
Abstract
The progressive myoclonic epilepsies (PMEs) represent a rare but devastating group of syndromes characterized by epileptic myoclonus, typically action-induced seizures, neurological regression, medically refractory epilepsy, and a variety of other signs and symptoms depending on the specific syndrome. Most of the PMEs begin in children who are developing as expected, with the onset of the disorder heralded by myoclonic and other seizure types. The conditions are considerably heterogenous, but medical intractability to epilepsy, particularly myoclonic seizures, is a core feature. With the increasing use of molecular genetic techniques, mutations and their abnormal protein products are being delineated, providing a basis for disease-based therapy. However, genetic and enzyme replacement or substrate removal are in the nascent stage, and the primary therapy is through antiepileptic drugs. Epilepsy in children with progressive myoclonic seizures is notoriously difficult to treat. The disorder is rare, so few double-blinded, placebo-controlled trials have been conducted in PME, and drugs are chosen based on small open-label trials or extrapolation of data from drug trials of other syndromes with myoclonic seizures. This review discusses the major PME syndromes and their neurogenetic basis, pathophysiological underpinning, electroencephalographic features, and currently available treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Progressive myoclonic epilepsy (PME) represents a clinically and genetically heterogeneous complex group of neurodegenerative diseases associated with spontaneous or action-induced myoclonus and progressive neurological deterioration. |
While disease-specific therapy using genetic treatment or enzyme replacement or substrate reduction is the ultimate therapeutic goal, most children with PME are treated symptomatically with antiepileptic drugs (AEDs). |
Efficacy and safety information for AEDs in PME is based on limited and weak data because of the rarity of the conditions and the lack of placebo-controlled, blinded, randomized studies. |
Some AEDs may exacerbate myoclonic epilepsy or worsen the underlying disease state. |
1 Introduction
Progressive myoclonic epilepsy (PME), sometimes called progressive myoclonus epilepsy, represents a clinically and genetically heterogeneous complex group of neurodegenerative diseases associated with spontaneous or action-induced myoclonus and progressive neurological deterioration [1,2,3,4,5]. Neurologic deterioration may include progressive cognitive decline, ataxia, neuropathy, and myopathy. While myoclonic seizures are a core feature, other seizure types often occur, including generalized tonic–clonic, tonic, and atypical absence seizures. The other seizure types are often more problematic than the myoclonus.
Typically, presentation is in late childhood or adolescence, but PME may affect all ages. The initial presentation is often epilepsy, which may begin in a benign manner and then progressively worsen. Very early on, distinguishing these conditions from more common forms of genetic generalized epilepsy, particularly juvenile myoclonic epilepsy or benign childhood myoclonic epilepsy, can be challenging. Likewise, the electroencephalogram (EEG) may initially be normal. Features suggesting PME are the presence or evolution of progressive neurological disability, failure to respond to antiepileptic drug (AED) therapy and background slowing on the EEG.
Over the last two decades, considerable advances have occurred in deciphering the neurogenetic basis of the PMEs, and they are now being recognized as a group of syndromes with specific genetic etiologies [6,7,8]. PMEs may have an autosomal dominant, recessive, or mitochondrial inheritance. While gene mutations are providing insight into the pathophysiology of the disorders, diagnosis of specific forms of PME is challenging because of genetic heterogeneity, phenotypic similarities, and an overlap of signs and symptoms with other epileptic and neurodegenerative diseases [5]. Although molecular genetics has changed the landscape for diagnosis, many patients still do not have a genetic mutation identified [5].
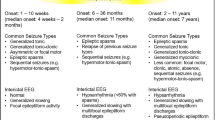
Given the heterogeneity in clinical and genetic findings in the group of syndromes, there is a lack of consensus on which disorders should be included under the category of PME. For the purposes of this review, a selected number of PME syndromes that begin in children are covered. In all disorders described, myoclonus is a predominant feature and neurological deterioration, most often cognitive, occurs. Table 1 lists the conditions and specific gene mutations of the conditions reviewed. Disease-specific therapy, such as genetic manipulation, enzyme replacement, or substrate reduction, are discussed with each syndrome. Symptomatic therapy of the epilepsy, a mainstay of therapy in PME, is discussed in a single section since the efficacy of AEDs is not syndrome specific.
2 Definition and Mechanisms of Myoclonus
Myoclonus is a sudden brief (20–250 ms) contraction (positive myoclonus) or a brief and sudden cessation (negative myoclonus) of tonic muscle inducing a lightening-like muscle jerk arising abnormally from the nervous system [9, 10]. Myoclonic movements are the most sudden and brief muscle jerks that can be produced from involuntary nervous system stimulation. While myoclonus can occur spontaneously, myoclonus exacerbated or triggered by muscle activation—action myoclonus—is common in PME, and some consider it essential for the diagnosis [4]. The brevity of the myoclonus means there is no apparent loss of consciousness.
Epileptic myoclonus occurs as a result of descending neuronal firing of action potentials, whose spatial (spread) and temporal amplification can trigger overt epileptic activity and be classified as cortical (positive and negative) or thalamocortical, also termed cortical-subcortical [9,10,11]. Myoclonus in PME is typically either cortical or cortical-subcortical although both processes may occur in many patients. Other seizure types may be seen in both forms of PME, usually generalized seizures such as absence or generalized tonic–clonic, tonic, or atonic seizures.
The cerebral cortex is the most common origin for myoclonus [3]. Cortical myoclonus is usually action induced or sensitive to somatosensory—or occasionally visual—stimuli or emotional cues and is of epileptic origin [12]. Cortical myoclonus can be focal, multifocal, or generalized arrhythmical jerks that often involve the face or distal extremities or can be generalized because of thalamic involvement. The movements are often multifocal because of intra- and interhemispheric spread. The myoclonus involves agonist and antagonist muscles simultaneously and shows a rostrocaudal recruitment of neurons. Cortical myoclonus tends to be of shorter duration than thalamocortical myoclonus. Surface electromyography (EMG) shows multifocal short duration (20–70 ms) muscle activity [13]. In cortical myoclonus, the EEG epileptiform discharges precede the myoclonus. While this may be difficult to detect during EEG monitoring because of muscle artifact, back-averaging EEG-EMG can show time-locked cortical spikes, polyspikes, or spike-and-wave preceding the myoclonic jerk by 10–40 ms [14, 15]. In high-frequency myoclonus, cortically driven synchronization demonstrates a coherence between EEG and EMG activity of different muscles [16,17,18]. The conduction of epileptiform discharges results from rapid firing of action potentials that are conducted to the muscles through the fast-conducting corticospinal pathways. The somatosensory evoked potential (SSEP) may also show an enlarged cortical amplitude (giant potential, > 50 mV) [16, 19]. Taken together, these findings show that in PME there is pronounced cortical hyperexcitability [20].
The hyperexcitability in cortical myoclonus results from an imbalance between cortical excitation and inhibition. Whereas the excitatory/inhibitory balance maintains normal neurological function, any alteration in this balance can result in seizures. Excessive excitation through primarily glutamatergic neurotransmission or impaired γ-aminobutyric acid (GABA)-ergic inhibition can result in seizures. Inhibition occurs among different inhibitory neurons with different time scales, mediated by the GABA(A) and GABA(B) inhibitory projections, respectively. Excessive inhibition can result in seizures through enhanced synchronization of epileptic discharges [21]. Competing mechanisms among different neuronal populations can lead to periodic spike-and-wave discharges when GABA(A)—which mediates fast inhibition—and GABA(B), which mediates slow inhibition, are discoordinated. Like absence seizures (another seizure type involving thalamocortical circuits) in action-induced myoclonic seizures, afferent input into the cortex activates pyramidal neurons, which provides excitatory input onto thalamocortical relay cells (TRC) and nucleus reticularis thalami (NRT) cells (Fig. 1) [22, 23]. The thalamic cells project excitatory input into the NRT, which is composed primarily of GABAergic cells. The NRT in turn sends inhibitory inputs back to the TRC. TRC and NRT neurons possess low-threshold, transient Ca2+ channels (T channels) that allow them to exhibit a burst-firing mode, followed by an inactive mode. Mild hyperpolarization of these neurons activates these T channels and allows the influx of extracellular Ca2+, resulting in the firing of action potentials. After T channels are activated, they become inactivated quickly; hence, the name transient. T channels require lengthy, intense hyperpolarization to remove their inactivation (a process termed deinactivation) [24]. The requisite hyperpolarization can be provided by GABA(B) receptors that are present on the TRC [25]. With sufficient hyperpolarization, the thalamic neurons fire bursts of action potentials, resulting in propagation of excitatory fibers to the cortex [23]. The spike-and-wave activity seen during absence and myoclonic seizures reflects this excitatory activity into the cortex. Whereas the pathophysiologic underpinnings of myoclonic and absence seizures are likely somewhat different, resulting in distinctly different semiologies, the sudden onset and offset of absence and myoclonic seizures and their EEG correlates suggest a similar biological underpinning.

Schematic diagram of neuronal substrates involved in generalized seizures [22]. The key neuronal ensembles consist of the pyramidal cells (PC) and interneurons (IN) in the cortex and the thalamocortical relay cells (TRC) and nucleus reticularis thalami cells (NRT) in the thalamus. The model involves two dependent inhibitory neural populations, IN1 and IN2, which are mediated by the fast and slow time scales of the inhibitory receptors GABA(A) and GABA(B), respectively. Excitatory synaptic connections are shown in red lines with arrows. Inhibitory synaptic connections are shown in green lines with arrows and closed circles, where solid and dashed lines represent the fast and slow synaptic function mediated by the GABA(A) and GABA(B), respectively. GABA γ-aminobutyric acid
Some myoclonus arises from paroxysmal abnormal excessive oscillation in bidirectional connections between cortical and subcortical structures (thalamocortical or cortical-subcortical) [9, 11, 26]. The abnormal excessive reciprocal excitation of cortical and subcortical sites is bilateral and diffuse at the time of the myoclonus. Although there is subcortical involvement, the cephalad spread of excitation to the cortex triggers the myoclonus. This myoclonus is typically not action induced and usually occurs from rest. The EEG correlate consists of spike-and-wave or polyspikes-and-wave discharges. Surface EMG myoclonic discharges may be just as brief as in cortical myoclonus or a little longer (up to ~ 100 ms). Myoclonic absence and myoclonic-astatic seizures are examples of seizures of cortical-subcortical origin [27]. Cortical-subcortical pathology also likely accounts for the myoclonic-dystonia syndrome [28].
3 Electroencephalogram Features in Progressive Myoclonic Epilepsy (PME)
Early in the progression of PME, many children have a normal initial EEG. Once seizures begin in earnest, the interictal EEGs typically consist of polyspikes, polyspikes and waves, spike and waves, or multifocal spike and wave (Fig. 2) [1, 4, 29, 30]. In PME, myoclonus may be associated with spike-and-wave discharges or may occur without any accompanying cerebral discharges (Fig. 3), indicating that the myoclonus may be of cortical or subcortical origin. Photosensitivity with activation of epileptiform discharges may occur [5]. During myoclonus, there are typically brief paroxysms of polyspikes or polyspikes-and-wave or spike-and-wave discharges. Over time, in association with cognitive decline, there may be a slowing of the background activity. Some patients may experience improvement in their EEG over time that parallels an improvement in seizures. Giant SSEPs are variable and may or may not be present in the various forms of PME [1, 31]. In general, there are no distinguishing features on the EEG that separate the PME syndromes from one another. Occipital spikes during photic stimulation at low frequencies have been associated with neuronal ceroid lipofuscinosis but is not specific for the diagnosis [32].

Examples of spike-and-wave discharges seen during myoclonic seizures in three children with progressive myoclonic epilepsy. Note the generalized irregular bursts of spike- and polyspikes-and-wave discharges during the myoclonus. The morphology, frequency, and duration of the epileptiform discharges are not syndrome specific

Electroencephalogram (EEG) from a patient with Unverricht–Lundborg disease. The EEG demonstrates a mild slowing of the background with irregular spike-wave activity as designated by the asterisk. Myoclonus occurred with and without spike-wave discharges
4 Consequences of Myoclonic Epilepsy
Myoclonus can be the cause of disability. Whether it is present at rest, with muscle activation, or occurs with external stimulation, myoclonic jerks interfere with performing or initiating the desired correct movement for a given significant task. As a result, impairment in activities of daily living and intolerable frustration occur. Some myoclonus is so severe that the patient falls without any ability to protect themselves. In severe cases, action myoclonus may be so severe that the patient requires a wheelchair. While myoclonic seizures are so brief there appears to be no impairment of consciousness or awareness, myoclonic seizures occurring in flurries may impair the ability of the person to adequately respond.
5 PME Syndromes
The diagnosis of specific forms of PME is challenging because of genetic heterogeneity, phenotypic similarities, and an overlap of symptoms with other epileptic and neurodegenerative diseases, leading to nosological confusion [4]. The specific diseases that cause PME are diagnosed by recognition of their age of onset, the associated clinical symptoms, the clinical course, the pattern of inheritance, and by special investigations such as enzyme measurement, skin/muscle biopsy, or gene testing. In this review, PMEs that usually begin in childhood are reviewed. Although many authors do not include Dravet syndrome under the PME syndromes [1, 4, 5], we discuss it here because affected children have severe myoclonic epilepsy with action myoclonus and deteriorate cognitively. Dravet syndrome is the one PME in which a well-done randomized, placebo-controlled clinical trial has been conducted [33].
5.1 Lafora Disease
Lafora disease is a fatal PME that strikes previously healthy adolescents [34, 35]. It typically starts with epilepsy in adolescence in otherwise neurologically healthy individuals. The seizures include myoclonic seizures, which usually are action- or stimulus-sensitive myoclonus, in addition to generalized tonic–clonic, absence, and atonic seizures. Neuropsychiatric symptoms, such as behavioral changes, mood disturbances, and apathy, are also often present. These symptoms are followed by rapidly progressing dementia, medically intractable epilepsy, psychosis, cerebellar ataxia, dysarthria, loss of language, and respiratory failure [36, 37]. Death is rapid, usually occurring within a decade from symptom onset, often due to status epilepticus [38].
Lafora disease is an autosomal recessive PME caused by mutations in the EPM2A or EPM2B genes, encoding the laforin dual-specificity phosphatase and the malin ubiquitin E3 ligase, respectively [39, 40]. These enzymes play essential roles in glycogen metabolism, specifically in ensuring the intact spherical architecture of glycogen. With loss of function of the gene, glycogen becomes malformed, with reduced branching and excessively long chains and is insoluble. It gradually precipitates, aggregates, and accumulates to form Lafora bodies in many cell types, including in the cell bodies and dendrites, likely resulting in the relentless progression of the epilepsy [41]. An additional gene, PRDM8, the mutation of which causes a variant of early childhood-onset phenotype in a single family, has been reported [35, 42].
Currently, only symptomatic therapies are available for Lafora disease. However, studies in virus-mediated gene replacement, degradation of Lafora bodies, and reducing brain glycogen synthesis through antisense oligonucleotides, RNA interference, genome engineering, and small-molecule therapies are ongoing [43].
5.2 Unverricht–Lundborg Disease
Unverricht–Lundborg disease, sometimes called Baltic or Mediterranean myoclonus or PME type 1 (EPM1), is an autosomal recessive disorder characterized by myoclonus, often action and stimulus induced, and generalized tonic–clonic seizures [4, 30, 44, 45]. It is more prevalent in some geographic regions than in others, with the highest reported prevalence in Finland [30]. EPM1 is characterized by onset at age 6–16 years, progressively incapacitating myoclonus, generalized tonic–clonic epileptic seizures, and only mild cognitive dysfunction [44, 46,47,48]. Patients frequently develop cerebellar findings such as ataxia, incoordination, intention tremor, and dysarthria. Individuals with Unverricht–Lundborg disease are mentally alert but show emotional lability, depression, and mild decline in intellectual performance over time [30]. However, the action myoclonus in this disorder can be so severe that patients cannot walk or gain meaningful employment.
EPM1 is caused by mutations in the gene encoding cystatin B (CSTB), a cysteine protease inhibitor [49,50,51,52,53]. This protein reduces the activity of cathepsins, which are involved in the degradation of proteins in the lysosomes.
Presently, only symptomatic pharmacological treatment of the epilepsy is available for patients with Unverricht–Lundborg disease, but genetic therapies are being investigated [54].
5.3 Neuronal Ceroid Lipofuscinoses (NCLs)
The neuronal ceroid lipofuscinoses (NCLs) are a group of heterogeneous inherited, neurodegenerative, lysosomal storage disorders characterized by progressive intellectual and motor deterioration, seizures, and early death [55, 56]. NCLs are named for the histological appearance of storage material containing autofluorescent lipopigments [57, 58]. The term Batten disease has been applied collectively to this group of disorders, although more recent understanding of the molecular basis of disease has led to a system of nomenclature whereby subtypes are designated a number based on the associated gene and phenotype, e.g., CLN2 [59]. Currently, 13 genes are known to cause NCL [60]. Most have an autosomal recessive inheritance pattern, but autosomal dominant inheritance can be seen in one of the adult-onset forms, CLN4.
All patients with NCLs, except for those with a rare congenital form (NCL type 10 [CLN10] disease), have normal mental and motor development before the onset of the first symptoms [61]. Disease onset is in childhood for most patients. The sequence in which symptoms occur varies and depends on the combination of the underlying mutations, which can affect age at onset and disease phenotype [61]. The main symptoms are a combination of at least two of the following: dementia, epilepsy, motor deterioration, and visual loss [61]. Symptoms can also be outside the central nervous system. For example, cardiac involvement has been reported in adolescent and adult patients with CLN3 disease [62,63,64].
Even though all types of NCLs share a similar set of clinical features (e.g., dementia, epilepsy, motor deterioration, and visual loss), their clinical severity and presentation often differ, even for those caused by mutations in the same gene. Increasing knowledge about the natural history of the different forms of NCLs has shown that, for some genes, the phenotype severity can vary substantially, even between siblings [64, 65].
5.3.1 Late-Infantile NCL
One of the most prevalent types of NCL is type 2 (CLN2) disease [57]. Infants with CLN2 develop normally for the first year or two of life. Language delay then becomes apparent, and seizures—often the first sign of serious disease—begin around 3 years of age. Delay in expressive language development is the first sign of regression of psychomotor function in 83% of patients with classic late-infantile (CLN2) disease and is a key feature of early diagnosis [66, 67]. Seizure semiology is varied and includes generalized tonic–clonic, partial, atonic, myoclonic, and status epilepticus [68]. Myoclonic seizures are observed in all patients by 3–4 years of age. Medically intractable epilepsy is almost universal in children with NCLs, with especially high seizure frequency, duration, and severity in CLN2 [67]. Ataxia and developmental regression follow, with rapid loss of language and motor milestones. Children are nonverbal and nonambulatory by 4–6 years [68, 69]. Retinopathy and blindness also become apparent during this period, leading to total disability. Children rarely survive beyond early adolescence. Atypical phenotypes are characterized by later onset and, in some instances, longer life expectancies.
Mutations of the CLN2 gene encoding tripeptidylpeptidase 1 (TPP1) underlie the pathogenesis of late-infantile NCL or CLN2 disease [70]. TPP1 is a lysosomal protease that requires acidic pH for its activation. Inactivation of the aminopeptidase activity of TPP1 impairs the removal of tripeptides from the N-terminus of small proteins, leading to CLN2 disease [71, 72].
Currently, disease-specific treatment is only available for CLN2. Cerliponase alfa (Brineura®) is an intracerebroventricular enzyme-replacement therapy that was approved for the treatment of CLN2 in 2017. In clinical trials, cerliponase alfa, administered via a surgically implanted intraventricular access device, slowed the loss of ambulation [73]. Cerliponase alfa is approved in the USA for children aged ≥ 3 years and in the EU for all age groups. Clinical trials to determine long-term efficacy are ongoing.
5.3.2 Juvenile NCL
CLN3 first presents in juveniles (age of onset 5–7 years), and the first symptoms are usually visual loss, followed by dementia, behavioral changes, loss of motor skills, and epilepsy by early adolescence [74]. Myoclonic seizures are one of the seizure types seen in CLN3. In patients with classic juvenile CLN3 disease, seizures are infrequent, with only mild worsening in later stages of disease [75, 76].
CLN3 disease is caused by mutations in the CLN3 gene, which provides instructions for making the battenin protein [77]. The battenin protein is primarily located in the membranes surrounding lysosomes and endosomes, which are compartments within the cell that digest and recycle materials. This mutation results in a substantial decrease in messenger RNA expression and stability. Therefore, it is likely that the mutant gene expresses a low level of a truncated CLN3 protein [78]. The function of battenin is not currently known.
5.4 Gaucher Disease
Gaucher disease is one of the most common lysosomal storage diseases [79]. It is an autosomal recessive disorder in which the metabolic defect is an inherited deficiency of glucocerebrosidase due to mutations in the GBA1 (acid-b-glucosidase) gene [79]. Three types have been identified: Type 1 is the non-neuronopathic form and is characterized by hepatosplenomegaly and anemia; type 2 is a neuronopathic form of disease with severe neurological disease and is usually fatal by 2 years of age [80]; type 3 is the chronic neuronopathic form, which has a later onset than type 2. Type 3 is characterized by a milder neurological involvement than type 2 and is associated with the visceral and bone marrow involvement seen in type 1. The earliest central nervous system involvement can be picked up on a detailed ophthalmologic examination revealing defects in horizontal saccades, strabismus, and bulbar palsy or paresis [81]. Neurologic progression is marked by severe hypertonia, rigidity, opisthotonus, dysphagia, and medically intractable seizures [82].
Mutations in the glucocerebrosidase (GBA) gene cause Gaucher disease, as it results from deficiency of a lysosomal enzyme glucocerebrosidase (also known as acid beta-glucosidase [GBA]) [83]. This enzyme breaks down glucocerebroside into a sugar (glucose) and a simpler fat molecule (ceramide). Mutations in the GBA gene greatly reduce or eliminate the activity of beta-glucocerebrosidase. Without this enzyme, glucocerebroside and related substances increase to toxic levels within cells. The brain and other organs are damaged by the abnormal accumulation and storage of these substances, causing the characteristic features of Gaucher disease. Definitive diagnosis is made by assessing the serum glucocerebrosidase assay.
Enzyme-replacement and substrate-reduction therapy have been used to treat type 1 disease [84, 85] but have not been effective in treating neurological problems in type 2 disease [86, 87].
5.5 Myoclonic Epilepsy with Ragged Red Fibers Syndrome
Myoclonic epilepsy with ragged red fibers (MERRF) is a multisystem disorder characterized by myoclonus and other seizure types, ataxia, weakness, and dementia [88]. MERRF is a mitochondrial syndrome associated with various mitochondrial DNA point mutations [89]. Clinically, MERRF manifests with not only epilepsy and myopathy but also with multiorgan abnormalities, including endocrine, cardiovascular, dermatological, hearing, and vision. The clinical diagnosis of MERRF is based on the following four “canonic” features: myoclonus, generalized epilepsy, cerebellar ataxia, and ragged red fibers on muscle biopsy [90,91,92]. Although the onset of clinical manifestation typically occurs in childhood and early adulthood, onset in adults is not uncommon in patients with MERRF [91, 93, 94]. Psychomotor development in children is usually normal in almost all patients with MERRF until the onset of the myoclonus [91]. Cerebellar ataxia is one of the most common clinical manifestations of MERRF and is supportive of the diagnostic criteria of MERRF, occurring in over 80% of cases [95]. Cerebellar ataxia occurs frequently in patients with MERRF, although it may not be present in the early phase of the disease [96, 97]. Other common findings include hearing loss, short stature, optic atrophy, and cardiomyopathy with Wolff-Parkinson-White syndrome [88, 98].
The myoclonus may occur alone or in association with generalized seizures [91, 92]. Myoclonic seizures are more frequent in the chronic than in the initial phase of the disease [99]. In MERRF, the semiology of the myoclonus is clinically indistinguishable from the manifestations of myoclonus in other patients with myoclonic disease. While cortical myoclonus is common, individuals with MERRF may have the thalamocortical type of myoclonus [96].
Point mutations in the mitochondrially encoded transfer ribonucleic acid-lysine (tRNA(Lys3) gene (MT-TK) are responsible for over 80% of cases [100]. The MT-TK gene is a tRNA gene affiliated with the noncoding RNA class and is critical in the formation of proteins involved in oxidative phosphorylation. Less frequently, mutations in the MT-TL1, MT-TH, and MT-TS1 genes have been reported in MERRF. Myoclonus is not strictly linked to MERRF syndrome, having been detected in other typical mitochondrial encephalopathies, such as in POLG (DNA polymerase gamma catalytic subunit) mutations that are associated with Alpers–Huttenlocher syndrome and myoclonic epilepsy, myopathy, and sensory ataxia (MEMSA) syndrome [101, 102].
No disease-specific therapy currently exists. While vitamins and food supplements, such as coenzyme Q10 and carnitine, have been proposed, there is no evidence from clinical studies that these alter the course of the disease or aid in seizure control [103]. Investigation of effective mitochondria-targeting gene delivery systems designed to reverse mitochondrial disorders is ongoing [104].
5.6 Action Myoclonus Renal Failure Syndrome
Action myoclonus renal failure syndrome, also called PME type 4 (EPM4), is a distinctive form of PME associated with renal dysfunction [1, 105]. The syndrome typically presents at ages 15–25 years, either with neurologic symptoms (including tremor, action myoclonus, and other generalized seizures, and ataxia) or with proteinuria that progresses to renal failure [106]. Despite severe neurologic disability due mainly to action myoclonus, cognition is preserved in patients who survive after renal transplantation. The movement problems associated with this syndrome typically begin with a resting tremor of the fingers and hands and increases with movements such as writing. Over time, tremors can affect other parts of the body, such as the head, torso, legs, and tongue. Eventually, the tremors evolve into more obvious myoclonic seizures that are exacerbated or induced by movement. The myoclonic jerks are typically multifocal and involve the trunk, extremities, and face. Kidney failure typically begins with proteinuria and ultimately results in end-stage renal disease.
The syndrome is autosomal recessive related to loss-of-function mutations in the scavenger receptor class B member 2 (SCARB2) gene [107, 108]. Onset is in the second and third decades, but a late-onset form also starts in the fifth and sixth decades without accompanying renal failure [106, 109]. Genotype–phenotype heterogeneity occurs in the syndrome, and affected family members can present differently despite identical gene mutations,: some have neurological symptoms, whereas others develop renal impairment [110].
There is no current disease-specific therapy for the neurological aspects of this condition, and treatment remains primarily symptomatic.
5.7 PRICKLE1-Gene-Related PME with Ataxia
PRICKLE1-gene-related PME with ataxia, also called PME type 5 (EPM5), is characterized by myoclonic seizures, generalized tonic–clonic seizures (often sleep related), and ataxia but with normal cognition [1, 111]. The age of onset is 5–10 years. Action myoclonus may affect the limbs or bulbar muscles, sometimes with spontaneous myoclonus of facial muscles causing marked dysarthria.
The disorder is caused by mutations in the planar cell polarity protein 1 (PRICKLE) gene. The gene encodes proteins, such as PRICKLE1, that are core constituents of the planar cell polarity-signaling pathway that establishes cell polarity during embryonic development [112].
There are no current disease-specific therapies, and treatment remains primarily symptomatic.
5.8 North Sea PME
North Sea PME is a rare disorder characterized by progressive myoclonus, seizures, early-onset ataxia, and areflexia. The condition bears the clinical and electrophysiological hallmarks of a PME due to a homozygous p.G144W mutation in GOSR2, termed “North Sea” PME (EPM6) because of the proximity of the patient’s families to the shores of the North Sea [113]. The disorder appears to have arisen as a founder mutation in Northern Europe. In addition to the PME phenotype of myoclonus and other seizure types, there is also early-onset ataxia (average 2 years of age), areflexia, and elevated serum creatine kinase. Independent ambulation is lost in the second decade, and affected individuals develop scoliosis by adolescence. There may also be skeletal deformities, including pes cavus and syndactyly.
The condition is caused by mutations in the Golgi SNAP receptor complex 2 gene (GOSR2) [114]. The gene encodes a trafficking membrane protein that transports proteins among the Golgi compartments. Cognition is typically spared in this syndrome.
There is no current disease-specific therapy, and treatment remains symptomatic.
5.9 Sialidosis
Sialidosis, also called mucolipidosis type 1, is an autosomal recessive lysosomal storage disease caused by a deficiency of the enzyme α-N-acetyl neuraminidase-1 [1, 115]. Sialidosis is classified into two main clinical variants: type 1, the milder variant, and type 2, usually more severe and with an earlier onset [116]. Patients with the late and milder type 1, which is known as “cherry-red spot myoclonus syndrome,” typically develop myoclonic epilepsy, visual impairment, and ataxia in the second or third decade of life [117]. The infantile sialidosis (type 2) is characterized by dysmorphic features and cognitive delay, followed by myoclonus starting during the second decade of life [118]. Action myoclonus leads to severe disability in both types of sialidosis [119, 120].
Although patients with sialidosis often have macular cherry-red spots, this is not a pathognomonic finding, since they also occur in central retinal artery occlusion and metabolic storage diseases such as Tay–Sachs disease, Sandhoff’s disease, Niemann–Pick disease, Fabry’s disease, and Gaucher’s disease, some of which can also have a PME phenotype [121].
In human lysosomes, the degradation of complex macromolecular substrates requires the synergistic action of multiple hydrolases that act synergistically to carry out the degradation process of complex macromolecular substrates efficiently. One such efficient catalytic team is formed by three hydrolases that are ubiquitous but differentially expressed: the serine carboxypeptidase, protective protein/cathepsin A (PPCA), the sialidase, neuraminidase-1 (NEU1), and the glycosidase β-galactosidase (β-GAL) [122]. Type 1 sialidosis is caused by the genetic deficiency of the enzyme α-N-acetylneuraminidase-1 (coded by the neuraminidase-1 [NEU1] gene on chromosome 6p21) [123]. The characteristic pathology of sialidosis reflects tissue accumulation and urinary excretion of sialylated oligosaccharides [124]. The clinical diagnosis is usually supported by increased urine-bound sialic acid excretion and confirmed by genetic analysis or the demonstration of neuraminidase enzyme deficiency in cultured fibroblasts [120]. While children with sialidosis type 1 have some functional NEU1 activity, children with type II sialidosis have mutations that severely reduce or eliminate NEU1 enzyme activity [125].
There is no specific therapy for sialidosis, and treatment is currently symptomatic.
5.10 Dravet Syndrome
Dravet syndrome is an early childhood-onset epilepsy syndrome characterized by drug-resistant seizures, frequent episodes of status epilepticus, and the development of neurocognitive impairment [126,127,128,129,130]. The syndrome was first described by Dravet as severe myoclonic epilepsy [131]. Subsequently, it was noted that myoclonic seizures are not the most predominant seizure type in many of the children, and the name was then changed to Dravet syndrome in recognition of the physician who brought this seizure syndrome to the attention of the medical community [132]. Seizure freedom in this condition is rare, and the rate of sudden unexpected death in epilepsy patients (SUDEP) is higher than in other epilepsy syndromes.
Beginning around 5 months of age, affected children typically present with generalized or unilateral prolonged febrile seizures [129, 130]. However, a variety of other seizure types later appear, including myoclonic, atonic, generalized tonic–clonic, absence, complex partial, and, less often, tonic. The myoclonus is like action-induced cortical myoclonus similar to those characterizing PME [133]. Between the age of 1 and 4 years, episodes of status epilepticus with fever, as well as episodes of nonconvulsive status epilepticus, are frequent. Frequent episodes of status epilepticus and ongoing susceptibility to hyperthermia-induced seizures are key clinical features. Reflex- or stimulus-provoked seizures, defined as seizures that are reliably evoked by a stimulus, are also very common, and > 50% of patients have photosensitive seizures [134]. After 5 years of age, convulsive status epilepticus tends to be less frequent, and nocturnal generalized tonic–clonic seizures predominate, with seizures evolving over time. In adults, nocturnal generalized tonic–clonic is the most common seizure type, and far fewer absence and myoclonic seizures are typically seen [135, 136].
While early-life seizures are perhaps the most striking feature of Dravet syndrome, the most debilitating consequences of the condition are the associated cognitive and behavioral impairments. During the first year, prior to increases in the frequency of febrile and afebrile seizures, infants appear to develop normally. However, during the second year, progressive decline is observed in multiple domains of cognitive function: Psychomotor, visuospatial, language, and social development are all impaired.
In most cases, Dravet syndrome is caused by mutations in the Na2+ voltage-gated channel α subunit 1 (SCN1A) gene, resulting in loss of function of the type I voltage-gated Na2+ channel (Nav1.1). Nav1.1 is one of four Na2+ channels expressed in the brain that are critical for initiating and propagating action potentials in neurons, and deficits in Nav1.1 have been linked to multiple neurological disorders associated with cognitive impairment.
There are no current disease-specific therapies for Dravet syndrome, and pharmacological therapy remains the mainstay of treatment [137]. There is considerable interest in gene therapy for this condition, and several studies are in early development [138].
6 Treatment of PME
The treatment of PME is one of the major therapeutic challenges in neurology. As described in the discussions of the specific syndromes, therapies targeting the underlying etiology for these genetic conditions are in their infancy and primarily symptomatic, relying primarily on AEDs and—to a far lesser degree—on dietary therapy, vagal nerve stimulation, and deep brain stimulation. For the most part, these therapies are used to treat the epilepsy, myoclonic, and other seizures. PME is not amenable to surgical resection.
Because of the rarity of these disorders, randomized controlled double-blind trials comparing the efficacy, tolerability, and safety of AEDs in PME have been limited, and evidence for efficacy is of a low grade [9, 26]. As discussed in the following sections, only five randomized placebo-controlled trials have been conducted in PME [33, 139,140,141]. Even when AED treatment in PME is studied, the myoclonic seizures are difficult to assess because of their frequency, brevity, and sometimes subtle semiology [33]. Rating scales such as the Unified Myoclonus Rating Scale have been used to assess action-induced myoclonus but may not be optimal in assessing AED efficacy [139]. Much of the information used for treating the epilepsy in the PMEs comes from small open-label studies using adjunctive therapy or from reports from patients with other forms of myoclonic epilepsy, such as juvenile myoclonic epilepsy.
Despite the introduction of many new AEDs in the last two decades, the treatment of these symptoms, particularly myoclonus, remains challenging, because of the incomplete efficacy of most drugs [54]. While seizures such as generalized tonic–clonic seizures in PME often respond to AEDs, myoclonic seizures are often refractory to treatment [33, 142]. The efficacy of AEDs used to treat the seizures in PME does not appear to be syndrome related, i.e., AEDs that are effective in controlling myoclonic seizures in Lafora disease are likely to be effective in controlling seizures in Unverricht–Lundborg disease. However, this generality does not apply to tolerance or safety, as some AEDs can be harmful in MERRF [143].
Valproate is often the first choice to treat myoclonic seizures because of its broad spectrum of antiepileptic action and its effectiveness in treating myoclonus in a variety of epileptic syndromes [54, 75, 76]. Valproate is efficacious in juvenile myoclonic epilepsy, a myoclonic epilepsy with a comparatively benign prognosis [144]. In adults with PME, older drugs such as clonazepam and phenobarbital have far better efficacy than phenytoin and carbamazepine [54, 145]. Clonazepam is the only drug approved by the US FDA as monotherapy for the treatment of myoclonic seizures [146].
Newer AEDs that have been shown to be effective in some studies include piracetam [147,148,149], levetiracetam [149, 150], topiramate [137, 149, 151], zonisamide [152, 153], clobazam [137, 154], stiripentol [155,156,157], and perampanel [158, 159]. Vagal nerve stimulation, deep brain stimulation, and dietary therapy have resulted in some improvements but have not had a major impact on seizure control [160,161,162]. Table 2 provides a list of commonly used AEDs in PME.
Two prospective, multicenter, double-blind, phase III trials have studied drug efficacy in Unverricht–Lundborg disease (EPM1) [139]. In these studies, 103 patients (aged ≥ 16 years) with genetically ascertained EPM1, showing moderate–severe myoclonus (action myoclonus score ≥ 30/160), were randomized to twice-daily brivaracetam 5, 50, or 150 mg/day or placebo. Both studies comprised a 2-week baseline period, 2-week up-titration period, 12-week stable-dose maintenance period, and down-titration or entry into a long-term follow-up study. Symptoms of myoclonus were assessed using the Unified Myoclonus Rating Scale. The primary efficacy endpoint was percent reduction from baseline in action myoclonus score at last treatment visit. Estimated differences versus placebo were not statistically significant. However, the authors questioned whether the Unified Myoclonus Rating Scale was an appropriate measure for myoclonus in PME.
In a double-blind, placebo-controlled trial, 20 children and young adults with Dravet syndrome and drug-resistant seizures received either cannabidiol oral solution 20 mg per kilogram of body weight daily or placebo as adjunctive therapy [33]. The primary endpoint was the change in convulsive seizure frequency over a 14-week treatment period compared with a 4-week baseline period. The median frequency of convulsive seizures per month decreased from 12.4 to 5.9 with cannabidiol compared with a decrease from 14.9 to 14.1 with placebo. The percentage of patients with at least a 50% reduction in convulsive seizure frequency was 43% with cannabidiol and 27% with placebo. There was no significant reduction in nonconvulsive seizures, including myoclonic seizures. The authors concluded that, among patients with Dravet syndrome, cannabidiol resulted in a greater reduction in convulsive seizure frequency than did placebo and was associated with higher rates of adverse events. Cannabidiol is now approved by the FDA for treatment of seizures associated with Dravet syndrome and Lennox–Gastaut syndrome.
In a double-blind, randomized, placebo-controlled trial, Lagae et al. [141] reported a substantial response to fenfluramine in regard to convulsive seizures, although myoclonic seizures were not formally assessed. Fenfluramine demonstrated significant improvements in monthly convulsive seizure frequency in patients with Dravet syndrome whose conditions were insufficiently controlled with stiripentol-inclusive antiepileptic drug regimens [140]. Stiripentol administered in conjunction with clobazam and valproate reduced convulsive seizures in Dravet syndrome, but myoclonic seizures were not measured [163].
Thus, even well-done clinical trials provide little evidence that cannabidiol or brivaracetam reduces the number of myoclonic seizures. Clearly, new and more efficacious drugs are necessary for PME. Orphan drug development is of increasing interest in the medically refractory epilepsies because of legislation enabling facilitated support of orphan drugs by regulatory agencies such as the FDA and the European Medicines Agency. Orphan designations for rare epilepsies have increased in the past 10 years, but the number of approved drugs for the PMEs remains limited [164].
As with other seizure disorders, it is important that care be given to precipitating factors for seizures, such as sleep deprivation and stress. It is commonly observed that intractable myoclonic seizures are more frequent and/or more severe when patients are undergoing physical or emotional stress, and this can become a vicious cycle [165, 166].
6.1 Adverse Effects of Antiepileptic Drugs (AEDs) in PME
Some AEDs may exacerbate or even induce myoclonus [167] and should be avoided, particularly Na2+ channel blockers (phenytoin, carbamazepine, oxcarbazepine, lamotrigine), certain GABAergic drugs (tiagabine, vigabatrin), and gabapentin and pregabalin [30, 75, 76, 168, 169]. Why these drugs exacerbate myoclonus is unclear. Tiagabine, a GABA uptake inhibitor [170], and vigabatrin, a GABA transaminase inhibitor [171], likely enhance GABA(B) slow inhibition, which would increase the likelihood of hyperpolarization of TRC and subsequent burst firing. The mechanism by which gabapentin and pregabalin, which have selective inhibitory effects on voltage-gated Ca2+ channels containing the α2Δ1 subunit [172], induces myoclonus is unknown. Likewise, the mechanism responsible for increased myoclonus in Na2+ channel blockers is unknown. These drugs should not be used initially in treatment but could be helpful in individual patients; however, close clinical and EEG monitoring is necessary to detect exacerbations.
Some of the AEDs recommended for myoclonic epilepsy have been proven to be mitochondrial toxic and should be avoided, if possible, in MERRF [89, 143]. AEDs with the strongest mitochondrion-toxic effect include valproate, carbamazepine, phenytoin, and phenobarbital [143]. Valproic acid causes complex-I and complex-IV dysfunction [173, 174], reduces adenosine triphosphate (ATP) production, sequesters cytochrome-aa3 and coenzyme-A, inhibits key enzymes of beta oxidation, and results in impaired organization of the inner mitochondrial membrane and secondary carnitine deficiency [143, 173, 174]. Carbamazepine, phenytoin, and phenobarbital reduce ATP production, the mitochondrial membrane potential, and impair Ca2+ uptake/release [143, 175]. Valproate is contraindicated in mitochondrial disease because of pathogenic variants of the POLG gene [143, 176, 177], where numerous adverse events and deaths have been recorded. However, in classical MERRF, the evidence against the use of valproate appears to be largely theoretical and not definitively backed by clinical evidence [103].
6.2 Monitoring of AED Efficacy and Safety in PME
As with other severe epilepsies, polytherapy with AEDs can result in increased adverse effects, including lethargy, which may exacerbate the epilepsy [61, 178]. With disease progression, AEDs that have been previously tolerated may cause new side effects, and therapy should be reconsidered if symptoms of the disease worsen [76]. In PME, it is important not to mistake drug toxicity as disease-related cognitive declines.
While AED therapy can be quite helpful in reducing seizure frequency, duration, and severity, most patients with PME are medically intractable to medical or dietary therapy. It is often not realistic to aim for total seizure control at the risk of drug toxicity with ensuing declines in quality of life. Balancing seizure control with AED toxicity is challenging and requires input from the child’s family and caregivers. Judicious use of rescue medications such as rectal, nasal, or sub-buccal benzodiazepines can be very helpful in treating flurries of seizures without leading to long-term toxicity. In addition, seizure control, while clearly important to individuals with PME, is only one aspect of the patient’s care. Goals and therapeutic interventions evolve as the disease progresses, and may shift in focus from maintaining function early in the course of the disorder to maintaining quality of life. A multidisciplinary approach involving many healthcare providers is critical for optimal patient care [67].
7 Conclusions
PME constitutes a heterogenous group of different syndromes that have myoclonic seizures and neurological deterioration at their core. While disease-specific therapy is the future of this devastating group of disorders, most children with PME are managed symptomatically with AEDs. Goals of therapy should be reduction or elimination of seizures but not at the risk of severe side effects that can affect quality of life. This vulnerable population of patients is at risk for AEDs that worsen the myoclonus or affect the underlying disease progression, particularly in MERRF.
In addition to disease-specific genetic or enzyme-replacement or -depletion therapy, more effective AEDs are greatly needed in PME. New therapies need to be stringently evaluated, preferably with double-blind, placebo-controlled studies.
References
Malek N, Stewart W, Greene J. The progressive myoclonic epilepsies. Pract Neurol. 2015;15(3):164–71.
Satishchandra P, Sinha S. Progressive myoclonic epilepsy. Neurol India. 2010;58(4):514–22.
Dijk JM, Tijssen MA. Management of patients with myoclonus: available therapies and the need for an evidence-based approach. Lancet Neurol. 2010;9(10):1028–36.
Kalviainen R. Progressive myoclonus epilepsies. Seminars in neurology. 2015;35(3):293–9.
Franceschetti S, Michelucci R, Canafoglia L, Striano P, Gambardella A, Magaudda A, et al. Progressive myoclonic epilepsies: definitive and still undetermined causes. Neurology. 2014;82(5):405–11.
Genton P, Striano P, Minassian BA. The history of progressive myoclonus epilepsies. Epileptic Disord. 2016;18(S2):3–10.
Minassian BA. Post-modern therapeutic approaches for progressive myoclonus epilepsy. Epileptic Disord. 2016;18(S2):154–8.
Minassian BA, Striano P, Avanzini G. Progressive myoclonus epilepsy: the gene-empowered era. Epileptic Disord. 2016;18(S2):1–2.
Caviness JN. Pathophysiology and treatment of myoclonus. Neurol Clin. 2009 Aug;27(3):757-77, vii.
Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2004;3(10):598–607.
Guerrini R, Takahashi T. Myoclonus and epilepsy. Handb Clin Neurol. 2013;111:667–79.
Eberhardt O, Topka H. Myoclonic Disorders. Brain Sci. 2017 Aug 14;7(8).
Shibasaki H, Hallett M. Electrophysiological studies of myoclonus. Muscle Nerve. 2005;31(2):157–74.
Avanzini G, Shibasaki H, Rubboli G, Canafoglia L, Panzica F, Franceschetti S, et al. Neurophysiology of myoclonus and progressive myoclonus epilepsies. Epileptic Disord. 2016;18(S2):11–27.
Shibasaki H, Yamashita Y, Kuroiwa Y. Electroencephalographic studies myoclonus. Brain. 1978;101(3):447–60.
Brown P, Farmer SF, Halliday DM, Marsden J, Rosenberg JR. Coherent cortical and muscle discharge in cortical myoclonus. Brain. 1999;122(Pt 3):461–72.
Brown P, Ridding MC, Werhahn KJ, Rothwell JC, Marsden CD. Abnormalities of the balance between inhibition and excitation in the motor cortex of patients with cortical myoclonus. Brain. 1996;1996(119):309–17.
Grosse P, Cassidy MJ, Brown P. EEG-EMG, MEG-EMG and EMG-EMG frequency analysis: physiological principles and clinical applications. Clin Neurophysiol. 2002;113(10):1523–31.
Shibasaki H, Yamashita Y, Kuroiwa Y. Electroencephalographic studies of myoclonus: myoclonus related cortical spike and high amplitude somatosensory evoked potential. Brain. 1978;1978(101):447–60.
Reutens DC, Puce A, Berkovic SF. Cortical hyperexcitability in progressive myoclonus epilepsy: a study with transcranial magnetic stimulation. Neurology. 1993;43(1):186–92.
Khazipov R. GABAergic synchronization in epilepsy. Cold Spring Harb Perspect Med. 2016;6(2):a022764.
Fan D, Liu S, Wang Q. Stimulus-induced epileptic spike-wave discharges in thalamocortical model with disinhibition. Sci Rep. 2016;23(6):37703.
Snead OC III. Basic mechanisms of generalized absence seizures. Ann Neurol. 1995;1995(37):146–57.
Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3(5):371–82.
Crunelli V, Leresche N. A role for GABAB receptors in excitation and inhibition of thalamocortical cells. TINS. 1991;1991(14):16–21.
Caviness JN. Treatment of myoclonus. Neurotherapeutics. 2014;11(1):188–200.
Oguni H, Fukuyama Y, Tanaka T, Hayashi K, Funatsuka M, Sakauchi M, et al. Myoclonic-astatic epilepsy of early childhood–clinical and EEG analysis of myoclonic-astatic seizures, and discussions on the nosology of the syndrome. Brain Dev. 2001;23(7):757–64.
Nardocci N. Myoclonus-dystonia syndrome. Handb Clin Neurol. 2011;100:563–75.
Panzica F, Canafoglia L, Franceschetti S, Binelli S, Ciano C, Visani E, et al. Movement-activated myoclonus in genetically defined progressive myoclonic epilepsies: EEG-EMG relationship estimated using autoregressive models. Clin Neurophysiol. 2003;114(6):1041–52.
Kalviainen R, Khyuppenen J, Koskenkorva P, Eriksson K, Vanninen R, Mervaala E. Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia. 2008;49(4):549–56.
Sinha S, Satishchandra P, Gayathri N, Yasha TC, Shankar SK. Progressive myoclonic epilepsy: A clinical, electrophysiological and pathological study from South India. J Neurol Sci. 2007;252(1):16–23.
Specchio N, Bellusci M, Pietrafusa N, Trivisano M, de Palma L, Vigevano F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia. 2017;58(8):1380–8.
Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011–20.
Parihar R, Rai A, Ganesh S. Lafora disease: from genotype to phenotype. J Genet. 2018;97(3):611–24.
Turnbull J, Tiberia E, Striano P, Genton P, Carpenter S, Ackerley CA, et al. Lafora disease. Epileptic Disord. 2016;18(S2):38–62.
Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25(1):21–9.
Striano P, Zara F, Turnbull J, Girard JM, Ackerley CA, Cervasio M, et al. Typical progression of myoclonic epilepsy of the Lafora type: a case report. Nat Clin Pract Neurol. 2008;4(2):106–11.
Ganesh S, Agarwala KL, Ueda K, Akagi T, Shoda K, Usui T, et al. Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum Mol Genet. 2000;9(15):2251–61.
Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171–4.
Chan EM, Bulman DE, Paterson AD, Turnbull J, Andermann E, Andermann F, et al. Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J Med Genet. 2003;40(9):671–5.
Minassian BA, Andrade DM, Ianzano L, Young EJ, Chan E, Ackerley CA, et al. Laforin is a cell membrane and endoplasmic reticulum-associated protein tyrosine phosphatase. Ann Neurol. 2001;49(2):271–5.
Turnbull J, Girard JM, Lohi H, Chan EM, Wang P, Tiberia E, et al. Early-onset Lafora body disease. Brain. 2012;135(Pt 9):2684–98.
Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease—from pathogenesis to treatment strategies. Nat Rev Neurol. 2018;14(10):606–17.
Hypponen J, Aikia M, Joensuu T, Julkunen P, Danner N, Koskenkorva P, et al. Refining the phenotype of Unverricht–Lundborg disease (EPM1): a population-wide Finnish study. Neurology. 2015;84(15):1529–36.
Lehesjoki AE, Gardiner M. Progressive myoclonus epilepsy: Unverricht–Lundborg disease and neuronal ceroid lipofuscinoses. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s basic mechanisms of the epilepsies. Bethesda (MD); 2012.
Berkovic SF, Andermann F, Carpenter S, Wolfe LS. Progressive myoclonus epilepsies: specific causes and diagnosis. N Engl J Med. 1986;315(5):296–305.
Berkovic SF, Andermann F. The progressive myoclonus epilepsies. In: Pedley TA, Meldrum BS, editors. Recent advances in epilepsy, vol. 3. Edinburgh: Churchill Livingstone; 1986. p. 157–87.
Classification of progressive myoclonus epilepsies and related disorders. Marseille consensus group. Ann Neurol. 1990;28(1):113–6.
Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, Miao J, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science. 1996;271(5256):1731–4.
Pennacchio LA, Myers RM. Isolation and characterization of the mouse cystatin B gene. Genome Res. 1996;6(11):1103–9.
Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, et al. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386(6627):847–51.
Lalioti MD, Scott HS, Genton P, Grid D, Ouazzani R, M’Rhabet A, et al. A PCR amplification method reveals instability of the dodecamer repeat in progressive myoclonus epilepsy (EPM1) and no correlation between the size of the repeat and age at onset. Am J Hum Genet. 1998;1998(62):842–7.
Lafreniere RG, Rochefort DL, Chretien N, Rommens JM, Cochius JI, Kalviainen R, et al. Unstable insertion in the 5’ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat Genet. 1997;15(3):298–302.
Michelucci R, Pasini E, Riguzzi P, Andermann E, Kalviainen R, Genton P. Myoclonus and seizures in progressive myoclonus epilepsies: pharmacology and therapeutic trials. Epileptic Disord. 2016;18(S2):145–53.
Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA); 1993.
Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, et al. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegener. 2019;14(1):4.
Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018;6(24):476.
Fietz M, AlSayed M, Burke D, Cohen-Pfeffer J, Cooper JD, Dvorakova L, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119(1–2):160–7.
Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79(2):183–91.
Alcalde-Cabero E, Almazan-Isla J, Garcia Lopez FJ, Ara-Callizo JR, Avellanal F, Casasnovas C, et al. Guillain–Barre syndrome following the 2009 pandemic monovalent and seasonal trivalent influenza vaccination campaigns in Spain from 2009 to 2011: outcomes from active surveillance by a neurologist network, and records from a country-wide hospital discharge database. BMC Neurol. 2016;16(1):75.
Schulz A, Kohlschutter A, Mink J, Simonati A, Williams R. NCL diseases—clinical perspectives. Biochim Biophys Acta. 2013;1832(11):1801–6.
Ostergaard JR, Rasmussen TB, Molgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. 2011;76(14):1245–51.
Dilaveris P, Koutagiar I, Aggeli C, Sideris S, Gatzoulis K, Stefanadis C. Severe sinus node dysfunction in a patient with juvenile neuronal ceroid lipofuscinosis. Int J Cardiol. 2014;174(1):143–6.
Lebrun AH, Moll-Khosrawi P, Pohl S, Makrypidi G, Storch S, Kilian D, et al. Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease. Mol Med. 2011;17(11–12):1253–61.
Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta. 2015;1852(10 Pt B):2237-41.
Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, Van de Graaf B, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. 2018;2(8):582–90.
Williams RE, Adams HR, Blohm M, Cohen-Pfeffer JL, de Los Reyes E, Denecke J, et al. Management strategies for CLN2 disease. Pediatr Neurol. 2017;69:102–12.
Perez-Poyato MS, Marfa MP, Abizanda IF, Rodriguez-Revenga L, Sanchez VC, Gonzalez MJ, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28(4):470–8.
Worgall S, Kekatpure MV, Heier L, Ballon D, Dyke JP, Shungu D, et al. Neurological deterioration in late infantile neuronal ceroid lipofuscinosis. Neurology. 2007;69(6):521–35.
Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower-Brooks S, Pullarkat RK, et al. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am J Hum Genet. 1999;64(6):1511–23.
Golabek AA, Kida E, Walus M, Wujek P, Mehta P, Wisniewski KE. Biosynthesis, glycosylation, and enzymatic processing in vivo of human tripeptidyl-peptidase I. J Biol Chem. 2003;278(9):7135–45.
Vines DJ, Warburton MJ. Classical late infantile neuronal ceroid lipofuscinosis fibroblasts are deficient in lysosomal tripeptidyl peptidase I. FEBS Lett. 1999;443(2):131–5.
Markham A. Cerliponase alfa: first global approval. Drugs. 2017;77(11):1247–9.
Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016;18(S2):73–88.
Augustine EF, Adams HR, Beck CA, Vierhile A, Kwon J, Rothberg PG, et al. Standardized assessment of seizures in patients with juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2015;57(4):366–71.
Johannsen J, Nickel M, Schulz A, Denecke J. Considering valproate as a risk factor for rapid exacerbation of complex movement disorder in progressed stages of late-infantile CLN2 disease. Neuropediatrics. 2016;47(3):194–6.
Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell. 1995;82(6):949–57.
Chan CH, Mitchison HM, Pearce DA. Transcript and in silico analysis of CLN3 in juvenile neuronal ceroid lipofuscinosis and associated mouse models. Hum Mol Genet. 2008;17(21):3332–9.
Nagral A. Gaucher disease. J Clin Exp Hepatol. 2014;4(1):37–50.
Mignot C, Doummar D, Maire I, De Villemeur TB, French Type 2 Gaucher Disease Study G. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006;28(1):39–48.
Harris CM, Taylor DS, Vellodi A. Ocular motor abnormalities in Gaucher disease. Neuropediatrics. 1999;30(6):289–93.
Blom S, Erikson A. Gaucher disease–Norrbottnian type. Neurodevelopmental, neurological, and neurophysiological aspects. Eur J Pediatr. 1983;140(4):316–22.
Messner MC, Cabot MC. Glucosylceramide in humans. Adv Exp Med Biol. 2010;688:156–64.
Shemesh E, Deroma L, Bembi B, Deegan P, Hollak C, Weinreb NJ, et al. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev. 2015;27(3):CD010324.
Pastores GM, Hughes DA. Gaucher Disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA): University of Washington.
Tezuka Y, Fukuda M, Watanabe S, Nakano T, Okamoto K, Kuzume K, et al. Histological characterisation of visceral changes in a patient with type 2 Gaucher disease treated with enzyme replacement therapy. Blood Cells Mol Dis. 2018;68:194–9.
Li M. Enzyme replacement therapy: a review and its role in treating lysosomal storage diseases. Pediatr Ann. 2018;47(5):e191–7.
DiMauro S, Hirano M. MERRF. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle (WA): University of Washington.
Finsterer J, Zarrouk-Mahjoub S. Management of epilepsy in MERRF syndrome. Seizure. 2017;50:166–70.
Lorenzoni PJ, Scola RH, Kay CS, Silvado CE, Werneck LC. When should MERRF (myoclonus epilepsy associated with ragged-red fibers) be the diagnosis? Arq Neuropsiquiatr. 2014;72(10):803–11.
DiMauro S, Hirano M, Kaufmann P, Tanji K, Sano M, Shungu DC, et al. Clinical features and genetics of myoclonic epilepsy with ragged red fibers. Adv Neurol. 2002;89:217–29.
Fukuhara N. Clinicopathological features of MERRF. Muscle Nerve Suppl. 1995;3:S90–4.
Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Silvado CE, Werneck LC. MERRF: clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion. 2011;11(3):528–32.
Finsterer J. Mitochondriopathies. Eur J Neurol. 2004;11(3):163–86.
Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain. 1997;120 (Pt 10):1713–21.
Berkovic SF, Carpenter S, Evans A, Karpati G, Shoubridge EA, Andermann F, et al. Myoclonus epilepsy and ragged-red fibres (MERRF). 1. A clinical, pathological, biochemical, magnetic resonance spectrographic and positron emission tomographic study. Brain. 1989;112 (Pt 5):1231–60.
Ozawa M, Goto Y, Sakuta R, Tanno Y, Tsuji S, Nonaka I. The 8,344 mutation in mitochondrial DNA: a comparison between the proportion of mutant DNA and clinico-pathologic findings. Neuromuscul Disord. 1995;5(6):483–8.
DiMauro S. Mitochondrial encephalomyopathies. In: Rosenberg RN, Prusiner SB, DiMauro S, Barchi RL, Kunkel LM, editors. The molecular and genetic basis of neurological disease. Boston: Butterworth-Heinemann; 1993. p. 665–94.
Canafoglia L, Franceschetti S, Antozzi C, Carrara F, Farina L, Granata T, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;56(10):1340–6.
Mancuso M, Orsucci D, Angelini C, Bertini E, Carelli V, Comi GP, et al. Phenotypic heterogeneity of the 8344A > G mtDNA “MERRF” mutation. Neurology. 2013;80(22):2049–54.
Mancuso M, Orsucci D, Angelini C, Bertini E, Catteruccia M, Pegoraro E, et al. Myoclonus in mitochondrial disorders. Mov Disord. 2014;29(6):722–8.
Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle (WA): University of Washington.
Rahman S. Mitochondrial disease and epilepsy. Dev Med Child Neurol. 2012;54(5):397–406.
Jang YH, Lim KI. Recent advances in mitochondria-targeted gene delivery. Molecules. 2018;23(9).
Badhwar A, Berkovic SF, Dowling JP, Gonzales M, Narayanan S, Brodtmann A, et al. Action myoclonus-renal failure syndrome: characterization of a unique cerebro-renal disorder. Brain. 2004;127(Pt 10):2173–82.
Dibbens LM, Michelucci R, Gambardella A, Andermann F, Rubboli G, Bayly MA, et al. SCARB2 mutations in progressive myoclonus epilepsy (PME) without renal failure. Ann Neurol. 2009;66(4):532–6.
Hopfner F, Schormair B, Knauf F, Berthele A, Tolle TR, Baron R, et al. Novel SCARB2 mutation in action myoclonus-renal failure syndrome and evaluation of SCARB2 mutations in isolated AMRF features. BMC Neurol. 2011;27(11):134.
Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82(3):673–84.
Higashiyama Y, Doi H, Wakabayashi M, Tsurusaki Y, Miyake N, Saitsu H, et al. A novel SCARB2 mutation causing late-onset progressive myoclonus epilepsy. Mov Disord. 2013;28(4):552–3.
Perandones C, Pellene LA, Micheli F. A new SCARB2 mutation in a patient with progressive myoclonus ataxia without renal failure. Mov Disord. 2014;29(1):158–9.
Fox MH, Bassuk AG. PRICKLE1-Related Progressive Myoclonus Epilepsy with Ataxia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA): University of Washington.
Liu C, Lin C, Whitaker DT, Bakeri H, Bulgakov OV, Liu P, et al. Prickle1 is expressed in distinct cell populations of the central nervous system and contributes to neuronal morphogenesis. Hum Mol Genet. 2013;22(11):2234–46.
Boisse Lomax L, Bayly MA, Hjalgrim H, Moller RS, Vlaar AM, Aaberg KM, et al. ‘North Sea’ progressive myoclonus epilepsy: phenotype of subjects with GOSR2 mutation. Brain. 2013;136(Pt 4):1146–54.
Corbett MA, Schwake M, Bahlo M, Dibbens LM, Lin M, Gandolfo LC, et al. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet. 2011;88(5):657–63.
Bonten EJ, Arts WF, Beck M, Covanis A, Donati MA, Parini R, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet. 2000;9(18):2715–25.
Caciotti A, Di Rocco M, Filocamo M, Grossi S, Traverso F, d’Azzo A, et al. Type II sialidosis: review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. J Neurol. 2009;256(11):1911–5.
d’Azzo A, Machado E, Annunziata I. Pathogenesis, emerging therapeutic targets and Treatment in Sialidosis. Expert Opin Orphan Drugs. 2015;3(5):491–504.
Federico A, Cecio A, Battini GA, Michalski JC, Strecker G, Guazzi GC. Macular cherry-red spot and myoclonus syndrome. Juvenile form of sialidosis. J Neurol Sci. 1980;48(2):157–69.
Canafoglia L, Franceschetti S, Antozzi C, Carrara F, Farina L, Granata T, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;2001(22):1340–6.
Canafoglia L, Franceschetti S, Uziel G, Ciano C, Scaioli V, Guerrini R, et al. Characterization of severe action myoclonus in sialidoses. Epilepsy Res. 2011;94(1–2):86–93.
Heroman JW, Rychwalski P, Barr CC. Cherry red spot in sialidosis (mucolipidosis type I). Arch Ophthalmol. 2008;126(2):270–1.
Bonten EJ, Annunziata I, d’Azzo A. Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci. 2014;71(11):2017–32.
Lowden JA, O’Brien JS. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet. 1979;31(1):1–18.
Seyrantepe V, Poupetova H, Froissart R, Zabot MT, Maire I, Pshezhetsky AV. Molecular pathology of NEU1 gene in sialidosis. Hum Mutat. 2003;22(5):343–52.
Khan A, Sergi C. Sialidosis. A review of morphology and molecular biology of a rare pediatric disorder. diagnostics (Basel). 2018 25;8(2).
Eschbach K, Knupp KG. Stiripentol for the treatment of seizures in Dravet syndrome. Expert Rev Clin Pharmacol. 2019;12(5):379–88.
Connolly MB. Dravet syndrome: diagnosis and long-term course. Can J Neurol Sci. 2016;43(Suppl 3):S3–8.
Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini PP. SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. 2012;23(3):177–86.
Dravet C. Dravet syndrome history. Dev Med Child Neurol. 2011;53 Suppl 2:1–6.
Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52 Suppl 2:3–9.
Dravet C. Severe myoclonic epilepsy in infants and its related syndromes. Epilepsia. 2000;2000(41 Suppl 9):7.
Dravet C, Bureau M, Roger J. Severe myoclonic epilepsy in infants. In: Roger J, Dravet C, Bureau M, Dreifuss FE, Wolf P, editors. Epileptic syndromes in infancy, childhood, and adolescence. London: John Libbey Eurotext, Ltd.; 1985. p. 58–67.
Canafoglia L, Ragona F, Panzica F, Piazza E, Freri E, Binelli S, et al. Movement-activated cortical myoclonus in Dravet syndrome. Epilepsy Res. 2017;130:47–52.
Bureau M, Dalla BB. Electroencephalographic characteristics of Dravet syndrome. Epilepsia. 2011;52 Suppl 2:13–23.
Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol. 2005;2005(95):71–102.
Jansen FE, Sadleir LG, Harkin LA, Vadlamudi L, McMahon JM, Mulley JC, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology. 2006;67(12):2224–6.
Wallace A, Wirrell E, Kenney-Jung DL. Pharmacotherapy for Dravet syndrome. Paediatr Drugs. 2016;18(3):197–208.
Perucca P, Perucca E. Identifying mutations in epilepsy genes: impact on treatment selection. Epilepsy Res. 2019;152:18–30.
Kalviainen R, Genton P, Andermann E, Andermann F, Magaudda A, Frucht SJ, et al. Brivaracetam in Unverricht–Lundborg disease (EPM1): results from two randomized, double-blind, placebo-controlled studies. Epilepsia. 2016;57(2):210–21.
Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. JAMA Neurol. 2019.
Lagae L, Sullivan J, Cross H. Fenfluramine HCl oral solution in Dravet syndrome: results of a phase 3, randomized, double-blind, placebo-controlled trial. Lancet. 2020.
Uthman BM, Reichl A. Progressive myoclonic epilepsies. Curr Treat Options Neurol. 2002;4(1):3–17.
Finsterer J. Mitochondrion-toxic drugs given to patients with mitochondrial psychoses. Behav Brain Funct. 2012;29(8):45.
Brodie MJ. Modern management of juvenile myoclonic epilepsy. Expert Rev Neurother. 2016;16(6):681–8.
Iivanainen M, Himberg JJ. Valproate and clonazepam in the treatment of severe progressive myoclonus epilepsy. Arch Neurol. 1982;39(4):236–8.
Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4(4):239–48.
Fedi M, Reutens D, Dubeau F, Andermann E, D’Agostino D, Andermann F. Long-term efficacy and safety of piracetam in the treatment of progressive myoclonus epilepsy. Arch Neurol. 2001;58(5):781–6.
Koskiniemi M, Van Vleymen B, Hakamies L, Lamusuo S, Taalas J. Piracetam relieves symptoms in progressive myoclonus epilepsy: a multicentre, randomised, double blind, crossover study comparing the efficacy and safety of three dosages of oral piracetam with placebo. J Neurol Neurosurg Psychiatry. 1998;64(3):344–8.
Roivainen R, Karvonen MK, Puumala T. Seizure control in Unverricht–Lundborg disease: a single-centre study. Epileptic Disord. 2014;16(2):191–5.
Magaudda A, Gelisse P, Genton P. Antimyoclonic effect of levetiracetam in 13 patients with Unverricht–Lundborg disease: clinical observations. Epilepsia. 2004;45(6):678–81.
Aykutlu E, Baykan B, Gurses C, Bebek N, Buyukbabani N, Gokyigit A. Add-on therapy with topiramate in progressive myoclonic epilepsy. Epilepsy Behav. 2005;6(2):260–3.
Henry TR, Leppik IE, Gumnit RJ, Jacobs M. Progressive myoclonus epilepsy treated with zonisamide. Neurology. 1988;38(6):928–31.
Italiano D, Pezzella M, Coppola A, Magaudda A, Ferlazzo E, Bramanti P, et al. A pilot open-label trial of zonisamide in Unverricht–Lundborg disease. Mov Disord. 2011;26(2):341–3.
Ng YT, Collins SD. Clobazam. Neurotherapeutics. 2007;4(1):138–44.
Myers KA, Lightfoot P, Patil SG, Cross JH, Scheffer IE. Stiripentol efficacy and safety in Dravet syndrome: a 12-year observational study. Dev Med Child Neurol. 2018;60(6):574–8.
Chiron C, Marchand MC, Tran A, Rey E, d’Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000;356(9242):1638–42.
Buck ML, Goodkin HP. Stiripentol: a novel antiseizure medication for the management of Dravet syndrome. Ann Pharmacother. 2019;6:1060028019856008.
Goldsmith D, Minassian BA. Efficacy and tolerability of perampanel in ten patients with Lafora disease. Epilepsy Behav. 2016;62:132–5.
Crespel A, Gelisse P, Tang NP, Genton P. Perampanel in 12 patients with Unverricht–Lundborg disease. Epilepsia. 2017;58(4):543–7.
Wheless JW. Nonpharmacologic treatment of the catastrophic epilepsies of childhood. Epilepsia. 2004;45(Suppl 5):17–22.
van Egmond ME, Weijenberg A, van Rijn ME, Elting JW, Gelauff JM, Zutt R, et al. The efficacy of the modified Atkins diet in North Sea progressive myoclonus epilepsy: an observational prospective open-label study. Orphanet J Rare Dis. 2017;12(1):45.
Vesper J, Steinhoff B, Rona S, Wille C, Bilic S, Nikkhah G, et al. Chronic high-frequency deep brain stimulation of the STN/SNr for progressive myoclonic epilepsy. Epilepsia. 2007;48(10):1984–9.
Inoue Y, Ohtsuka Y. Effectiveness of add-on stiripentol to clobazam and valproate in Japanese patients with Dravet syndrome: additional supportive evidence. Epilepsy Res. 2014;108(4):725–31.
Auvin S, Avbersek A, Bast T, Chiron C, Guerrini R, Kaminski RM, et al. Drug development for rare paediatric epilepsies: current state and future directions. Drugs. 2019;79(18):1917–35.
van Campen JS, Jansen FE, Pet MA, Otte WM, Hillegers MH, Joels M, et al. Relation between stress-precipitated seizures and the stress response in childhood epilepsy. Brain. 2015;138(Pt 8):2234–48.
Koepp MJ, Caciagli L, Pressler RM, Lehnertz K, Beniczky S. Reflex seizures, traits, and epilepsies: from physiology to pathology. Lancet Neurol. 2016;15(1):92–105.
Striano P, Belcastro V. Treatment of myoclonic seizures. Expert Rev Neurother. 2012;12(12):1411–7 (quiz 8).
Vossler DG. Exacerbation of seizures in Lennox–Gastaut syndrome by gabapentin. Neurology. 1996;1996(46):852–3.
Genton P. When antiepileptic drugs aggravate epilepsy. Brain Dev. 2000;22(2):75–80.
Schmidt D, Gram L, Brodie M, Kramer G, Perucca E, Kalviainen R, et al. Tiagabine in the treatment of epilepsy—a clinical review with a guide for the prescribing physician. Epilepsy Res. 2000;41(3):245–51.
Sills GJ. Pre-clinical studies with the GABAergic compounds vigabatrin and tiagabine. Epileptic Disord. 2003;5(1):51–6.
Sills GJ. The mechanisms of action of gabapentin and pregabalin. Curr Opin Pharmacol. 2006;6(1):108–13.
Ponchaut S, van Hoof F, Veitch K. Cytochrome aa3 depletion is the cause of the deficient mitochondrial respiration induced by chronic valproate administration. Biochem Pharmacol. 1992;43(3):644–7.
Ponchaut S, van Hoof F, Veitch K. In vitro effects of valproate and valproate metabolites on mitochondrial oxidations. Relevance of CoA sequestration to the observed inhibitions. Biochem Pharmacol. 1992;43(11):2435–42.
Santos NA, Medina WS, Martins NM, Mingatto FE, Curti C, Santos AC. Aromatic antiepileptic drugs and mitochondrial toxicity: effects on mitochondria isolated from rat liver. Toxicol In Vitro. 2008;22(5):1143–52.
Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55(5):706–12.
Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323–38.
Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39(1):5–17.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by National Institutes of Health grants NS108765 and NS1089296.
Conflict of interest
Gregory L. Holmes has no conflicts of interest that are directly relevant to the content of this article.
Rights and permissions
About this article

Cite this article
Holmes, G.L. Drug Treatment of Progressive Myoclonic Epilepsy. Pediatr Drugs 22, 149–164 (2020). https://doi.org/10.1007/s40272-019-00378-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-019-00378-y