
Abstract
Background
Enteral ibuprofen was first approved as a prescription drug in 1974 for the US market. An intravenous (IV) ibuprofen formulation is approved for use in children older than 6 months of age, but there are limited studies specifically evaluating the pharmacokinetics and safety in children 1–6 months of age.
Aims
The primary purpose of this study was to evaluate the pharmacokinetics of IV ibuprofen in infants younger than 6 months of age. The secondary objective was to evaluate the safety of single and repeated doses of IV ibuprofen in infants younger than 6 months of age.
Methods
This was an industry-sponsored multi-center study. Institutional Review Board approval and informed parental consent were obtained prior to enrollment. Hospitalized neonates and infants younger than 6 months of age with fever or expected postoperative pain were eligible. Enrolled patients received 10 mg/kg of IV ibuprofen every 6 h, with up to four doses per day. Patients were randomized to two sparse sampling technique pharmacokinetic sample time groups. Group 1 samples were drawn at 0, 30 min, and 2 h, while group 2 samples were drawn at 0 min, 1, and 4 h after administration.
Results
A total of 24 children were enrolled in the study, with 15 male patients and 9 female patients. The median age of the cohort was 4.4 months (range 1.1–5.9 months), and the median weight was 5.9 kg (range 2.3–8.8 kg). The arithmetic mean and standard error for peak plasma ibuprofen concentration was 56.28 ± 2.77 µg/mL. Plasma levels declined rapidly with a mean elimination half-life of 1.30 h. Time to peak ibuprofen effect and concentration were similar when compared with older pediatric patients. Clearance and volume of distribution were also similar to those reported in older pediatric patients. No drug-related adverse events were reported.
Conclusions
The pharmacokinetic and short-term safety profiles of IV ibuprofen in pediatric patients 1–6 months of age are comparable to those in children older than 6 months of age.
Trial Registration
Clinicaltrials.gov Trial Registration number and date: NCT02583399—Registered July 2017.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Previous studies have shown that intravenous ibuprofen is safe and effective in treating both pain and fever in adults and in older children. |
Little is known about the pharmacokinetics or short-term safety of intravenous ibuprofen in children 1–6 months of age. |
In this study, the pharmacokinetic profile of intravenous ibuprofen in children 1–6 months of age was similar to previous findings in older children. |
1 Background
Ibuprofen is a nonsteroidal anti-inflammatory drug (NSAID) first approved for adult use in the USA in 1974. Oral ibuprofen has long been used for treating mild-to-moderate pain due to multiple causes in children and adults [1,2,3]. Pediatric indications have focused on the benefits of NSAIDs as adjuncts for analgesia and fever relief with a recent meta-analysis finding that ibuprofen exhibited superior efficacy in reducing fever and pain when compared with acetaminophen [4, 5]. Administration of oral analgesics and antipyretics may not be feasible in hospitalized pediatric patients, necessitating the use of parenteral formulations for treatment of pain and/or fever in these settings [6]. Although a multitude of studies have demonstrated the effectiveness and safety of oral and rectal ibuprofen in the treatment of fever and pain in both adults and children, studies on parenteral ibuprofen are only recently becoming available, as manufacturing constraints for parenteral forms are being overcome [7,8,9,10,11,12,13].
Caldolor® (Cumberland Pharmaceuticals Inc., Nashville, TN, USA), is an IV formulation of ibuprofen. It received approval from the US Food and Drug Administration (FDA) in 2009 for use in adults and in 2016 for use in children older than 6 months of age [14, 15]. Pharmacokinetic (PK) data of IV ibuprofen in adults reveal that IV infusions result in maximum plasma concentrations double that of oral ibuprofen without a difference in the elimination half-life [6]. IV ibuprofen is well tolerated in both adult and pediatric patients older than 6 months of age, and PK data in children older than 6 months of age are similar to those reported in adults [16, 17]. The use of NSAIDs for analgesia has increased as the reduction of opioid administration has become a priority across clinical settings due to the potential risks and side effects associated with drug class [18]. Recent studies have demonstrated the safety and efficacy of IV ibuprofen to treat both pain and fever in children older than 6 months of age and in young and preterm infants [17, 19,20,21,22,23,24], however, there are limited studies evaluating the PK of IV ibuprofen in children 1–6 months of age. The objectives of this study were to evaluate both the PK profile and the short-term safety profile of single and repeat doses of IV ibuprofen in children aged birth to 6 months of age.
2 Methods
This protocol was approved by the Western Institutional Review Board (WIRB) and the study was approved by site-specific institutional review boards (IRB). This study was conducted under an Investigational New Drug Application and performed in accordance with the Declaration of Helsinki, Title 21 of the CFR Parts 50, 56, and 312 and International Conference on Harmonisation (ICH) E6. The study was registered at ClinicalTrials.gov (Study identifier: NCT02583399) on 20 October 2015, prior to the enrollment of the first patient.
Eligible patients included hospitalized male or female pediatric patients between the ages of birth [> 37 weeks (+ 0 days) gestational age at birth] to less than 6 calendar months of age with a clinical indication of pain or fever with written informed consent provided by the parent or legal guardian of the patient. The study was conducted at four clinical centers in the USA.
Patients were excluded from the study if they had (1) inadequate intravenous access for obtaining laboratory samples, (2) an uncorrected ductus-dependent congenital heart defect, (3) history of allergy or hypersensitivity to NSAIDs or acetylsalicylic acid (ASA), (4) current history of uncorrected hypovolemia, or acute renal or liver disease, (5) received NSAIDs, acetaminophen, or ASA drug therapy within 4 h prior to study drug dosing or another investigational drug within the previous 30 days, and (6) if the patient was considered unsuitable for the study in the opinion of the investigator.
This in-patient study utilized a prospective, open-label single- or multiple-dose study design that consisted of a screening/baseline period (up to 48 h prior to the first dose of the study drug), a treatment period [beginning with the first dose of the study drug (study hour 0) and continuing for up to 48 h] and a post-treatment period [beginning at the end of the treatment period and continuing for 24 h (study hour 72)] for a maximum of 120 h from screening to end of study.
During the screening/baseline period, informed consent was documented, and a standard physical examination, including vital signs [temperature, heart rate (HR), blood pressure (BP), and respiratory rate (RR)], was performed to document each patient’s baseline vital signs and symptoms. Demographic data, medical history, concomitant medications, and safety laboratory data (complete blood count, renal and hepatic function, coagulation profile) were also obtained.
On the basis of the current FDA-approved dosing for IV ibuprofen in pediatric patients older than 6 months of age (10 mg/kg every 4–6 h as necessary for pain or fever) and additional published results [24], the dose selected for this study was 10 mg/kg of IV ibuprofen administered over 10 min. The total daily dose did not exceed 40 mg/kg or four doses per day for a maximum of eight total doses during the 48-h treatment period [17]. Use of other NSAIDs or acetaminophen was restricted between 4 h prior to and 48 h following the first dose of IV ibuprofen for safety assessments. Use of these medications was restricted for 4 h prior to the initial IV ibuprofen dose to account for the minimum time recommended between doses of these medications for pain and/or fever.
The treatment period began at study hour 0, when the initial 10 mg/kg dose of IV ibuprofen was administered. Subsequent doses of study drug were administered at the investigator’s discretion every 6–8 h as needed during the remainder of the treatment period. Vital signs were taken immediately pre-dose, post-dose, and at 30, 60, 120, and 240 min following each dose of study drug as well as 24 and 48 h following initial dosing. Blood samples for safety monitoring were taken at study hour 24. To minimize laboratory testing, samples obtained for routine care within the allowable time window (± 6 h) could be used for study-specific safety testing.
The post-treatment period began 48 h after the first dose of IV ibuprofen and continued through 72 h following the initial dose. During this period, vital signs and laboratory assessments were performed 72 h following the initial dose of ibuprofen or at the time of hospital discharge, whichever came first.
2.1 Pharmacokinetic Analysis
Blood samples were drawn in K-2 EDTA tubes for PK profiling. To minimize individual patient lab sampling, a sparse sampling model was utilized. Patients were randomized 1:1 to 2 PK sample time groups with group 1 samples drawn at 0, 30, and 120 min and group 2 samples were drawn at 0, 60, 240 min after first dose administration. An allowance of ± 5 min was permissible for sampling times. Collection of samples per venipuncture was preferred; however, if the patient had indwelling venous or arterial catheters, collection from these devices was permitted provided that a sufficient waste sample was extracted per the site’s standard protocol prior to PK sample collection. Blood samples collected for PK analysis were 2 mL each for a total of 6 mL collected from each patient for PK. Patients that withdrew before completion of the PK sample collection were replaced.
Plasma samples were separated by refrigerated centrifuge, frozen at − 80 °C, and kept frozen until analyzed. Ibuprofen and its internal analytical standard, ibuprofen-d3, were extracted from 0.025 mL aliquots of plasma. Samples were extracted utilizing a liquid–liquid extraction procedure, then injected into a liquid chromatograph equipped with a tandem mass spectrometry detector. Quantitation is based on peak area ratio of the analyte versus its stable labeled internal standard. A weighted (\(1/C2\)) linear regression was performed to determine the concentration of the analyte.
The extracted samples were analyzed by liquid chromatography mass spectrometry/mass spectrometry (LC-MS/MS) using an AB Sciex API 4000 equipped with a TurboIon Spray using a gradient at 25 °C at a flow rate of 1.000 mL/min. Study samples were analyzed once using a calibration curve composed of four sets of quality control (QC) samples [low (3.00 µg/mL), intermediate (7.50 µg/mL), medium 50.00 (µg/mL), and high (100.00 µg/mL)] analyzed at least in duplicate. The calibration range of the assay was 1.00–100.00 µg/mL. There were no reassays in this project.
Of the 70 samples analyzed, 50 samples were selected for the incurred sample reproducibility test to demonstrate that results obtained from study sample analysis are reproducible. The bioanalytical method was considered reproducible if at least 67% of the incurred sample reanalysis data were within 20% of the difference between the two values divided by the average of both values [25]. All 50 reanalyzed samples met the criteria of assay reproducibility definition.
The performance of the analytical method was successfully demonstrated during the validation and also by the in-study method performance. The analyte and the internal standard were stable under all conditions tested. The long-term stability of analyte in matrix at − 80° C (641 days) was extended to cover the storage period of the study samples (699 days).
2.2 Safety Monitoring
Vital signs, including temperature, heart rate, respiratory rate, and systolic and diastolic blood pressures, were measured and recorded as described above.
Blood samples for safety monitoring were taken at screening, then at study hour 24 (± 6 h) and 72 (± 6 h) or hospital discharge, whichever occurred first. The safety labs consisted of serum electrolytes (sodium, potassium, chloride, carbon dioxide content), glucose, renal function (blood urea nitrogen, creatinine), liver function (total bilirubin, albumin, total protein, aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase), complete blood count with differential, and coagulation profile (prothrombin time and partial thromboplastin time). If any of the laboratory assessments were taken as part of the standard of care testing within the allowable time window for data collection, these values were used for safety monitoring and additional research assessments were not collected. Concomitant medications were collected through study hour 48 (end of the treatment period) or discharge, whichever occurred first. Adverse events (AEs) were monitored and recorded continuously throughout the study. If the patient was discharged prior to study hour 72, a post-discharge follow-up phone call was made to assess for any AEs following discharge up to study hour 72.
2.3 Statistical Analysis
A sample size of 24 patients was chosen for adequate PK and safety evaluation. No power calculations were performed. Patients who were enrolled into the study but did not receive any study drug were replaced. Patients who were withdrawn from the study after receiving study drug but did not complete the PK portion of the study within the first 4 h were also replaced. Patients who received the initial dose of study drug and had the primary PK assessments in the 48-h study period were included in the PK-evaluable population. All concentration values Below the Lower Limit of Quantitation (BLQ) were set to zero. Samples with no reportable value occurring prior to dosing were replaced by “0.00”, otherwise, they were set to missing for tabulation, graphical representation, and calculation purposes; all samples with no reportable value observed after dosing were set to missing. For the PK calculation, only observed data were used in the analysis except for concentration values BLQ and samples with no reportable value occurring prior to dosing as described above. There was no extrapolation or imputation of missing data.
Descriptive statistics including number of observations (N), mean, median, standard deviation (SD), standard error of the mean (SEM), minimum, and maximum were calculated from plasma concentrations for each patient at the collected time points. Continuous data were summarized as the mean, median, SD or SE, minimum (Min), maximum, and number of patients.
The following PK parameters were calculated from the mean ibuprofen plasma drug concentration–time data using standard non-compartmental methods consistent with the IV infusion route of administration:
AUC0–t: area under the concentration–time curve from time zero until the last measurable concentration calculated using the trapezoidal method.
AUC0–inf: area under the concentration-time curve from time zero to infinity (extrapolated), calculated as \({AUC}_{0-t}+ \left(\frac{{C}_{\mathrm{last}}}{{K}_{\mathrm{el}}}\right)\), where Clast is the last measurable concentration and Kel is the terminal elimination rate constant (the negative of the estimated slope of the linear regression of log-transformed concentration versus time profile in the terminal elimination phase).
Cmax: maximal measured plasma concentration, taken from the plasma concentration–time profile.
Tmax: time when the maximal serum concentration was observed, taken from the serum concentration–time profile.
T1/2 el: terminal elimination half-life, calculated as \(\mathrm{ln}(2)/{K}_{\mathrm{el}}\).
Cl: apparent clearance, calculated as \(\frac{\mathrm{Dose}}{{\mathrm{AUC}}_{0-\mathrm{inf}}}\).
Cl/WT: apparent clearance, calculated as \(\left(\frac{\mathrm{Dose}}{{\mathrm{AUC}}_{0-\mathrm{inf}}}\right)/\mathrm{weight}\). The mean body weight was used.
Vz: apparent volume of distribution, calculated as \(\frac{\mathrm{Dose}}{\left({K}_{\mathrm{el}}\times {\mathrm{AUC}}_{0-\mathrm{inf}}\right)}\).
Vz/WT: apparent volume of distribution, calculated as \(\left(\frac{\mathrm{Dose}}{{{K}_{\mathrm{el}}\times \mathrm{AUC}}_{0-\mathrm{inf}}}\right)/\mathrm{weight}\). The mean body weight was used. Standard error (SE) was calculated for observed parameters measured (AUC0–t and Cmax). The nominal time of infusion (10 min) and nominal dose were used for the derivation of PK parameters calculated using Phoenix WinNonlin software. The non-compartmental analysis sparse sampling methodology employed by Phoenix WinNonlin software calculates PK parameters on the basis of the mean profile for all the subjects in the dataset. Therefore, Cl, Vz, and T1/2 el are derived data from this modeling rather than calculated for each individual patient, so individual weights were not used as the denominator for the Cl/WT and Vz/WT parameters. All statistical computations were performed using the SAS® Software version 9.4 (SAS Institute Inc., Cary, NC, USA).
3 Results
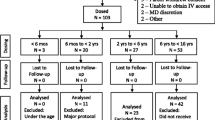
A total of 30 hospitalized patients were enrolled between 8 August 2017 and 21 February 2019, and 24 patients received at least one dose of IV ibuprofen at 10 mg/kg. Enrollment by center is summarized in Table S1. Consent was obtained for six patients who did not receive IV ibuprofen for the following reasons: attending physician decision (n = 3), research team not available for study procedures following consent (n = 2), and an inability to obtain adequate vascular access for PK sampling (n = 1). Of the 24 patients enrolled, 22 were hospitalized for a surgical procedure, 1 was hospitalized for peritoneal dialysis, and 1 was hospitalized for myocarditis secondary to influenza A/B infection. A total of 23 patients were treated with IV ibuprofen for the indication of post-procedural pain and 1 patient was treated for both pain and fever. Primary diagnoses and intervention for treated patients are presented in Table 1.
Of the 24 patients who received IV ibuprofen, 15 (62.5%) were male and 9 (37.5%) were female. The majority of treated patients were white (17/24, 70.8%) and non-Hispanic or non-Latino (19/24, 79.2%). The median age of the cohort was 4.4 months (range 1.1–5.9 months), and the median weight was 5.9 kg (range 2.3–8.8 kg). Baseline demographics are presented in Table 2.
Patients were administered IV ibuprofen as a single dose or every 6–8 h as needed for the treatment of pain or fever. Of the 24 patients who received treatment, 20 received a single dose, and 4 received multiple doses. One patient had undetectable plasma drug concentrations at all time points and was excluded from the PK analysis. Therefore, the PK population was composed of 23 total patients. Plots of mean plasma ibuprofen concentrations versus time profile are presented using linear scales in Fig. 1. IV ibuprofen in pediatric patients under the age of 6 months displayed a similar PK profile to its PK profile in both adults and in older pediatric populations [17].

Mean plasma concentrations over time following a single dose of IV ibuprofen in pediatric patients aged 1–6 months. Error bars represent the standard deviation.
Consistent with findings in older children, peak ibuprofen concentration (Tmax) was observed at the end of the 10-min infusion period (Table 3). Following a single 10 mg/kg IV dose, the mean peak ibuprofen concentration Cmax was 56.28 µg/mL. This is similar to levels reported for all pediatric age groups: 59.2 µg/mL for patients aged 6 months to less than 2 years; 64.2 µg /mL for 2 years to less than 6 years, and 61.9 µg/mL for those 6–16 years of age (Table 3). The AUC0–t and AUC0–inf were 75.74 h·µg/mL and 85.87 h·µg/mL, respectively. Ibuprofen plasma levels declined rapidly with an elimination half-life (T1/2 el) of 1.30 h. The apparent volume of distribution (Vz) in children was 0.22 L/kg.
Absolute clearance and volume of distribution were lower in the current cohort compared with pediatric patients > 6 months of age, however, when normalized by body weight, clearance and volume of distribution were similar between the age groups (Table 3).
3.1 Safety
Of the 30 enrolled patients, 6 were not dosed, 20 received a single dose of ibuprofen, 1 received four doses, and 3 received eight doses. Of the 24 patients who received the drug, 17 completed all three study periods (i.e., screening, treatment, post-treatment). There were seven patients who were prematurely withdrawn prior to the end of the 48-h treatment period, one of whom was withdrawn prior to 24 h at 6 h following the initial dose of IV ibuprofen. Of those prematurely withdrawn, four were discharged home prior to completion of the 72-h study dosing and observation periods, and three were withdrawn due to the primary team’s desire to introduce study-restricted medications (acetaminophen and acetaminophen-hydrocodone). Of the 17 patients who completed all study procedures, 14 were discharged prior to study hour 72, and 3 remained hospitalized at 72 h. No patients withdrew due to study-related adverse events (AE).
A total of eight patients experienced 14 AEs during the study period; of these, 8 were considered mild and 6 moderate. There were three serious AEs in a single patient in the study: two events of chylothorax and one event of pericardial effusion. One patient experienced an event of chylothorax that was not deemed serious. No events were considered related to study drug administration, and all resolved without apparent sequelae. There were no severe AEs or deaths in the study. A summary of the events reported is included in Table 4.
A listing of concomitant medications recorded from screening through study hour 48 (or discharge, whichever occurred first) is presented in Table S2. No significant safety concerns or trends arose from the safety laboratory measurements collected at screening, 24, and 72 h or time of discharge, which included complete blood count, coagulation, hepatic function, and renal function. A summary of laboratory values is presented in Tables S3–S5.
4 Discussion
Oral ibuprofen and acetaminophen have a long history of use for the treatment of fever and mild pain in pediatric patients. While evaluation of pain in infants and older children has advanced in recent years, adequate pain control remains an elusive goal; however, as up to 40% of hospitalized children still experience moderate-to-severe pain following surgery [2, 26, 27]. Oral analgesic administration has limitations, as a significant number of hospitalized patients may have difficulty ingesting and/or tolerating enteral medications. Children under 6 months of age pose additional practice concerns given their reduced ability to communicate pain, concerns regarding physiologic immaturity, and longstanding dogma about gastrointestinal and renal safety with ibuprofen use in this cohort [28, 29]. Adding these concerns to the current opioid epidemic compels physicians to actively search for adjuncts that can be safely administered via multiple routes for pediatric patients.
Caldolor® (ibuprofen) injection was approved by the US Food and Drug Administration in 2016 for pediatric patients older than 6 months of age. This phase 4 study’s primary objective was to determine the PK profile of single and/or multiple doses of IV ibuprofen in children who were aged birth (> 37 weeks gestational age at birth) and less than 6 months of age. The PK profiles of single-dose IV ibuprofen in children 1–6 months of age were similar to those previously observed in older children [17] (Fig. 2) and consistent with previous findings examining both oral ibuprofen PK in patients 3 months to 10.4 years of age [30] and IV ibuprofen in a single 1-month-old patient [17]. The elimination half-life (T½ el) of IV ibuprofen in patients 1–6 months of age was 1.30 h, which is slightly lower than that observed for older pediatric patients (1.5–1.8 h) [17]. It is unclear if this difference in elimination half-life is due to variability observed in this age cohort, difference in overall condition, or other factors. The study examining IV ibuprofen PK in older children was conducted in febrile hospitalized patients [17], while the cohort in this study was primarily composed of hospitalized pediatric patients with an indication of pain, except for one patient treated for both pain and fever. Studies in adult patients have demonstrated that the PK of IV ibuprofen differs between critically ill and non-critically ill patients [31], and therefore it is possible that PK variability observed in this study and others is related to the individual patient condition. The volume of distribution and clearance were also slightly lower in this cohort compared with older children, although this difference disappears when adjusted for body weight. Oral and IV ibuprofen are commonly used in the treatment of premature neonates with patent ductus arteriosus (PDA). Previous studies evaluating the PK of the same dose of IV ibuprofen in preterm neonates with PDA have reported significantly slower elimination (T1/2 el of 30.5 h to 43.1 h) and reduced clearance (2.1 to 9.5 ml/kg/h) with consequent increased exposure to IV ibuprofen compared with infants older than 1 month and older children [17, 32, 33]. Exposure to ibuprofen appears similar between oral and IV formulations in preterm neonates, infants, and older children [30, 34, 35].

The secondary objective for this study was to assess the short-term safety of IV dosing of ibuprofen in patients 1–6 months of age. No deaths or severe organ injuries occurred during this study. While we noted several serious AEs that were moderate in severity during the study period, including chylothorax and pericardial effusion, none of these events were deemed related to ibuprofen administration.
There are several limitations to these findings. The study was designed to assess the PK profile and safety of ibuprofen in pediatric patients under 6 months of age. While the mean age of the patients enrolled was 4.2 months, only four patients were under the age of 4 months and none were under the age of 1 month. Consequently, the current data provided limited information of the PK profile in pediatric patients under the age of 3 months and no safety or PK information in pediatric patients under the age of 1 month. Previous findings in preterm neonates treated with IV ibuprofen for PDA highlight a significantly different PK profile in patients under 1 month of age compared with older infants and children [17, 23]. This is consistent with the developmental changes that occur in hepatic oxidative mechanisms and glomerular filtration rate during early postnatal life [36, 37]. Furthermore, patients with renal or hepatic disease were also excluded from this study, and these data may also not be reflective of ibuprofen PK in patients with these organ dysfunctions. While we report no AEs that were deemed to be related to ibuprofen administration, the sample size of the current study is inadequate to be confident regarding safety profile, and larger safety-based studies are warranted in this age group. Related to this, the duration of drug exposure in this study was short, as only four patients received more than one dose. Therefore, we cannot comment on safety following longer-term exposure. The study was not designed to assess pain efficacy.
5 Conclusion
This study demonstrates that a single 10 mg/kg dose of IV ibuprofen has a similar PK profile in pediatric patients aged 1–6 months when compared with older pediatric age groups. The volume of distribution and clearance are also similar when adjusted for body weight. While adverse events were noted in this study, none were attributed or related to the administration of ibuprofen. IV ibuprofen is well tolerated in pediatric patients older than 1 month and younger than 6 months of age administered the standard recommendation of 10 mg/kg. The PK profiles in this age group reflect similar available data for older pediatric patients, thus no adjustment in dosing is warranted when children 1–6 months of age require IV ibuprofen for the control of fever or pain.
References
Rainsford KD. History and development of ibuprofen. In: Ibuprofen: discovery, development and therapeutics. New York: Wiley Blackwell; 2015. p. 1–21. https://doi.org/10.1002/9781118743614.ch1.
Mencía S, Alonso C, Pallás-Alonso C, López-Herce J. Evaluation and treatment of pain in fetuses, neonates and children. Children. 2022. https://doi.org/10.3390/children9111688.
Doria M, Careddu D, Iorio R, Verrotti A, Chiappini E, Barbero GM, et al. Paracetamol and ibuprofen in the treatment of fever and acute mild–moderate pain in children: Italian experts’ consensus statements. Children. 2021. https://doi.org/10.3390/children8100873.
Pierce CA, Voss B. Efficacy and safety of ibuprofen and acetaminophen in children and adults: a meta-analysis and qualitative review. Ann Pharmacother. 2010;44(3):489–506. https://doi.org/10.1345/aph.1M332.
Tan E, Braithwaite I, Mckinlay CJD, Dalziel SR. Comparison of acetaminophen (paracetamol) with ibuprofen for treatment of fever or pain in children younger than 2 years: a systematic review and meta-analysis. JAMA Netw Open. 2020. https://doi.org/10.1001/jamanetworkopen.2020.22398.
Pavliv L, Voss B, Rock A. Pharmacokinetics, safety, and tolerability of a rapid infusion of i.v. ibuprofen in healthy adults. Am J Health-Syst Pharm. 2011;68(1):47–51. https://doi.org/10.2146/ajhp100120.
Vilenchik R, Berkovitch M, Jossifoff A, Ben-Zvi Z, Kozer E. Oral versus rectal ibuprofen in healthy volunteers. J Popul Ther Clin Pharmacol. 2012;19(2):179–86.
Schwartz JI, Chan CC, Mukhopadhyay S, McBride KJ, Jones TM, Adcock S, et al. Cyclooxygenase-2 inhibition by rofecoxib reverses naturally occurring fever in humans. Clin Pharmacol Ther. 1999;65(6):653–60. https://doi.org/10.1016/S0009-9236(99)90087-5.
Smith HS, Voss B. Pharmacokinetics of intravenous ibuprofen: implications of time of infusion in the treatment of pain and fever. Drugs. 2012;72(3):327–37. https://doi.org/10.2165/11599230-000000000-00000.
Southworth S, Peters J, Rock A, Pavliv L. A multicenter, randomized, double-blind, placebo-controlled trial of intravenous ibuprofen 400 and 800 mg every 6 hours in the management of postoperative pain. Clin Ther. 2009;31(9):1922–35. https://doi.org/10.1016/j.clinthera.2009.08.026.
Litalien C, Jacqz-Aigrain E. Risks and benefits of nonsteroidal anti-inflammatory drugs in children. Pediatr Drugs. 2001;3(11):817–58. https://doi.org/10.2165/00128072-200103110-00004.
Bookstaver B, Miller, Norris, Rudisill. Intravenous ibuprofen: the first injectable product for the treatment of pain and fever. J Pain Res. 2010;3:67–79. https://doi.org/10.2147/jpr.s6993.
Capparelli EV. Pharmacologic, pharmacodynamic, and pharmacokinetic considerations with intravenous ibuprofen lysine. J Pediatr Pharmacol Ther. 2007;12(3):158–70. https://doi.org/10.5863/1551-6776-12.3.158.
Fields E. Summary review for regulatory action. Deputy Division Director Summary Review. NDA 22348 Supplement 005. Caldolor pediatric indication [Internet]. 2015 [cited 2023 May 2]. Available from: https://fda.report/media/95403/22348-Ibuprofen-DD-Clinical-PREA.pdf
Caldolor [package insert]. Nashville: Cumberland Pharmaceuticals Inc.; 2021.
Southworth SR, Sellers JA. Narrative summary of recently published literature on intravenous ibuprofen. Clin Ther. 2020;42:1210–21. https://doi.org/10.1016/j.clinthera.2020.05.004.
Khalil SN, Hahn BJ, Chumpitazi CE, Rock AD, Kaelin BA, Macias CG. A multicenter, randomized, open-label, active-comparator trial to determine the efficacy, safety, and pharmacokinetics of intravenous ibuprofen for treatment of fever in hospitalized pediatric patients. BMC Pediatr. 2017;17(1):42. https://doi.org/10.1186/s12887-017-0795-y.
Lovecchio F, Premkumar A, Stepan JG, Albert TJ. fighting back: institutional strategies to combat the opioid epidemic: a systematic review. HSS J. 2019;15(1):66–71. https://doi.org/10.1007/s11420-018-09662-y.
Cui X, Zhang J, Gao Z, Sun L, Zhang F. A randomized, double-blinded, placebo-controlled, single dose analgesic study of preoperative intravenous ibuprofen for tonsillectomy in children. Front Pediatr. 2022. https://doi.org/10.3389/fped.2022.956660.
Patel NK, Shah SJ, Lee NK, Gao Q, Carullo VP, Yang CJ. Intraoperative intravenous ibuprofen use is not associated with increased post-tonsillectomy bleeding. Int J Pediatr Otorhinolaryngol. 2020. https://doi.org/10.1016/j.ijporl.2020.109965.
Abdelbaser I, Abo-Zeid M, Hayes S, Taman HI. The analgesic effects of the addition of intravenous ibuprofen to a multimodal analgesia regimen for pain management after pediatric cardiac surgery: a randomized controlled study. J Cardiothorac Vasc Anesth. 2022. https://doi.org/10.1053/j.jvca.2022.11.016.
Gokmen T, Erdeve O, Altug N, Oguz SS, Uras N, Dilmen U. Efficacy and safety of oral versus intravenous ibuprofen in very low birth weight preterm infants with patent ductus arteriosus. J Pediatr. 2011;158(4):549-554.e1. https://doi.org/10.1016/j.jpeds.2010.10.008.
Aranda JV, Thomas R. Systematic review: intravenous ibuprofen in preterm newborns. Semin Perinatol. 2006;30:114–20. https://doi.org/10.1053/j.semperi.2006.04.003.
Ohlsson A, Walia R, Shah SS. Ibuprofen for the treatment of patent ductus arteriosus in preterm or low birth weight (or both) infants. Cochrane Database Syst Rev. 2020. https://doi.org/10.1002/14651858.CD003481.pub8.
US Department of Health and Human Services. Guidance for industry bioanalytical method validation. [Internet]. 2018 [cited 2023 Jan 30]. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf.
Kozlowski LJ, Kost-Byerly S, Colantuoni E, Thompson CB, Vasquenza KJ, Rothman SK, et al. Pain prevalence, intensity, assessment and management in a hospitalized pediatric population. Pain Manag Nurs. 2014;15(1):22–35. https://doi.org/10.1016/j.pmn.2012.04.003.
Poddighe D, Brambilla I, Licari A, Marseglia GL. Ibuprofen for pain control in children new value for an old molecule [Internet]. 2018. Available from: http://www.pec-online.com.
Walsh P, Rothenberg SJ, Bang H. Safety of ibuprofen in infants younger than six months: a retrospective cohort study. PLoS ONE. 2018;13(6): e0199493. https://doi.org/10.1371/journal.pone.0199493.
Ziesenitz VC, Welzel T, van Dyk M, Saur P, Gorenflo M, van den Anker JN. Efficacy and safety of NSAIDs in infants: a comprehensive review of the literature of the past 20 years. Pediatr Drugs. 2022;24:603–55. https://doi.org/10.1007/s40272-022-00514-1.
Kauffman RE, Nelson MV. Effect of age on ibuprofen pharmacokinetics and antipyretic response. J Pediatr. 1992;121(6):969–73. https://doi.org/10.1016/s0022-3476(05)80354-3.
Morris PE, Promes JT, Guntupalli KK, Wright PE, Arons MM. A multi-center, randomized, double-blind, parallel, placebo-controlled trial to evaluate the efficacy, safety, and pharmacokinetics of intravenous ibuprofen for the treatment of fever in critically ill and non-critically ill adults. Crit Care. 2010. https://doi.org/10.1186/cc9089.
Aranda J, Varvarigou A, Beharry K, Bansal R, Bardin C, Modanlou H, et al. Pharmacokinetics and protein binding of intravenous ibuprofen in the premature newborn infant. Acta Paediatr. 1997;86(3):289–93. https://doi.org/10.1111/j.1651-2227.1997.tb08892.x.
Van Overmeire B, Touw D, Schepens PJC, Kearns GL, Van Den Anker JN. Ibuprofen pharmacokinetics in preterm infants with patent ductus arteriosus. Clin Pharmacol Ther. 2001;70(4):336–43.
Nahata MC, Durrell DE, Powell DA, Gupta N. Pharmacokinetics of ibuprofen in febrile children. Eur J Clin Pharmacol. 1991. https://doi.org/10.1007/BF00265858.
Edison PE, Chen S, Yeo CL, Allen JC, Poon WB, Baral VR, et al. Pharmacokinetics of oral versus intravenous ibuprofen for closure of patent ductus arteriosus: a pilot randomised controlled study. J Paediatr Child Health. 2022;58(3):397–403. https://doi.org/10.1111/jpc.15720.
Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther. 2008;118(2):250–67. https://doi.org/10.1016/j.pharmthera.2008.02.005.
Brion LP, Fleischman AR, McCarton C, Schwartz GJ. A simple estimate of glomerular filtration rate in low birth weight infants during the first year of life: Noninvasive assessment of body composition and growth. J Pediatr. 1986;109(4):698–707. https://doi.org/10.1016/s0022-3476(86)80245-1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This clinical trial was sponsored by Cumberland Pharmaceuticals Inc.
Conflict of interest
CDG, JWB, MBT, NVP, and JZ were investigators on this clinical trial. BK and BHYG are employees of Cumberland Pharmaceuticals Inc.
Ethics approval
All studies were conducted following approval by site Institutional Review Boards and performed in accordance with the Declaration of Helsinki.
Consent
Prior to any study-related procedures, written informed consent was obtained from the parent or legal guardian of the patient.
Author contributions
CDG, JWB, MBT, NVP, and JZ were investigators in the clinical study. BK developed study design and BHYG prepared the data. All authors contributed to drafting the manuscript, providing critical revisions, and approving the final manuscript.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Glover, C.D., Berkenbosch, J.W., Taylor, M.B. et al. A Multi-Center Evaluation of the Pharmacokinetics and Safety of Intravenous Ibuprofen in Infants 1–6 Months of Age. Pediatr Drugs 25, 585–593 (2023). https://doi.org/10.1007/s40272-023-00576-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-023-00576-9