also present with severe cognitive impairment, macrophage activation syndrome, dysmorphic features, and failure to thrive [5, 44, 50].
Systemic inflammation is present and reflected by elevated acute phase reactants, leukocytosis, and neutrophilia in laboratory workups. Persistent elevation of serum immunoglobulin D (IgD) level is observed in the majority of HIDS patients. Serum IgD levels can be normal in patients with HIDS, especially in children younger than 3 years. IgD can also be elevated in other autoinflammatory conditions. Therefore, IgD levels have low diagnostic sensitivity and specificity [5, 50, 54]. On the other hand, during disease flares, mevalonate levels increase in urine secondary to reduced MVK enzyme activity leading to the accumulation of mevalonic acid. Urine mevalonate level is a more specific test for MKD and can be used to aid diagnosis. Nevertheless, a definitive diagnosis can only be confirmed through genetic testing [5, 55].
3.4 DIRA
Patients with DIRA present with early onset neutrophilic pustular rashes, multifocal sterile osteomyelitis [e.g., chronic recurrent multifocal osteomyelitis (CRMO)-like disease, rib flaring and cloaking of the femoral head, odontoid lesions/osteomyelitis], periostitis, and nail changes (onychomadesis). Hypoxemia and dyspnea are frequently seen in infants secondary to interstitial pneumonia, areas of atelectasis or gastroesophageal reflux. Pustular skin lesions resemble pustular psoriasis and can be triggered by mechanical stress (pathergy). Lesions can be sparse, occur in clusters, or present as severe generalized pustular dermatosis. In contrast to CAPS, TRAPS and MKD, fever is rarely present in DIRA disease flares, although marked elevation in inflammatory markers with neutrophilia is typical [7, 22, 44, 56].
In patients with presumed DIRA, a diagnostic workup should be initiated to assess peripheral neutrophilia and elevated inflammatory markers. To determine bone involvement, X-ray or bone magnetic resonance imaging (MRI) should be performed. Typical radiographic manifestations show widened ribs and clavicles, as well as multifocal osteolytic lesions mainly observed in long bones. Computerized tomography (CT) or MRI can be used to screen for vertebral involvement presenting with odontoid osteomyelitis, vertebral block formation, and gibbus-like spinal changes. However, even with all of these presentations, genetic testing is crucial for a definite diagnosis [5, 22].
4 Genetic Testing
As mentioned above, while clinical features and laboratory tests typically suggest specific SAIDs, genetic testing needs to be performed to confirm the exact diagnosis and provide accurate genetic counseling [57,58,59]. Although this step is crucial for diagnosis, accessing appropriate genetic testing can be challenging [5]. There are important points that need to be considered when ordering genetic testing, and these are outlined in Table 3. If available, next-generation sequencing (NGS) is recommended over Sanger sequencing. The initial genetic test should be an NGS–autoinflammatory diseases (AIDs) panel [57]. Sanger sequencing should be limited to patients with a clear family history and classic clinical phenotypes, or in areas where NGS is not available or is prohibitively expensive [60]. Care providers can find worldwide available panels in the Genetic Testing Registry database (https://www.ncbi.nlm.nih.gov/gtr/). In any NGS report, detected variants are classified as follows: pathogenic mutation; variant, likely pathogenic (VLP); variant, unknown significance (VUS), variant, likely benign (VLB); and benign. Some databases, such as MOLGENIS and INFEVERS offer clinicians clear, accurate, clinically relevant details about those variants [61, 62]. However, it can still be challenging to interpret genetic testing results for some variants as they may cause a less severe clinic phenotype than a low-penetrance mutation. Therefore, collaboration among rheumatologists, immunologists, and medical geneticists is necessary to achieve an accurate diagnosis.
Although NGS should be the first step of genetic evaluation where available, it comes with some limitations such as inability to detect somatic mosaicism. Therefore, deep sequencing with a much greater NGS depth (e.g., > 1000×) [60] may be necessary for CINCA/NOMID patients since somatic NLRP3 mosaicism can be present in mutation-negative NOMID/CINCA, which was identified in 69.2% of patients with mutation-negative NOMID/CINCA in one study [63]. Another genetic diagnostic challenge with NGS is seen in DIRA patients. A large deletion in IL1RN gene is one of the genetic reasons for DIRA, and large deletions sometimes cannot be determined by NGS; therefore, resequencing may be necessary [64, 65]. If the targeted NGS panel returns negative results, consideration should be given to whole-exome sequencing (WES) and whole-genome sequencing (WGS) to enhance the likelihood of making a genetic diagnosis by discovering novel variants [66]. If the genetic diagnosis cannot be made with NGS, patients should be referred to a SAIDs center of excellence.
5 Treatment
Early diagnosis is the first step of treatment as it will guide us to implement etiological anti-inflammatory treatment with biologics (see below). A multidisciplinary team-based approach is crucial for accurate and prompt diagnosis, initiating treatment early in the disease course, minimizing disease complications, and improving prognosis. The implementation of outcome instruments, quality-of-life (QOL) measures, and patient-reported outcomes (PRO) will help identify patients’ needs and prospectively evaluate the quality of service provided.
5.1 Pharmacological Treatment
Specific inhibitors of IL-1 signaling is the standard therapy for patients with CAPS, TRAPS, MKD, and DIRA. Early and sustainable normalization of all clinical signs and symptoms of inflammation and inflammatory markers is the primary treatment goal to reduce inflammation burden, organ damage, and the need for other therapies. Strong clinical suspicion may authorize starting anti-IL-1 therapy before a definitive genetic diagnosis to prevent organ damage secondary to treatment delays [5].
Anakinra (Kineret), rilonacept (Arcalyst), and canakinumab (Ilaris) are three IL-1 inhibitors that have been shown to be effective and safe in controlling disease activity. These drugs are approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) [67]. Anakinra, the recombinant human IL-1 receptor antagonist (IL-1Ra), efficiently blocks IL-1α and IL-1β signaling by competitively binding to the IL-1 receptor. A daily subcutaneous injection is required due to its short half-life, which is a disadvantage in many cases. However, its good central nervous system (CNS) penetration may make it the therapy of choice in cases with CNS involvement [68,69,70]. Rilonacept, a dimeric fusion protein, also blocks IL-1α/β with longer half-life than Anakinra [71]. Canakinumab is a fully human IgG monoclonal antibody selective to IL-1β so it does not block the IL-1α signaling pathway [72]. The significant advantage of canakinumab over anakinra and rilonacept is monthly/bimonthly administration due to its longer half-life [73]. All three anti IL-1 treatments can be used subcutaneously. Dosing regimens are summarized in Table 4.
Anakinra has shown to be a safe and effective treatment for all CAPS phenotypes [74,75,76,77,78]. Although the recommended anakinra dosage for FCAS patients is 1–2 mg/kg/daily, higher dosages are recommended (1–8 mg/kg/daily) for patients with more severe phenotypes, such as MWS and CINCA/NOMID. Kullenberg T et al. showed that anakinra treatment was safe and well tolerated for up to 5 years in 43 patients with severe CAPS [75]. The most frequently reported adverse events (AEs) were typical CAPS symptoms such as headache and arthralgia, or injection site reactions, which resolved mainly after the first month of anakinra treatment. Infections including pneumonia and gastroenteritis were the most common types of severe adverse events (SAEs) observed. There were no permanent treatment discontinuations due to AEs.
The efficacy and safety of rilonacept in MWS and FCAS have been shown in three randomized controlled trials [71, 79, 80]. A loading dose of 4.4 mg/kg (max 320 mg) is administered subcutaneously once, followed by the weekly maintenance doses of 2.2 mg/kg (max 160 mg). Rilonacept is not yet approved by FDA for the severe phenotype of CAPS (i.e., CINCA/NOMID). The most common AEs were injection site reactions and upper respiratory tract infections during the rilonacept treatment course. No SAEs have been documented in CAPS patients [71, 80].
Several studies showed the long-term safety and efficacy of canakinumab in all CAPS phenotypes [81,82,83,84]. Yokota S. et al. [83] enrolled 19 patients (12 NOMID, 7 MWS) for a median of 109 weeks. All patients achieved a complete response by week 48. At the end of the study, 11 (57.9%) patients had no disease activity according to the physician global assessment (PGA). All patients experienced at least one AE, the most common being infections (100%) and a total of five SAEs were reported. Brogan PA et al. [84] showed that canakinumab was safe and effective in 17 children ages 5 years and younger (12 MWS, 4 CINCA, 1 FCAS). In addition, no negative impact was found regarding postvaccination antibody production following the administration of non-live vaccines in nine pediatric patients treated with canakinumab.
Limited data suggest that canakinumab may be insufficient for neutralization of IL-1β in the central nervous system (CNS) [70]. Patients with severe CAPS phenotypes require higher and more frequent dosing (up to 8 mg/kg/q4w) to achieve remission [81, 83].
Canakinumab is the only anti-IL-1 treatment currently approved by the FDA [85] and EMA [86] for patients with TRAPS or MKD, on the basis of data from clinical trials and real-world effectiveness and safety [87,88,89]. Anakinra has also been used in both diseases, but effectiveness data remains scarce, mostly from small case series and a patient registry [67, 90,91,92]. Anti-IL-1 treatment can be sufficient when used at the beginning of a flare in patients with mild MKD [67]. In cases of anti-IL-1 treatment failure or lack of availability, anti-TNF or anti-IL-6 therapies may be considered, especially for those with TRAPS and MKD [93,94,95,96,97]. Finally, TRAPS and MKD flares may also respond to short-term glucocorticoid therapy [97, 98].
To conclude on pharmacological treatment, anakinra and rilonacept are FDA-approved treatments for DIRA [99, 100]. Both drugs are useful in controlling disease flares and preventing complications [22, 64, 65, 101]. Anakinra is the drug of choice for initial DIRA treatment, followed by maintenance therapy with rilonacept. Canakinumab exhibits a failure to control bone disease in patients with DIRA, likely because it only blocks IL-1β and IL-1α blockade may be additionally required to control osteitis. The recommended anakinra dosage for DIRA is 1–8 mg/kg/day, and rilonacept dosage is 4.4 mg/kg once weekly or 320 mg once weekly [99, 100].
In addition to targeting downstream cytokines in the IL-1 pathway (IL-1β, IL-1R, and IL-1α), studies have been aiming to directly inhibit a protein that is altered in a monogenic disease by developing small-molecule inhibitors. The most prevalent small-molecule target for treating autoinflammatory diseases has been NLRP3. Multiple companies are currently studying their own NLRP3 antagonists for CAPS [e.g., DFV90 (NCT04868968), ZYIL1 (NCT05186051)]. Small-molecule inhibitors with targets upstream and downstream of the inflammasome pathway are similarly under investigation such as selective PTX (7) receptor antagonist. Finally, other drugs that block downstream IL-1R signaling, including IRAK4 and MK2, are in development.
5.2 Treatment Monitoring
Anti-IL-1 therapies have overall excellent safety profiles but patients on anti-IL-1 therapy still require safety monitoring (Table 5). Furthermore, physicians should be aware of increased risks of infection, especially respiratory tract infections with streptococcus pneumonia and staphylococcal skin infections [81, 87]. Patients should be immunized according to local immunization guidelines. Although Brogan et al. showed that anti-IL-1 therapies do not affect the ability to produce antibodies against standard childhood inactivated vaccines [84], it is recommended that scheduled immunizations should be completed (particularly attenuated vaccines) before starting anti-IL-1 therapy, if possible. This recommendation includes MKD patients with increased disease flare risk by vaccination and CAPS patients with the potential risk of local and systemic reactions [84, 102, 103]. Live vaccines should not be administered concurrently with anti-IL-1 therapy. Coronavirus disease 2019 (COVID-19) mRNA vaccines are non-live vaccines so they can be administered to patients on anti-IL-1 therapy, but they may cause disease flares, and the immunogenicity of the COVID-19 vaccine may be decreased [104].
6 Disease Monitoring
Disease activity and end-organ damage should be regularly monitored by a multidisciplinary team consisting of primary health care providers as well as experts in SAIDs, ideally based in a center of excellence. Rheumatologists, immunologists, hepatologists, ophthalmologists, otolaryngologists, nephrologists, neurologists, genetic counselors, physiatrists, occupational therapists, speech therapists, nutrition team, social support experts, and psychological health experts may be involved in the patients’ care according to individual patient needs. The frequency of monitoring depends on the disease activity, disease severity, treatment status, and timing of the treatment. Another crucial aspect of monitoring is the necessity for treatment adjustments based on patients’ changing needs. The multidisciplinary patient care team should be attentive to the varying needs of children, adolescents, adults, reproductive-age women, and seniors while monitoring these patients [105, 106].
Clinical symptoms, patient-reported outcomes (PROs), acute phase reactants, growth, and development should be evaluated at every visit to monitor disease activity and prevent long-term morbidity and mortality by using a treat-to-target approach. The most commonly used laboratory workup to evaluate systemic inflammation includes neutrophil count, CRP, ESR, and, if available, SAA and S100 protein. Overall disease activity and patient weight are important parameters for adjusting anti-IL-1 drug doses. Systemic inflammation may cause amyloidosis, so patients should be regularly monitored for kidney amyloidosis through urinalysis. Growth and development should also be assessed regularly at every visit, as chronic inflammation can significantly impact these parameters in children.
7 Conclusions
The diagnosis of IL-1-mediated SAIDs requires a high index of suspicion due to their rarity. Early recognition can lead timely treatment with appropriate biologic medications and referral to specialized health care teams, with the goal of improving patient outcomes and reducing morbidity and mortality. A coordinated care approach can decrease the burden secondary to multiple subspecialty visits and may assist decreasing hospitalizations for these medically complex patients. Moreover, streamlining access to specific genetic testing and treatment not only makes the diagnostic process easier but also proves to be more cost-effective. This comprehensive approach is instrumental in diagnosing these patients early, thereby preventing morbidity and mortality and improving overall healthcare outcomes.
References
Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621–68.
Clarke JT, Coyle D, Evans G, Martin J, Winquist E. Toward a functional definition of a “rare disease” for regulatory authorities and funding agencies. Value Health. 2014;17(8):757–61.
Piskin D, Romano M, Aletaha D, Feldman BM, Goldbach-Mansky R, Carmona L, Demirkaya E. Developing guidelines for ultrarare rheumatic disorders: a bumpy ride. Ann Rheum Dis. 2022. https://doi.org/10.1136/ard-2022-222538
McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97(1):133–44.
Romano M, Arici ZS, Piskin D, Alehashemi S, Aletaha D, Barron KS, et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis. 2022;81(7):907–21.
ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. 2015;74(9):1636–44.
Broderick L, Hoffman HM. IL-1 and autoinflammatory disease: biology, pathogenesis and therapeutic targeting. Nat Rev Rheumatol. 2022;18(8):448–63.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26.
Challagundla N, Saha B, Agrawal-Rajput R. Insights into inflammasome regulation: cellular, molecular, and pathogenic control of inflammasome activation. Immunol Res. 2022;70(5):578–606. https://doi.org/10.1007/s12026-022-09286-9.
Challagundla N, Saha B, Agrawal-Rajput R. Insights into inflammasome regulation: cellular, molecular, and pathogenic control of inflammasome activation. Immunol Res. 2022;70(5):578–606.
Harapas CR, Steiner A, Davidson S, Masters SL. An update on autoinflammatory diseases: inflammasomopathies. Curr Rheumatol Rep. 2018;20(7):40.
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–5.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. https://doi.org/10.3390/ijms20133328.
Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med. 2011;208(3):519–33.
Savic S, McDermott MF. Tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS). In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of autoinflammation. Cham: Springer Nature Switzerland AG; 2019. p. 329–45.
Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF receptor-associated periodic syndrome (TRAPS): current perspectives. Int J Mol Sci. 2020;21(9):3263. https://doi.org/10.3390/ijms21093263.
Frenkel J, Houten SM, Waterham HR, Wanders RJ, Rijkers GT, Duran M, et al. Clinical and molecular variability in childhood periodic fever with hyperimmunoglobulinaemia D. Rheumatology (Oxford). 2001;40(5):579–84.
Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22(2):175–7.
Politiek FA, Waterham HR. Compromised protein prenylation as pathogenic mechanism in mevalonate kinase deficiency. Front Immunol. 2021;12: 724991.
Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17(8):914–21.
Naik HB, Cowen EW. Autoinflammatory pustular neutrophilic diseases. Dermatol Clin. 2013;31(3):405–25.
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360(23):2426–37.
Welzel T, Benseler SM, Kuemmerle-Deschner JB. Management of monogenic IL-1 mediated autoinflammatory diseases in childhood. Front Immunol. 2021;12: 516427.
Johnstone RF, Dolen WK, Hoffman HM. A large kindred with familial cold autoinflammatory syndrome. Ann Allergy Asthma Immunol. 2003;90(2):233–7.
Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108(4):615–20.
Yildiz M, Haslak F, Adrovic A, Barut K, Kasapcopur O. Autoinflammatory diseases in childhood. Balkan Med J. 2020;37(5):236–46.
Nakagawa K, Gonzalez-Roca E, Souto A, Kawai T, Umebayashi H, Campistol JM, et al. Somatic NLRP3 mosaicism in Muckle-Wells syndrome. A genetic mechanism shared by different phenotypes of cryopyrin-associated periodic syndromes. Ann Rheum Dis. 2015;74(3):603–10.
Kümmerle-Deschner JB, Tyrrell PN, Reess F, Kötter I, Lohse P, Girschick H, et al. Risk factors for severe Muckle-Wells syndrome. Arthritis Rheum. 2010;62(12):3783–91.
Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50(2):607–12.
Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56(4):1273–85.
Caroli F, Pontillo A, D’Osualdo A, Travan L, Ceccherini I, Crovella S, et al. Clinical and genetic characterization of Italian patients affected by CINCA syndrome. Rheumatology (Oxford). 2007;46(3):473–8.
Welzel T, Kuemmerle-Deschner JB. Diagnosis and management of the cryopyrin-associated periodic syndromes (CAPS): what do we know today? J Clin Med. 2021;10(1):128. https://doi.org/10.3390/jcm10010128.
Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep. 2011;13(2):123–31.
Wittkowski H, Kuemmerle-Deschner JB, Austermann J, Holzinger D, Goldbach-Mansky R, Gramlich K, et al. MRP8 and MRP14, phagocyte-specific danger signals, are sensitive biomarkers of disease activity in cryopyrin-associated periodic syndromes. Ann Rheum Dis. 2011;70(12):2075–81.
Pastore S, Paloni G, Caorsi R, Ronfani L, Taddio A, Lepore L, et al. Serum amyloid protein A concentration in cryopyrin-associated periodic syndrome patients treated with interleukin-1 beta antagonist. Clin Exp Rheumatol. 2014;32(Supplement84):S63–6.
Haas N, Küster W, Zuberbier T, Henz BM. Muckle-Wells syndrome: clinical and histological skin findings compatible with cold air urticaria in a large kindred. Br J Dermatol. 2004;151(1):99–104.
Cekic S, Yalcinbayir O, Kilic SS. Ocular Involvement in Muckle-Wells Syndrome. Ocul Immunol Inflamm. 2020;28(1):70–8.
Alejandre N, Ruiz-Palacios A, Garcia-Aparicio AM, Blanco-Kelly F, Bermudez S, Fernandez-Sanz G, et al. Description of a new family with cryopyrin-associated periodic syndrome: risk of visual loss in patients bearing the R260W mutation. Rheumatology (Oxford). 2014;53(6):1095–9.
Dollfus H, Hafner R, Hofmann HM, Russo RA, Denda L, Gonzales LD, et al. Chronic infantile neurological cutaneous and articular/neonatal onset multisystem inflammatory disease syndrome: ocular manifestations in a recently recognized chronic inflammatory disease of childhood. Arch Ophthalmol. 2000;118(10):1386–92.
Nakanishi H, Yamada S, Kita J, Shinmura D, Hosokawa K, Sahara S, Misawa K. Auditory and vestibular characteristics of NLRP3 inflammasome related autoinflammatory disorders: monogenic hearing loss can be improved by anti-interleukin-1 therapy. Front Neurol. 2022;13:865763. https://doi.org/10.3389/fneur.2022.865763.
Iida Y, Wakiguchi H, Okazaki F, Nakamura T, Yasudo H, Kubo M, et al. Early canakinumab therapy for the sensorineural deafness in a family with Muckle-Wells syndrome due to a novel mutation of NLRP3 gene. Clin Rheumatol. 2019;38(3):943–8.
Klein AK, Horneff G. Improvement of sensoneurinal hearing loss in a patient with Muckle-Wells syndrome treated with anakinra. Klin Padiatr. 2010;222(4):266–8.
Federici S, Sormani MP, Ozen S, Lachmann HJ, Amaryan G, Woo P, et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015;74(5):799–805.
Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol. 2013;147(3):155–74.
Lachmann HJ, Papa R, Gerhold K, Obici L, Touitou I, Cantarini L, et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis. 2014;73(12):2160–7.
Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore). 2002;81(5):349–68.
Sangiorgi E, Rigante D. The clinical chameleon of autoinflammatory diseases in children. Cells. 2022;11(14):2231. https://doi.org/10.3390/cells11142231.
Romano M, Arici ZS, Piskin D, Alehashemi S, Aletaha D, Barron K, et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Arthritis Rheumatol. 2022;74(7):1102–21.
Li Y, Yu M, Lu M. Pathophysiology, clinical manifestations and current management of IL-1 mediated monogenic systemic autoinflammatory diseases, a literature review. Pediatr Rheumatol Online J. 2022;20(1):90.
Ter Haar NM, Jeyaratnam J, Lachmann HJ, Simon A, Brogan PA, Doglio M, et al. The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the Eurofever Registry. Arthritis Rheumatol. 2016;68(11):2795–805.
Lainka E, Neudorf U, Lohse P, Timmann C, Bielak M, Stojanov S, et al. Incidence and clinical features of hyperimmunoglobulinemia D and periodic fever syndrome (HIDS) and spectrum of mevalonate kinase (MVK) mutations in German children. Rheumatol Int. 2012;32(10):3253–60.
Bader-Meunier B, Florkin B, Sibilia J, Acquaviva C, Hachulla E, Grateau G, et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics. 2011;128(1):e152–9.
van der Hilst JCH, Bodar EJ, Barron KS, Frenkel J, Drenth JPH, van der Meer JWM, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore). 2008;87(6):301–10.
Ammouri W, Cuisset L, Rouaghe S, Rolland MO, Delpech M, Grateau G, et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology (Oxford). 2007;46(10):1597–600.
Cuisset L, Drenth JP, Simon A, Vincent MF, van der Velde VS, van der Meer JW, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet. 2001;9(4):260–6.
Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–44.
Rusmini M, Federici S, Caroli F, Grossi A, Baldi M, Obici L, et al. Next-generation sequencing and its initial applications for molecular diagnosis of systemic auto-inflammatory diseases. Ann Rheum Dis. 2016;75(8):1550–7.
Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019;78(8):1025–32.
Aktas B, Gumus D, Tunali A, Kunter Z, Adrovic A. Mevalonate kinase deficiency/hyperimmunoglobulin D syndrome (MVK/HIDS) in a differential diagnosis of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome and familial Mediterranean fever (FMF): a case report. Turk Arch Pediatr. 2022;57(3):365–7.
Shinar Y, Ceccherini I, Rowczenio D, Aksentijevich I, Arostegui J, Ben-Chetrit E, et al. ISSAID/EMQN best practice guidelines for the genetic diagnosis of monogenic autoinflammatory diseases in the next-generation sequencing era. Clin Chem. 2020;66(4):525–36.
Van Gijn ME, Ceccherini I, Shinar Y, Carbo EC, Slofstra M, Arostegui JI, et al. New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet. 2018;55(8):530–7.
Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. 2008;29(6):803–8.
Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011;63(11):3625–32.
Mendonca LO, Malle L, Donovan FX, Chandrasekharappa SC, Montealegre Sanchez GA, Garg M, et al. Deficiency of interleukin-1 receptor antagonist (DIRA): report of the first indian patient and a novel deletion affecting IL1RN. J Clin Immunol. 2017;37(5):445–51.
Jesus AA, Osman M, Silva CA, Kim PW, Pham TH, Gadina M, et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome: description of two unrelated cases from Brazil. Arthritis Rheum. 2011;63(12):4007–17.
Schnappauf O, Aksentijevich I. Current and future advances in genetic testing in systemic autoinflammatory diseases. Rheumatology (Oxford). 2019;58(Suppl 6):vi44–55.
Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. 2011;70(12):2155–8.
Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92.
Fox E, Jayaprakash N, Pham TH, Rowley A, McCully CL, Pucino F, et al. The serum and cerebrospinal fluid pharmacokinetics of anakinra after intravenous administration to non-human primates. J Neuroimmunol. 2010;223(1–2):138–40.
Rodriguez-Smith J, Lin YC, Tsai WL, Kim H, Montealegre-Sanchez G, Chapelle D, et al. Cerebrospinal fluid cytokines correlate with aseptic meningitis and blood-brain barrier function in neonatal-onset multisystem inflammatory disease: central nervous system biomarkers in neonatal-onset multisystem inflammatory disease correlate with central nervous system inflammation. Arthritis Rheumatol. 2017;69(6):1325–36.
Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58(8):2443–52.
Caorsi R, Lepore L, Zulian F, Alessio M, Stabile A, Insalaco A, et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis Res Ther. 2013;15(1):R33.
Dhimolea E. Canakinumab. MAbs. 2010;2(1):3–13.
Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, Wittkowski H, Bialkowski A, Tzaribachev N, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 2011;63(3):840–9.
Kullenberg T, Lofqvist M, Leinonen M, Goldbach-Mansky R, Olivecrona H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology (Oxford). 2016;55(8):1499–506.
Neven B, Marvillet I, Terrada C, Ferster A, Boddaert N, Couloignier V, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62(1):258–67.
Sibley CH, Plass N, Snow J, Wiggs EA, Brewer CC, King KA, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012;64(7):2375–86.
Ross JB, Finlayson LA, Klotz PJ, Langley RG, Gaudet R, Thompson K, et al. Use of anakinra (Kineret) in the treatment of familial cold autoinflammatory syndrome with a 16-month follow-up. J Cutaneous Med Surg. 2008;12(1):8–16.
Goldbach-Mansky R, Shroff SD, Wilson M, Snyder C, Plehn S, Barham B, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58(8):2432–42.
Hoffman HM, Throne ML, Amar NJ, Cartwright RC, Kivitz AJ, Soo Y, et al. Long-term efficacy and safety profile of rilonacept in the treatment of cryopryin-associated periodic syndromes: results of a 72-week open-label extension study. Clin Ther. 2012;34(10):2091–103.
Kuemmerle-Deschner JB, Hachulla E, Cartwright R, Hawkins PN, Tran TA, Bader-Meunier B, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. 2011;70(12):2095–102.
Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360(23):2416–25.
Yokota S, Imagawa T, Nishikomori R, Takada H, Abrams K, Lheritier K, et al. Long-term safety and efficacy of canakinumab in cryopyrin-associated periodic syndrome: results from an open-label, phase III pivotal study in Japanese patients. Clin Exp Rheumatol. 2017;35(Supplement108):S19–26.
Brogan PA, Hofer M, Kuemmerle-Deschner JB, Kone-Paut I, Roesler J, Kallinich T, et al. Rapid and sustained long-term efficacy and safety of canakinumab in patients with cryopyrin-associated periodic syndrome ages five years and younger. Arthritis Rheumatol. 2019;71(11):1955–63.
Highlights of prescribing Information: Ilaris. accessdata.fda.gov; 2020. p. 1–2. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125319s100lbl.pdf.
Ilaris: EPAR - prouduct information. 2023. p. 2. https://www.ema.europa.eu/en/documents/product-information/ilaris-epar-product-information_en.pdf.
De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. 2018;378(20):1908–19.
Arostegui JI, Anton J, Calvo I, Robles A, Iglesias E, López-Montesinos B, et al. Open-label, phase II study to assess the efficacy and safety of canakinumab treatment in active hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheumatol. 2017;69(8):1679–88.
Hosono K, Matsumoto K, Shimbo M, Tsumiyama I, Kato C. Real-world safety and effectiveness of canakinumab in patients with tumour necrosis factor receptor-associated periodic syndrome or hyperimmunoglobulinaemia D syndrome: Interim results from post-marketing surveillance in Japan. Mod Rheumatol. 2023;33(2):381–91. https://doi.org/10.1093/mr/roac041.
Obici L, Meini A, Cattalini M, Chicca S, Galliani M, Donadei S, et al. Favourable and sustained response to anakinra in tumour necrosis factor receptor-associated periodic syndrome (TRAPS) with or without AA amyloidosis. Ann Rheum Dis. 2011;70(8):1511–2.
Ter Haar N, Lachmann H, Özen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. 2013;72(5):678–85.
Gattorno M, Pelagatti MA, Meini A, Obici L, Barcellona R, Federici S, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58(5):1516–20.
Bulua AC, Mogul DB, Aksentijevich I, Singh H, He DY, Muenz LR, et al. Efficacy of etanercept in the tumor necrosis factor receptor-associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum. 2012;64(3):908–13.
Vaitla PM, Radford PM, Tighe PJ, Powell RJ, McDermott EM, Todd I, et al. Role of interleukin-6 in a patient with tumor necrosis factor receptor-associated periodic syndrome: assessment of outcomes following treatment with the anti-interleukin-6 receptor monoclonal antibody tocilizumab. Arthritis Rheum. 2011;63(4):1151–5.
Shendi HM, Devlin LA, Edgar JD. Interleukin 6 blockade for hyperimmunoglobulin D and periodic fever syndrome. J Clin Rheumatol. 2014;20(2):103–5.
Musters A, Tak PP, Baeten DL, Tas SW. Anti-interleukin 6 receptor therapy for hyper-IgD syndrome. BMJ Case Rep. 2015;2015:bcr2015210513. https://doi.org/10.1136/bcr-2015-210513.
Ozen S, Kuemmerle-Deschner JB, Cimaz R, Livneh A, Quartier P, Kone-Paut I, et al. International retrospective chart review of treatment patterns in severe familial Mediterranean fever, tumor necrosis factor receptor-associated periodic syndrome, and mevalonate kinase deficiency/hyperimmunoglobulinemia D syndrome. Arthritis Care Res (Hoboken). 2017;69(4):578–86.
Papa R, Lane T, Minden K, Touitou I, Cantarini L, Cattalini M, et al. INSAID variant classification and Eurofever criteria guide optimal treatment strategy in patients with TRAPS: Data from the Eurofever Registry. J Allergy Clin Immunol Pract. 2021;9(2):783-91.e4.
Administration UFaD. Highlights of Prescribing Information: Kineret; 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/103950s5189lbl.pdf.
Administration UFaD. Highlights of Prescribing Information: Arcalyst; 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125249s049lbl.pdf.
Garg M, de Jesus AA, Chapelle D, Dancey P, Herzog R, Rivas-Chacon R, Muskardin TLW, Reed A, Reynolds JC, Goldbach-Mansky R, Sanchez GAM. Rilonacept maintains long-term inflammatory remission in patients with deficiency of the IL-1 receptor antagonist. JCI Insight. 2017;2(16):e94838. https://doi.org/10.1172/jci.insight.94838.
Jaeger VK, Hoffman HM, van der Poll T, Tilson H, Seibert J, Speziale A, et al. Safety of vaccinations in patients with cryopyrin-associated periodic syndromes: a prospective registry based study. Rheumatology (Oxford). 2017;56(9):1484–91.
Jeyaratnam J, Ter Haar NM, Lachmann HJ, Kasapcopur O, Ombrello AK, Rigante D, Dedeoglu F, Baris EH, Vastert SJ, Wulffraat NM, Frenkel J. The safety of live-attenuated vaccines in patients using IL-1 or IL-6 blockade: an international survey. Pediatr Rheumatol Online J. 2018;16(1):19. https://doi.org/10.1186/s12969-018-0235-z.
Atagündüz P, Keser G, Soy M. Interleukin-1 inhibitors and vaccination including COVID-19 in inflammatory rheumatic diseases: a nonsystematic review. Front Immunol. 2021;12: 734279.
Chuamanochan M, Weller K, Feist E, Kallinich T, Maurer M, Kummerle-Deschner J, et al. State of care for patients with systemic autoinflammatory diseases - results of a tertiary care survey. World Allergy Organ J. 2019;12(3): 100019.
Erbis G, Schmidt K, Hansmann S, Sergiichuk T, Michler C, Kuemmerle-Deschner JB, et al. Living with autoinflammatory diseases: identifying unmet needs of children, adolescents and adults. Pediatr Rheumatol Online J. 2018;16(1):81.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Not applicable.
Conflicts of Interest
Authors reported no conflict of interest.
Availability of Data and Materials
Not applicable
Ethics Approval
Not applicable
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Author Contributions
The manuscript was drafted by KCG and ZSA. All authors were involved in revising it critically for important intellectual content and provided critical feedback on the manuscript. All authors approved the final manuscript for submission and are accountable for the accuracy and integrity of the article.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cetin Gedik, K., Arici, Z.S., Kul Cinar, O. et al. Practical Approach to Diagnosis and Management of IL-1-Mediated Autoinflammatory Diseases (CAPS, TRAPS, MKD, and DIRA). Pediatr Drugs 26, 113–126 (2024). https://doi.org/10.1007/s40272-023-00615-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-023-00615-5