
Abstract
Background
Pulmonary arterial hypertension is a hemodynamic disorder. Signs and symptoms are generally difficult to recognize because they are non-specific. The current treatment for pulmonary arterial hypertension offers no cure or prevention; therefore, it is important to explore treatment avenues for novel pulmonary arterial hypertension treatments. In this study, we tested the hypothesis: pulmonary vasodilator responses of adenosine are dependent on the activation of l-type calcium channels, independent of the synthesis of nitric oxide from l-arginine, activation of adenosine triphosphate-sensitive potassium channels, and the release of cyclooxygenase products.
Methods
We performed an isolated lobar lung preparation in mongrel cats. The thromboxane A2 analog U-46619 was used to increase lobar arterial pressure to a high steady level. We recorded responses to adenosine and other vasodepressor agents in the pulmonary vascular bed of a cat under conditions of controlled pulmonary blood flow and constant left atrial pressure.
Results
These data show that adenosine has significant vasodepressor activity in the pulmonary vascular bed of the cat. The data suggest that pulmonary vasodilator responses to adenosine are partially dependent on the activation of adenosine 1 and 2 receptor pathways, and independent of the activation of cyclooxygenase activation, adenosine triphosphate-sensitive K + channels, or synthesis of nitric oxide in the pulmonary vascular bed of the cat.
Conclusions
Vasodepressor effects of adenosine are species specific, and this species specificity will impact the development of future testing and treatments for pulmonary arterial hypertension. Clinical studies are warranted to see if adenosine moieties could play a therapeutic role in patients with pulmonary arterial hypertension and/or other pulmonary pathogeneses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The vasodepressor effects of adenosine are species specific. This species specificity will impact the development of future testing and the development of treatments for pulmonary arterial hypertension. |
More clinical studies are necessary to see if adenosine moieties could play a role in treating patients with pulmonary arterial hypertension and/or other pulmonary pathogeneses. |
1 Introduction
1.1 Pulmonary Arterial Hypertension
Numerous conditions give rise to pulmonary arterial hypertension (PAH) with most of them being idiopathic. Pulmonary arterial hypertension is a hemodynamic disorder defined as a mean pulmonary arterial pressure > 25 mmHg at rest during right heart catheterization [1]. The increase in mean pulmonary arterial pressure causes increased pulmonary vascular resistance that can cascade into a life-threatening condition [2]. The increased pulmonary pressures directly cause cardiac remodeling that eventually progresses to right-sided heart failure [1, 2]. Signs and symptoms are generally difficult to recognize initially because they present as non-specific and typically are mistaken for age-related physiologic processes or alternate medical conditions. Many advances have been made toward PAH-specific therapies that have led to advanced clinical management of the disease. The current treatment for PAH includes vasodilators, prostanoids, phosphodiesterase type 5 and 3 inhibitors, endothelin receptor antagonists, calcium channel blockers, digoxin, and some anticoagulants; however, many of these drugs have severe side effects, can cause permanent liver damage, and offer post-disease diagnosis treatment rather than a cure or prevention [3]. Therefore, it is important to continue to explore treatment avenues for novel PAH treatments.
1.2 Adenosine
Adenosine mediates vasodilation, vessel remodeling, cell proliferation, and antiplatelet and inflammatory responses [4], and is therefore important in pulmonary and cardiac pathogenesis. Adenosine also stimulates vasculogenesis and angiogenesis during wound healing and tumor growth [4]. The current clinical uses of adenosine are limited to (1) treatment of supraventricular tachycardia or (2) as a coronary vasodilator during radionuclide myocardial perfusion imaging [4]. Because adenosine is involved in various pathological conditions, the targeting of specific adenosine receptor (ADR) subtypes in the vasculature, using selective ADR agonists or antagonists, could have possible therapeutic benefits [4].
Adenosine has four G protein-coupled receptor subtypes, A1, A2A, A2B, and A3, and each of these receptor subtypes have widespread expression throughout the body that is species specific. In particular, the A2A receptor has been implicated in vascular tension, vascular stenosis, atherosclerosis, ischemic preconditioning, and metabolic disease. Specifically, A2A is highly expressed in the cardiovascular system and modulation of A2A receptors via agonists or antagonists can regulate heart rate, blood pressure, heart rate variability, and cardiovascular toxicity during both normal and hypoxic conditions [5]. However, the distribution of the ADRs differs between species [4]. Therefore, cross-species investigations involving ADR agonists or antagonists is essential to validate drug function [4].
Adenosine can also create tone-dependent responses, for example, in the feline pulmonary vascular bed. The pressor response is mediated by A1 activation under low-tone conditions, whereas vasodilation is mediated by A2 activation under elevated tone conditions [6]. Furthermore, A2 receptor activation produces vasodilation in the vascular beds of many different species [4, 6, 7]. For example, A2 receptor activation leads to vasodilation in the rat renal artery. On the contrary, A1 receptor activation in this same cohort produces constriction [8,9,10,11,12]. Furthermore, the mechanism of the vasodilation effects of adenosine also seem to vary within species. For example, the enhanced vasopressor effect of adenosine in rats induced by comakalim and nicorandil [adenosine triphosphate (ATP) channel openers] was attenuated by glibenclamide (ATP-sensitive K + channel blocker), suggesting an ATP-sensitive K + channel mechanism [13]. In contrast, another rat study suggested that CGS (A2AR agonist)-induced hypotension is independent of ATP-sensitive K + channels [14]. Bahreyni et al. have a comprehensive review on this subject with many mechanistic details still lacking in the literature [5].
Given the mechanistic discrepancy within rat and other studies involving adenosine signaling mechanisms, and recent dialogue suggesting rodent models are poor predictors of human disease states [15], it is important to explore disease modeling in different species. It is well established that there is significant translatability of the feline pulmonary vascular bed to humans. Elevated pulmonary vasculature tone, a prominent feature in many pathological states, is an important model to elucidate adenosine-mediated or -modulated vasodepressor mechanisms to understand potential therapeutic roles of adenosine and adenosine analogs.
In the present investigation, therefore, we studied under an elevated pulmonary vascular tone, adenosine-mediated effects in the pulmonary vascular bed of the cat. In this study, we tested the hypothesis that pulmonary vasodilator responses of adenosine are dependent on teh activation of l-type calcium channels, independent of synthesis of nitric oxide (NO) from l-arginine, activation of ATP-sensitive potassium channels, and the release of cyclooxygenase products.
2 Methods
This experiments in this article were performed with approval from the animal subject committees at Tulane School of Medicine in New Orleans, LA, USA and Texas Tech School of Medicine in Lubbock, TX, USA. The study conformed with the Helsinki Declaration of 1964, as revised in 2013, concerning human and animal rights. Thirty-six adult mongrel cats of either sex, weighing 2.9–4.5 kg, were heavily sedated with ketamine hydrochloride and anesthetized with sodium pentobarbital. The animals were restrained on a fluoroscopic table in the supine position, and a uniform level of anesthesia was maintained. The trachea was intubated, and the cats spontaneously breathed room air. Systemic arterial (aortic) pressure was measured, and intravenous injections were made via a catheter placed in the inferior vena cava.
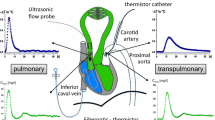
For perfusion of the lower left lung lobe, a triple-lumen catheter was passed from an external jugular vein into the artery to the lower left lung lobe under fluoroscopic guidance. The lower left lung lobar artery was isolated by a distending balloon cuff on the perfusion catheter, after heparinization. The lung lobe was perfused through the catheter lumen beyond the cuff with a perfusion pump. Blood was withdrawn from a femoral artery. Measurement of lobar arterial pressure occurred via a second catheter port that was beyond the cuff and on the perfusion catheter. The mean pressure in the main pulmonary artery was adjusted so that the perfusion rate approximated the pressure in the lower left lung lobar arterial perfusion, and once established, this perfusion rate was maintained. The flow range was 29–40 mL/min. A radiopaque catheter transseptally passed into the left atrium was used to measure left atrial pressure. Mean vascular pressures were measured with Spectromed (Oxford, CA, USA) DTX Plus transducers zeroed at the right atrial level. They were recorded on a Grass (Quincy, MA, USA) Model 7 recorder after characteristic waveforms had been established. This model has been previously described, and a schematic representing our experimental design is shown in Fig. 1.

Schematic of the experimental design. LAP left atrial pressure, Lo AP lobar arterial pressure, MAP mean arterial blood pressure
These experiments were divided into six groups. The first sequence of experiments assessed the effects of adenosine and other vasodepressor agents in the pulmonary circulation under low and elevated tone states. At all times, the agonists were injected into the perfusion circuit distal to the pump in a random sequence. To provide experimentally induced, elevated pulmonary lobar vascular resistance, an infusion of U-46619 (a thromboxane A2 analog) was infused until a steady-state level of 31–35 mm Hg was reached. These responses were compared with other known pulmonary vasodepressor drugs including acetylcholine, adenosine, isoproterenol, and lemakalim (a potassium channel agonist).
The second sequence of experiments investigated the hypothesis that adenosine exerts a vasodepressor action in the pulmonary circulation via a K + ATP-channel-sensitive mechanism. The effects of U-37883A on responses to adenosine and other vasodepressor agents were compared before and after administration of U-37883A, the selective K + ATP-channel antagonist, in the pulmonary vascular bed of the cat.
In the third and fourth set of experiments, the hypotheses that adenosine exerts a vasodepressor effect via either an NO or cyclooxygenase sensitive pathway was evaluated. The influence of l-N5-(1-iminoethyl)-ornithine (l-NIO; a NO synthase inhibitor) and sodium meclofenamate (cyclooxygenase inhibitor) on responses to adenosine and other vasodepressor agents was compared before and after the administration of l-NIO or sodium meclofenamate in the feline pulmonary vascular bed.
In the fifth and sixth set of experiments, the hypotheses that adenosine exerts a vasodepressor effect via an adenosine receptor-sensitive pathway was evaluated. The influence of the adenosine 1(A1) receptor antagonist 8-cyclopentyl-1,3-dimethylxanthine (8-CPT) and the adenosine 2 (A2) receptor antagonist 8-(3-chlorostyryl)caffeine (CSC) on responses to adenosine and other vasodepressor agents was compared before and after the administration of 8-CPT or CSC in the feline pulmonary vascular bed.
Adenosine was acquired from Crinos Biological Research Laboratories in Como, Italy. Pinacidil (Eli Lilly, Indianapolis, IN, USA) was prepared by dissolving the compound in 400 μL of 4 N HCl and 300 μL of ethanol and was diluted with standard saline. Lemakalim (levcromakalim, a potassium channel agonist) [SmithKline Beecham, Sussex, UK] was dissolved in a 20% ethanol-saline solution at a concentration of 1 mg/mL and diluted in standard saline. The solvents for the drugs in these experiments displayed no significant effect on baseline vascular pressure nor on responses to the vasoactive drugs. Acetylcholine chloride, sodium arachidonate, isoproterenol hydrochloride (Sigma, St. Louis, MO, USA), L-NIO (Alexis Biochemical, San Diego, CA, USA), sodium meclofenamate (Warner Lambert-Parke-Davis, Ann Arbor, MI, USA), and U-37883A (Upjohn, Kalamazoo, MI, USA) were dissolved in standard saline. N6-Cyclopentyladenosine (CPA) [Research Biochemicals International, Natick, MA, USA] was prepared in 1 N of acetic acid and diluted with standard saline. 8-Cyclopentyl-1,3-dimethylxanthine (Sigma) was diluted within 0.2 N NaOH. N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine (DPMA) [Research Biochemicals International] was diluted in 0.2% dimethyl sulfoxide. 8-(3-Chlorostyryl)caffeine (Research Biochemicals International) was diluted in 0.2% dimethyl sulfoxide.
All solutions were prepared on a regular basis and were kept on crushed ice during the experiments. Solutions were stored in a freezer at 0 °F (− 18 °C) in amber bottles. Injections of the compounds were randomized and were injected into the perfused lobar artery in fixed small volumes. U-46619 (Sigma), a thromboxane A2 analog, was dissolved in 100% ethanol to concentration of 10 mg/mL and was diluted in 0.9% normal saline. The perfused lobar artery was then infused with U-46619 (Sigma) using a Harvard (Holliston, MA, USA) infusion pump at a rate of (55–320 ng/min), which was required to increase the lobar arterial pressures to values of 31–35 mm Hg.
An analyzer, Corning (Palo Alto, CA, USA) Model 178, was used to measure arterial blood gas tensions and pH and were in the normal range. All hemodynamic data in this manuscript are expressed in absolute units and are presented as mean ± standard error. Responses represent peak changes. A one-way analysis of variance and the Scheffé’s test or a paired Student’s t test (30) were used to analyze the data. A p value of < 0.05 indicated statistical significance.
3 Results
Responses to adenosine, acetylcholine, isoproterenol, CPA, and DPMA were evaluated and compared in the pulmonary vascular bed of the cat. These data are summarized in Fig. 2. Under baseline conditions with tone in the pulmonary vascular bed at resting levels (0–6 mm Hg), injections of adenosine into the perfused lobar artery in doses of 10–100 µg had no significant effect on the lobar arterial pressure (data not shown). However, when the tone in the pulmonary vascular bed was increased to a high steady value (342 mm Hg) with an infusion of the thromboxane A2 analog U-46619, the administration of adenosine in doses of 10–100 µg caused a significant dose-related vasodepressor effect in the pulmonary arterial perfusion pressure. Systolic and diastolic, left atrial, and mean systemic arterial pressures were unchanged at the studied doses of adenosine.

a Bar graph illustrating the decrease in lobar arterial pressure in response to injections of adenosine in doses of 10–100 µg into the pulmonary vascular bed of the cat. b Dose–response curves comparing decreases in lobar arterial pressure in response to adenosine, isoproterenol, acetylcholine, N6-cyclopentyladenosine (CPA) adenosine-1 agonist, and N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine (DPMA) adenosine-2 agonist. Compounds were injected directly into the lobar artery and baseline pressure was increased to a high steady value with an infusion of U-46619; “n” indicates the number of experiments
On a nanomolar basis, adenosine demonstrated a less significant vasodepressor effect when compared to isoproterenol and acetylcholine; however, the agent was more potent on a nanomolar basis than CPA and DPMA. Vasodepressor responses to adenosine and other agents were compared under an elevated tone before and after administration of sodium meclofenamate (Fig. 3). After administration of sodium meclofenamate in a dose of 2.5 mg/kg intravenously, significant decreases in lobar arterial pressure in response to arachidonic acid were noted; however, meclofenamate did not significantly alter responses to both adenosine and acetylcholine (Fig. 3).

Influence of meclofenamate (2.5 mg/kg intravenously; hatched bars) on vasodepressor responses to adenosine (a, n = 8) and acetylcholine (b, n = 8). Influence of vasoconstrictor responses to arachidonic acid (c, n = 8). Solid bars, control. *p < 0.05
Vasodepressor responses to adenosine and other agents were compared before and after administration of L-NIO in a dose of 5 mg/kg intro arterial (IA) (Fig. 4). Under elevated tone conditions, decreases in lobar arterial pressure in response to all adenosine, acetylcholine, DPMA, and isoproterenol were not significantly reduced, whereas responses to acetylcholine were significantly reduced after the administration of CPA (Fig. 4).

Influence of NG-L-nitro-l-arginine methyl ester (5 mg/kg IA; hatched bars) on vasodepressor responses to adenosine (a, n = 6–8), acetylcholine (b, n = 7–8), N6-cyclopentyladenosine [CPA] (c, n = 4–6), DPMA (d, n = 4–6), and isoproterenol (e, n = 6–8). IA intro arterial. *p < 0.05. Solid bars, control
Vasodepressor responses to adenosine and other agents were compared under an elevated tone before and after administration of the K + ATP-channel antagonist U-37883A [2 mg/kg intravenously] (Fig. 5). Significant decreases in lobar arterial pressure in response to adenosine, CPA, DPMA, and isoproterenol were not seen. Significant decreases in the lobar arterial pressure in response to lemakalim were seen when compared with the treatment period (Fig. 5).

Influence of K + adenosine triphosphate (ATP) [potassium channel sensitive to intracellular ATP levels] channel inhibitor U-37883A in a dose-dependent manner (2 mg/kg intravenously) on vasodepressor responses to adenosine (a, n = 6–8), N6-cyclopentyladenosine [CPA] (b, n = 4–6), N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine [DPMA] (c, n = 4–6), lemakalim (d, n = 8), and isoproterenol (e, n = 8). *p < 0.05
Vasodepressor responses to adenosine and other agents were compared under an elevated tone before and after administration of the A1 receptor antagonist 8-CPT in a dose of 1 mg/kg IA. Responses to vasodilators were compared before and after the administration of 8-CPT. Significant decreases in lobar arterial pressure in response to adenosine and the A1 receptor agonist CPA were seen after administration of the A1 receptor antagonist 8-CPT, whereas responses to the A2 receptor agonist DPMA and pinacidil were not significantly altered (Fig. 6).

Influence of adenosine-1 receptor antagonist 8-cyclopentyl-1,3-dimethylxanthine (1.0 mg/kg IA) on vasodepressor responses to adenosine (a, n = 6–8), n6-cyclopentyladenosine (b, n = 5–6), N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine (DPMA) adenosine 2 agonist (c, n = 5–6), and pinacidil (d, n = 4–5). IA intro arterial. *p < 0.05
Vasodepressor responses to adenosine and other agents were compared under an elevated tone before and after administration of the A2 receptor antagonist CSC in a dose of 1 mg/kg IA (Fig. 7). Significant decreases in lobar arterial pressure in response to adenosine and the A2 receptor agonist DPMA were seen; however, responses to the A1 receptor agonist 8-CPT and pinacidil were not significantly altered (Fig. 7).

Influence of adenosine A2A receptor antagonist 8-(3-chlorostyryl)caffeine (0.5 mg/kg IA) on vasodepressor responses of adenosine (a, n = 6–8), n6-cyclopentyladenosine (b, n = 8), N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine (DPMA) adenosine 2 agonist (c, n = 8), and pinacidil (d, n = 8). IA intro arterial. *p < 0.05
4 Discussion
These data show that adenosine has significant vasodepressor activity in the pulmonary vascular bed of the cat. The data also suggest that pulmonary vasodilator responses to adenosine are partially dependent on both the activation of adenosine 1 and 2 receptor pathways, and independent of the activation of cyclooxygenase activation, ATP-sensitive K + channels, or synthesis of NO. The goal of these experiments was to explore the pulmonary vasodilator responses of adenosine in the pulmonary vasculature of the mongrel cat to ascertain if these effects were independent of synthesis of NO from l-arginine, activation of ATP-sensitive potassium channels, and/or the release of cyclooxygenase products. Importantly, these results from this study show that adenosine has moderate vasodepressor activity in the pulmonary vascular bed under increased tone conditions with the thromboxane analog, U-46619. Additionally, adenosine-induced decreases in lobar arterial pressure were dose dependent. Responses to adenosine were significantly altered after the administration of 8-CPT and CSC, indicating that the vasodepressor effect is mediated or modulated, in part, by both A1 and A2 pathways, respectively. Responses to adenosine were not altered after the administration of L-NIO or U-37883A. This suggests that the vasodepressor activity of adenosine is independent of the release of endothelium-derived NO and does not activate ATP-sensitive K + channels in the pulmonary vascular bed of the mongrel cat.
The present investigation further demonstrated that when tone in the pulmonary vascular bed is increased to a high steady-state level, adenosine induced dose-related decreases in lobar arterial pressure. Furthermore, the decreases in lobar arterial pressure reflected decreases in pulmonary vascular resistance as the pulmonary blood flow was maintained constant and the left atrial blood pressure was unchanged. With regard to relative vasodilator activity in the pulmonary vascular bed, the effect of adenosine was significantly more potent than both CPA and DPMA but less potent than isoproterenol and acetylcholine.
These data are in agreement with a previous rat study suggesting that CGS (A2AR agonist)-induced hypotension is independent of ATP-sensitive K + channels. When glibenclamide (ATP-sensitive K + antagonist) was administered as a pre-treatment, CGS-induced hypotension was no longer suppressed in the presence of 8-(p-sulfophenyl)theophylline, an A1-A2 receptor antagonist.
These data are in contrast to an experiment performed in spinally anesthetized dogs where glibenclamide (ATP-sensitive K + antagonist) did not antagonize an adenosine-induced decrease in blood pressure, suggesting that the opening of ATP-sensitive K + channels is recruited in adenosine-induced vasodepression [16]. Furthermore, an experiment in rats found similar results using glipizide (ATP-sensitive K + antagonist), which attenuated the depressor responses of CPCA (A2AR agonist), suggesting an ATP-sensitive K + channel mechanism [17]. The reasons for the differences between these studies are unclear. Given these inter- and within-species mechanistic differences in response to adenosine-induced vasodepression, it is clear that more studies are warranted to elucidate successful treatments for pulmonary and cardiovascular diseases where pulmonary hypertension is a prominent feature. This is additionally relevant because there is evidence that ADR-targeted treatments for diseases like PAH are emerging [3].
As mentioned, adenosine exerts its effects through the G protein-coupled receptor subtypes, A1, A2A, A2B, and A3. When adenosine binds A1 receptors in ventricular and atrial myocytes, it inhibits the activation of adenylyl cyclase and increases an inward rectifying K + current, which creates negative chronotropic and inotropic effects on the heart [18]. Adenosine displays high selectivity for both the A1 and A2 receptor sites, with the capability of triggering adenosine receptor-mediated events agonistically. A2 receptor activation causes stimulation of adenylyl cyclase, stimulation of NO formation, and activation of ATP-dependent K + (K + ATPATP +) channels [10, 19]. There is evidence, however, that inhibition of NO synthesis may not affect the dilator properties of adenosine in isolated canine coronary arteries [6]. The vasodepressor response to adenosine in coronary arteries has been shown to be mediated through A2 receptors [20,21,22]. A study evaluating the existence of the A2 receptor serves as a potential explanation for the potency of A2 receptors on coronary vasodepressor effects, finding unexpected coronary vasodilator responses to relatively selective A1 receptor agonists, such as CPA, which activate A2 receptors only at high concentrations [22]. In the present study, the selective adenosine agonists, CPA and DPMA, both caused dose-dependent vasodepressor effects. The respective adenosine agonists were selectively blocked with either the A1 antagonist, 8-CPT or the A2 antagonist, CSC, and not both. Further, there was an attenuation of adenosine-mediated vasodepressor effects with both the selective A1 and A2 antagonists in the feline pulmonary vascular bed. These data suggest that both adenosine receptor subtypes play a role in mediating or modulating adenosine-induced effects in the pulmonary vascular bed of the cat.
There is also evidence that adenosine has selective and protective effects on microvascular endothelium and vascular homeostasis [23]. As seen in myocardial ischemia, adenosine is released from the myocardium in response to a decrease in oxygen [23]. Adenosine induces its protective role through coronary and collateral vasodilation, which increases oxygen supply overall through multiple effects that act in unison to decrease myocardial oxygen demand (i.e., negative inotropism, chronotropism, and dromotropism). Oxygen deprivation causes adenosine to enhance energy production via an increase in glycolytic flux, which causes adenosine to act as a substrate for purine salvage, which then goes on to restore cellular energy charge during reperfusion. Adenosine inhibits oxygen radical release from activated neutrophils by limiting the degree of vascular injury during ischemia and reperfusion [23]. Therefore, adenosine attenuates ischemia–reperfusion injury to preserve endothelial cells [22]. There is evidence that patients with PAH have low levels of adenosine, caused by increased levels of the metabolic enzyme adenosine deaminase, suggesting that adenosine is important in the manifestation of PAH [24]. However, adenosine has not been used as a treatment for PAH owing to its extremely short half-life and its non-selective activity. However, the widespread expression of ADRs throughout the body, including cardiac and vascular cells, implores its importance in the normal physiological functioning of these body systems, and thus, their misfunctioning [25].
5 Conclusions
The results in this investigation show that adenosine has significant vasodepressor activity in the pulmonary vascular bed of the cat when the tone is increased experimentally. Although the exact mechanism by which adenosine induces vasodepressor effects in felines is uncertain, the results of this study suggest that the vasodilator response to adenosine is mediated, in part, by adenosine 1 and 2 receptor-mediated pathways and that this mixture of single-stranded oligonucleotides may be useful beyond its previously discovered pharmacological properties [22]. The limitations to this study (and comparable studies) includes inter- and within-species differences in response to adenosine-induced vasodepression. Therefore, more studies are warranted to elucidate successful treatments for pulmonary and cardiovascular diseases where pulmonary hypertension is a prominent feature.
References
Varela DL, Teleb M, El-Mallah W. Advanced therapies for the management of adults with pulmonary arterial hypertension due to congenital heart disease: a systematic review. Open Heart. 2018;5:e000744.
Rosenkranz S, Ghofrani H-A, Beghetti M, Ivy D, Frey R, Fritsch A, et al. Riociguat for pulmonary arterial hypertension associated with congenital heart disease. Heart. 2015;101:1792–9.
Alencar AKN, Montes GC, Barreiro EJ, Sudo RT, Zapata-Sudo G. Adenosine receptors as drug targets for treatment of pulmonary arterial hypertension. Front Pharmacol. 2017;8:858. http://www.ncbi.nlm.nih.gov/pubmed/29255415.
Ho M-F, Rose’Meyer RB. Vascular adenosine receptors; potential clinical applications. Curr Vasc Pharmacol. 2013;11:327–37. http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1570-1611&volume=11&issue=3&spage=327.
Bahreyni A, Avan A, Shabani M, Ryzhikov M, Fiuji H, Soleimanpour S, et al. Therapeutic potential of A2 adenosine receptor pharmacological regulators in the treatment of cardiovascular diseases, recent progress, and prospective. J Cell Physiol. 2019;234:1295–9. http://www.ncbi.nlm.nih.gov/pubmed/30146778.
Cheng DY, DeWitt BJ, Suzuki F, Neely CF, Kadowitz PJ. Adenosine A1 and A2 receptors mediate tone-dependent responses in feline pulmonary vascular bed. Am J Physiol Circ Physiol. 1996;270:H200–7. http://www.ncbi.nlm.nih.gov/pubmed/8769752.
Shryock JC, Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am J Cardiol. 1997;79:2–10. http://www.ncbi.nlm.nih.gov/pubmed/9223356.
Casellas D, Moore LC. Autoregulation and tubuloglomerular feedback in juxtamedullary glomerular arterioles. Am J Physiol. 1990;258:F660–9http://www.ncbi.nlm.nih.gov/pubmed/2316670.
Ghai G, Francis JE, Williams M, Dotson RA, Hopkins MF, Cote DT, et al. Pharmacological characterization of CGS 15943A: a novel nonxanthine adenosine antagonist. J Pharmacol Exp Ther. 1987;242:784–90. http://www.ncbi.nlm.nih.gov/pubmed/3656113.
Jarvis MF, Williams M, Do UH, Sills MA. Characterization of the binding of a novel nonxanthine adenosine antagonist radioligand, [3H]CGS 15943, to multiple affinity states of the adenosine A1 receptor in the rat cortex. Mol Pharmacol. 1991;39:49–54. http://www.ncbi.nlm.nih.gov/pubmed/1987452.
Holz FG, Steinhausen M. Renovascular effects of adenosine receptor agonists. Ren Physiol. 1987;10:272–82. http://www.ncbi.nlm.nih.gov/pubmed/3454474.
Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem. 1993;268:16887–90. http://www.ncbi.nlm.nih.gov/pubmed/8349579.
Saito K, Sakai K. Enhancement of the vasodepressor response to adenosine by nicorandil in rats: comparison with cromakalim. Fundam Clin Pharmacol. 1998;12:37–43. http://www.ncbi.nlm.nih.gov/pubmed/9523182.
Fozard J, Carruthers A. The cardiovascular effects of selective adenosine A1 and A2 receptor agonists in the pithed rat: no role for glibenclamide-sensitive potassium channels. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:192–6. http://springerlink.bibliotecabuap.elogim.com/10.1007/BF00169266.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–12. http://www.ncbi.nlm.nih.gov/pubmed/23401516.
Orito K, Satoh K, Taira N. ATP-sensitive K + channels are gradually recruited in the vasodepressor response to adenosine in spinally-anesthetized dogs. Jpn J Pharmacol. 1992;60(3):295–7. https://www.jstage.jst.go.jp/article/jphs1951/60/3/60_3_295/_pdf.
Kang MJ, Park MS, Shin IC, Koh HC. Modification of cardiovascular response of posterior hypothalamic adenosine A2 receptor stimulation by adenylate cylase, guanylate cyclase and by KATP channel blockade in anesthetized rats. Neurosci Lett. 2003;344:57–61. http://linkinghub.elsevier.com/retrieve/pii/S0304394003004026.
Belardinelli L, Isenberg G. Isolated atrial myocytes: adenosine and acetylcholine increase potassium conductance. Am J Physiol Circ Physiol. 1983;244:H734–7. http://www.ncbi.nlm.nih.gov/pubmed/6846564.
Abebe W, Hussain T, Olanrewaju H, Mustafa SJ. Role of nitric oxide in adenosine receptor-mediated relaxation of porcine coronary artery. Am J Physiol. 1995;269:H1672–8. http://www.ncbi.nlm.nih.gov/pubmed/7503264.
King AD, Milavec-Krizman M, Müller-Schweinitzer E. Characterization of the adenosine receptor in porcine coronary arteries. Br J Pharmacol. 1990;100:483–6. http://www.ncbi.nlm.nih.gov/pubmed/2390673.
Shryock JC, Snowdy S, Baraldi PG, Cacciari B, Spalluto G, Monopoli A, et al. A2A-adenosine receptor reserve for coronary vasodilation. Circulation. 1998;98:711–8. http://www.ncbi.nlm.nih.gov/pubmed/9715864.
Kaye AD, Skonieczny BD, Kaye AJ, Harris ZI, Luk EJ. An analysis of responses to defibrotide in the pulmonary vascular bed of the cat. Am J Ther. 2016;23:e757–65. http://www.ncbi.nlm.nih.gov/pubmed/24368612.
Ely SW, Berne RM. Protective effects of adenosine in myocardial ischemia. Circulation. 1992;85:893–904. http://www.ncbi.nlm.nih.gov/pubmed/1537125.
Tofovic SP, Jackson EK, Rafikova O. Adenosine deaminase–adenosine pathway in hemolysis-associated pulmonary hypertension. Med Hypotheses. 2009;72:713–9. http://www.ncbi.nlm.nih.gov/pubmed/19237250.
Nootens M, Schrader B, Kaufmann E, Vestal R, Long W, Rich S. Comparative acute effects of adenosine and prostacyclin in primary pulmonary hypertension. Chest. 1995;107:54–7. http://www.ncbi.nlm.nih.gov/pubmed/7813311.
Acknowledgments
The authors acknowledge Dr. Ikhlass N. Ibrahim, DVM, who performed the experiments in the present investigation and was critical with many significant contributions to Dr. Kaye’s PhD studies and basic science career over two decades.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the conduct of this study or the preparation of this article.
Conflict of interest
Alan David Kaye, Syed R. Baber, Mohammed T. Sharief, Rachel J. Kaye, and Elyse M. Cornett have no conflicts of interest that are directly relevant to the content of this article. The authors have no relationships with pharmaceutical companies or products to disclose, nor do they discuss off-label or investigative products in this manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kaye, A.D., Baber, S.R., Sharief, M.T. et al. Vasodepressor Effects of Adenosine in the Cat are Independent of Cyclooxygenase, Potassium Channels, and Nitric Oxide Pathways. Drugs R D 19, 319–328 (2019). https://doi.org/10.1007/s40268-019-00283-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-019-00283-2