
Abstract
Alpha-1 antitrypsin (AAT) protects the lung by inhibiting neutrophil proteinases, but AAT has many other non-proteolytic functions that are anti-inflammatory, antiviral and homeostatic. Approximately 1 in 1600 to 1 in 5000 people have the homozygous Z mutation, which causes AAT misfolding, accumulation in (predominantly) liver cells and low circulating levels of AAT, leading to AAT deficiency (AATD). AATD is classically a disease of neutrophilic inflammation, with an aggressive and damaging innate immune response contributing to emphysema and other pathologies. AATD is one of the most common genetic disorders but considerably under-recognised. Most patients are diagnosed later in life, by which time they may have accumulated significant lung, liver and multisystem damage. Disease presentation is heterogeneous and not fully explained by deficiency levels alone or exposure to cigarette smoking. This suggests other factors influence AATD-associated pathological processes. Aging itself is associated with organ dysfunction, including emphysema and airflow obstruction, inflammation, altered immune cell responses (termed immunosenescence) and a loss of proteostasis. Many of these processes are present in AATD but at an earlier age and more advanced stage compared with chronological aging alone. Augmentation therapy does not completely abrogate the manifold disease processes present in AATD. New approaches are needed. There is emerging evidence that both age- and AATD-related disease processes are amenable to correction by targeting proteostasis, autophagy, immunosenescence and epigenetic factors. This review explores the impact of the aging process on AATD presentation and discusses novel therapeutic strategies to mitigate low levels of AAT or misfolded AAT in an aging host.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Alpha-1 antitrypsin (AAT) deficiency (AATD) is the most well-established genetic cause of chronic obstructive pulmonary disease (COPD), accounting for up to 2% of COPD cases. The most common severe deficiency is characterised by a loss of function (reduction in functional AAT) and a gain of function (the formation of misfolded AAT polymers), both of which are pro-inflammatory states and associated with tissue damage. |
Many of the disease manifestations of AATD are only clinically apparent in older patients, and AATD patients have other chronic inflammatory conditions more frequently than would be expected. |
The inflammation present in AATD and some AATD-associated pathological processes share features of accelerated aging. |
Strategies to improve the inflammatory burden of AATD by enhancing AAT secretion, reducing polymer formation or clearance or targeting accelerated aging may improve disease outcomes. |
1 Introduction
“All diseases run into one, old age” Ralph Emerson
Most chronic inflammatory diseases have a strong association with age. For example, periodontitis [1], ischaemic heart disease [2] and most chronic respiratory conditions are all more common in older adults [3]. Initially, this was thought to reflect that diseases that are slow in progression might only become clinically apparent at an older age. However, epidemiological studies have highlighted that chronic conditions cluster in the same individuals with a prevalence higher than expected, even when shared risk factors are considered [4, 5]. Furthermore, studies across chronic inflammatory morbidities have identified potentially shared pathological mechanisms, including excessive reactive oxygen species (ROS) production, immune cell dysfunction and a loss of proteostasis, all of which are associated with age [6].
Many chronic inflammatory illnesses demonstrate biological features of aging that appear advanced compared with healthy individuals of the same age [7]. This is thought to represent a mechanistic continuum between aging and age-related disease, termed accelerated aging [8]. There appears to be a dose response between the level of chronic inflammation present, the burden of chronic disease and the presence of accelerated aging. Fortunately, there are also potential therapeutic strategies to combat accelerated aging, with early studies suggesting that mammalian aging can be delayed through dietary and pharmacological manipulation [9]. If there were links between heightened chronic inflammation, aging and chronic disease, one would expect the clearest evidence of this to be seen in conditions with the greatest burden of inflammation.
Alpha-1 antitrypsin (AAT) deficiency (AATD) is a chronic inflammatory condition associated most commonly with lung and liver disease. AATD lung disease shares features of chronic obstructive pulmonary disease (COPD), but patients with AATD are considered to have levels of pulmonary inflammation that are greater than those with non-AATD COPD [10]. This is because the loss of functional AAT and the gain of misfolded AAT are highly pro-inflammatory. COPD itself is considered a disease of accelerated aging, demonstrating all of the classical biological hallmarks of aging [11]. Were there a simple mechanistic link between chronic inflammation and accelerated aging, it should be more clearly identifiable in AATD and, indeed, certain manifestations of AATD are recognised features of aging. This review article discusses the evidence that links AATD with accelerated aging, including whether therapeutics designed to target aging might be beneficial in AATD.
2 Alpha-1 Antitrypsin (AAT) and its Deficiency
AAT encoded by the SERPINA1 gene on chromosome 14 is a serine proteinase inhibitor [12] produced principally by hepatocytes but also in small amounts by neutrophils, monocytes and endothelial and epithelial cells [13,14,15,16]. It is the classical circulating anti-proteinase in humans, and its central function is to inhibit free neutrophil proteinases [such as neutrophil elastase (NE) and proteinase 3 (PR3)] in the lungs to limit proteinase-associated inflammation and tissue damage. Damage is limited rather than prevented, as AAT binds proteinases on a one-to-one molar basis, and concentrations of NE and PR3 far exceed those of AAT at the site of a degranulating neutrophil. This leads to an area of obligate tissue damage around each degranulating cell until concentrations of the proteinases are reduced by diffusion into the local tissue environment [17]. In addition to the anti-proteinase function of AAT, it is increasingly recognised that AAT has multiple non-proteinase-mediated effects. These are anti-inflammatory and immunomodulatory. AAT reduces free radical production and associated damage. For example, AAT reduces neutrophil superoxide production through interactions with inflammatory mediator surface receptors and binds hemin (heme oxidized to the ferric state), which reduces hemin-associated free radical production [18,19,20]. AAT reduces neutrophil migration towards inflammation through steric binding of interleukin (IL)-8 and leukotriene B4 (LTB4) [21]. AAT reduces the inflammatory cytokine cascade by inhibiting the production of tumour necrosis factor (TNF)-α in macrophages [22] and reducing cytokine release from monocytes and neutrophils [23]. AAT also has antimicrobial properties (as shown in studies of pseudomonas aeruginosa [24]) and antiviral functions (being able to block HIV-1 virus from entering cluster of differentiation [CD]-4+ T cells [25]). AAT appears to mitigate auto-immunity and increase immune tolerance by enhancing the number and function of foxp3-positive regulatory T cells [26]. AAT has a role in diverse organ systems, such as preventing overt hyperglycaemia in diabetic mice [27] and reducing inflammation-mediated apoptosis and increasing insulin secretion of pancreatic B cells [28]. In keeping with this, trials of AAT therapy in new-onset type 1 diabetes mellitus to protect pancreatic B cells are ongoing, with dose-ranging studies recently reported [29]. AAT modulates microglial cells in inflammatory conditions, enhancing microglial survival from amyloid β-induced toxicity [30].
AATD is a condition of low circulating levels of AAT, caused by mutations in SERPINA1 and inherited in an autosomal and co-dominant pattern, with two copies of the gene (allele) contributing to the disease. The most common is the ‘M’ variant. PiMM (two copies of the M allele) is considered normal and corresponds to AAT blood levels of 20–53 μM. Over 150 mutations in SERPINA1 have been described, but the most common and most studied mutations are the ‘Z’ and ‘S’ variants. These are associated with misfolding of the AAT protein, especially the Z mutant protein, leading to its retention and accumulation in the endoplasmic reticulum (ER) of AAT-producing cells. This causes low circulating levels of AAT, decreasing the inhibition of neutrophil proteinases and impacting on the non-proteolytic activities of AAT, already described. Misfolded proteins may polymerise (a key feature of the Z protein), and these polymers are pro-inflammatory [31] and can act as an activating factor, potentially enhancing neutrophil recruitment [32]. Polymers are implicated in AATD-associated liver disease but may also impact on other cells where misfolded proteins accumulate, including monocytes and macrophages, activating the stress response. When homozygous, the Z allele (PiZZ) is associated with circulating AAT levels of 3–7 μM; however, with co-dominant inheritance, patients can also have heterozygote patterns, including PiSZ, which is also associated with a less severe deficiency (10–20 μM) [33].
Despite being considered a rare disease, AATD is relatively common, affecting between 1 in 1600 and 1 in 5000 people in screening studies, depending on geographical location [34, 35]. Extrapolation of data from population studies suggests approximately 3.4 million individuals globally have an AATD genotype that leads to a deficiency [36]. An estimated 250,000 PiZZ individuals exist worldwide, but the prevalence varies by region, with approximately 120,000 in Europe, 90,000 in America and the Caribbean, 4000 in Africa and 3200 in Asia [37]. PiSZ is more common: over 1,400,000 PiSZ individuals are estimated globally, with over 700,000 in Europe, over 500,000 in America and the Caribbean, over 85,000 in Africa and over 77,500 in Asia [38]. However, AATD is under-diagnosed, with epidemiological studies suggesting that < 0.5% of expected cases are detected [39].
The World Health Organization recommended AATD testing for anyone with a diagnosis of COPD or adult-onset asthma [40]. In 2003, the American Thoracic Society and European Respiratory Society (ERS) published a joint statement highlighting that a diagnosis of AATD should be considered in patients with, among other things, early-onset emphysema [41], recently reinforced by the updated ERS strategy document for AATD [42]. However, adoption of this guidance is variable, as confirmed in a recent UK-based study, where only 2.2% of 29,596 patients diagnosed with COPD before 60 years of age had any record of being tested for AATD; in those that had been tested, > 20% had a confirmed diagnosis [43], highlighting the benefit of screening in this population.
Patients with confirmed AATD frequently experienced significant delay to diagnosis. Surveys of patients recalling their disease journey described the first symptoms of disease being present in the fourth decade of life but the diagnosis being made almost a decade later and requiring, on average, three different specialist assessments [44, 45]. This underdiagnosis limits the ability of individuals to consider appropriate lifestyle changes, monitoring or therapeutic strategies to mitigate the impact of AATD.
3 The Heterogeneity of Disease Associations of AAT Deficiency (AATD)
Classically, AATD is associated with lung and liver disease and, less commonly, necrotising panniculitis, an inflammatory condition of subcutaneous fat tissue that presents as tender, erythematous or pigmented skin nodules and ulcers.
The most common lung manifestation is emphysema, which tends to have an earlier onset in AATD than in non-AATD smokers—as initially described in 1963 in patients aged 35–44 years [46] —and can occur in the absence of cigarette smoking (although it is enhanced by smoking). AATD-associated emphysema is classically panacinar and disproportionately lower zone (in contrast with the more apical distribution seen in non-AATD COPD). However, the presence of emphysema in AATD is not ubiquitous and, when present, its distribution can also be heterogeneous. A computed tomography study described 8.5% of patients with AATD as having no emphysema (including never and ex-smokers) [46]. In those with emphysema, 64% had basal-predominant emphysema and 36% had predominantly apical emphysema [47], more typical of non-deficient COPD. Similarly, airflow obstruction is very common in AATD but not universal, with a recent study of never-treated severely deficient individuals reporting normal airflow in 18% of the cohort on baseline assessment [48].
Interestingly, even in severely deficient PiZZ patients, the rate of decline in lung function parameters varies [49] (see Fig. 1), and studies suggest this variability cannot be explained by smoking status or pack-year exposure alone (with rapid decline seen in some never smokers and no decline seen in some smokers [48]). This strongly suggests that not all manifestations of disease can be explained by the level of deficiency or environmental exposures, and other explanations for the heterogeneity must be sought.

The heterogeneity of lung function decline in PiZZ AATD. A recent study of 482 never treated, severely deficient individuals with AATD were assessed annually with post bronchodilator spirometry and gas transfer on at least four occasions for ≥ 3 years. Patients were divided into those meeting spirometry criteria for COPD and those who did not. Patients were then divided into those with a change in lung function that reflected normal aging (‘no decline’: change < − 0.1% predicted per year), those with a slow decline (‘slow’: change of − 0.1 to < − 0.5% predicted per year), moderate decline (‘moderate’: change of − 0.5 to < − 1.0% predicted per year), and rapid decline (‘rapid’: change of > −1.0% predicted per year), in both FEV1 and KCO as previously described [49]. Percentage of individuals with an average change in a FEV1 % predicted and b KCO by groups. In patients without COPD at baseline, 43 and 71% had a faster than expected decline in FEV1 and KCO, respectively. Of those with COPD at baseline, 76 and 81% had a faster than expected decline in FEV1 and KCO, respectively. Adapted from Stockley et al. [48]. AATD alpha-1 antitrypsin deficiency, COPD chronic obstructive pulmonary disease, PiZZ homozygous for Z allele, FEV1 forced expiratory volume in 1 s, KCO the carbon monoxide transfer coefficient
The liver diseases associated with AATD include neonatal hepatitis and fulminant post-natal liver failure, adult-onset cirrhosis, and hepatocellular carcinoma. While this is mainly limited to AATD phenotypes associated with intrahepatocyte polymerisation, the exact mechanism remains unknown. Presentation is variable, with a recent systematic review [50] reporting that 10.5% of patients with PiZZ homozygotes and PiSZ allele described liver cirrhosis and 14.7% reported having had a liver transplantation. While it seems possible that concurrent liver injury from viral infections, alcohol or high body mass index accelerate the progression of liver disease, these risk factors are not identified in all adults [51].
These classically associated conditions are not the only diseases to cluster with AATD. Asthma and partially reversible airflow obstruction have been reported in 35% and 61% of patients with AATD, respectively [52], and clinically significant bronchiectasis has been described in 27% of PiZZ patients [53]. Vasculitis and, more specifically, anti-PR3 vasculitis is seen three- to ninefold more commonly than in non-AATD individuals [54], and outcomes are worse in patients with AATD, with a mortality rate of 39% for PiZZ patients compared with 16% for non-AATD patients [55]. Also, inflammatory bowel disease (particularly ulcerative colitis) and hypothyroidism have been identified more frequently in patients with AATD than would be expected in a general population according to a UK registry of AATD patients [56].
Without population screening, there is a risk of ascertainment bias when cohort studies describe disease associations with AATD, but these diseases readily ‘match’ the known functions of AAT (including a reduction in immune tolerance to promote auto-immunity and allergy and reduced inhibition of proteinases with associated tissue damage). However, AATD has also been shown to co-occur more commonly than expected with conditions less classically associated with AAT function and more associated with aging. These conditions include arterial hypertension, cardiovascular disease (CVD), chronic kidney disease, osteoporosis and type 2 diabetes mellitus (T2DM) [57,58,59]. A similar pattern of comorbidity is seen in non-AATD COPD and, although COPD, CVD and related conditions [4] share the common risks of smoking, obesity and a sedentary lifestyle, these cannot fully account for the increased burden of disease [60]. Furthermore, in AATD, behavioural risk factors such as smoking are less prevalent following diagnosis, with up to 78% of smoking patients with AATD undertaking smoking cessation measures upon diagnosis [61]. This is in contrast with the majority of patients with non-AATD COPD, who continue to smoke cigarettes, even in large clinical trials [62]. This raises the important question of what is driving the variable disease presentation and multimorbidity in AATD—is it AATD, the associated inflammation or age (as suggested by Ralph Emerson)?
4 The Aging Host and its Impact on Lung Function, Inflammation and Cell Function
Aging is associated with a general decline in organ systems, including the lungs. At the extremes of age, structural changes to the thoracic cage cause a reduction in chest wall compliance. Osteoporosis can lead to a reduced height of the thoracic vertebrae, and associated kyphosis can affect inspiratory capacity and diaphragmatic function [63]. Respiratory muscle function decreases, with one study describing a 12% reduction in respiratory muscle strength in older adults over 6 years of follow-up [64]. The structure of the lungs changes, with degeneration of the elastin fibres around the alveolar duct leading to enlargement of airspaces, termed senile emphysema [65]. The corresponding reduction in supporting tissue can lead to premature closure of small airways during normal breathing, with associated air trapping and hyperinflation.
These changes are associated with an age-related decline in lung function measurements, but lung function measures are more variable in ‘healthy’ older individuals than in younger adults, making it difficult to establish what is ‘normal’ in old age. Most cross-sectional studies showed a decline in forced expiratory volume in 1 s (FEV1) with age, with an estimated rate of decline in FEV1 of approximately 15–25 ml/year at age 40–50 years but greater after the age of 70 years [66]. Gas exchange also declines with age when corrected for alveolar volume, suggesting changes to the alveolar–capillary membrane [67] in addition to ‘senile emphysema’.
Aging is associated with increased inflammation, with most studies of older adults describing a low-grade inflammatory signal that can be measured systemically, even in ‘health’. Termed inflammaging by some, this includes measurable increases in proinflammatory cytokines such as IL-6, TNFα and IL-1β and reductions in some (e.g. IL-10) but not all anti-inflammatory mediators [68]. Although tissue inflammation with age is less well-studied, lung challenge animal models and studies of human synovium do suggest greater inflammation is also seen in aging tissues and organs [69, 70]. Systemic and local inflammation has been consistently associated with non-communicable chronic disease and frailty, and a recent prospective study showed that mid-life levels of inflammatory markers predicted frailty 21 years later, highlighting a potential causative link [71].
Aging is also associated with a decline in immune cell function (termed immunosenescence), with all facets of the immune system altered, including the neutrophil (the cell most implicated in the pathology of AATD) and its responses. Circulating neutrophil numbers do not alter with age [72], but most neutrophil functions show an age-related change. The ability of neutrophils to undertake chemotaxis accurately towards bacteria and other instigators of inflammation, through the extracellular matrix, appears impaired with age [73, 74]. This decline in chemotaxis is seen from middle age onwards, although the deficit is most apparent after people reach their sixth decade of life. The cause of this defect is related to excessive phosphoinositide 3 kinase activity [75], an enzyme associated with neutrophil directional sensing, ROS release and phagocytosis of large particles [76]. Neutrophil degranulation increases, and evidence exists of increased neutrophil proteinase activity systemically, as demonstrated by higher levels of CD63 on the surface of neutrophils (a marker of primary granule release, which contain proteinases and other bactericidal products) and higher concentrations of the NE-specific fibrinogen degradation product, Aα360VAL in the plasma [75]. The ability of neutrophils to phagocytose opsonised bacteria [77,78,79], especially staphylococcus aureus [72], reduces. Finally, neutrophil extracellular trap release also reduces with age [80].
These functions appear even more impaired during acute infections, with the degree reflecting the severity of the infectious insult [81]. Furthermore, during severe or repeated infections, older patients can develop a neutropenia because of two compounding factors. First, a reduced response to granulocyte colony-stimulating factor (G-CSF), which should usually increase the release of immature neutrophils from the bone marrow [82, 83], and, second, an inability to prolong the lifespan of activated neutrophils in response to survival signals (granulocyte macrophage colony-stimulating factor [GM-CSF], interferon-1) at the site of inflammation [84]. Although all of these features have been described during normal aging, this process appears accelerated in some individuals, most commonly in the presence of chronic disease [85].
The mechanisms that lead to inflammation and cellular senescence with aging have been identified, as described in a landmark review by Lopez-Otin et al. [86] in 2013. These include genetic instability (the accumulation of genetic damage throughout life and a reduction in DNA repair mechanisms [87]); telomere attrition (the progressive loss of telomere-protective sequences from chromosome ends [88]); epigenetic alteration (including histone modifications, DNA methylation, chromatin remodelling and aberrant production and maturation of many messenger RNAs [mRNAs] [89]) and loss of proteostasis (impaired ability to prevent the formation of or clear misfolded proteins [90]).
Associated with these are increased nutrient signalling through insulin/insulin-like growth factor and target of rapamycin (TOR)-signalling networks, and mitochondrial dysfunction, with increased ROS production and an increased presence of senescent cells that have entered irreversible cell cycle arrest, all of which are pro-inflammatory. For example, senescent cells exacerbate the release of a signature cocktail of pro-inflammatory cytokines called the senescence-associated secretory phenotype (SASP), induced in part by the TNF-receptor (TNFR) superfamily cytokine receptors [91], impairing immune cell function and accelerating tissue aging [92, 93]. However, they appear amenable to treatment with interventions that reduce the activity of these pathways (such as caloric restriction), extending lifespan and healthspan (the duration of adult life spent in health), in both murine and rhesus monkey models [94, 95].
The body of evidence indicating that many chronic, noncommunicable diseases might reflect a state of accelerated aging is growing, and AATD may also be considered in this light.
5 AATD as a Form of Accelerated Aging
5.1 Biological Processes and Ageing in AATD
AATD shares many features with accelerated aging. There is chronic inflammation, both systemically and locally in the lung, that is greater than seen in non-AATD COPD, especially for neutrophil chemoattractants such as LTB4 and IL-8 [96]. AAT can inhibit the effects of IL-8 by direct binding, isolating IL-8 from its cogent receptors, CXCR-1 (associated with phagocytosis) and CXCR2 (associated with neutrophil migration) [97]. NE can increase LTB4 production and associated BLT1 (LTB4’s receptor) expression on endothelial, epithelial and immune cells. In keeping with this, neutrophils from PiZZ AATD patients demonstrate increased adhesion and degranulation in response to LTB4 [98].
TNFα (heavily implicated in many chronic diseases and accelerated aging [99,100,101]) is raised and is particularly significant in AATD. AAT reduces TNFα activity by several mechanisms, including binding and directly inhibiting TNF-converting enzyme activity, preventing pro-TNFα from becoming the active cytokine and inhibiting TNF receptors on the surface of cells, preventing activation of nuclear factor (NF)-κB [102]. Conversely TNFα decreases AAT concentration in cells [102]; in turn, AAT inhibits genes upregulated by TNF-α, including TNF-α-induced self-expression. Interestingly, these effects are also induced by oxidized AAT, a modified form lacking significant serine protease inhibitor activity [103], indicating it is not a feature of the active inhibitory site (Met 358). In AATD, the accumulation of misfolded Z and related AAT proteins leads to ER-associated protein degradation and the unfolded protein response [104], increasing TNFα production and expression, which in turn increases cellular apoptosis and (when expressed on neutrophils) leads to deficient bacterial killing [105].
Although not fully studied, chronic inflammation in AATD may also contribute to the increasing presence of the primary hallmarks of aging, as exemplified by TNFα. Prolonged TNFα exposure has been shown to induce telomere attrition [106] and cause pro-inflammatory epigenetic alterations in immune cells [107] and is a potent inducer of DNA damage (genetic instability), both directly [108] and indirectly through ROS generation [109]. In keeping with this, mutations in telomerase genes have been found to induce susceptibility to young-onset severe emphysema at a similar rate to that of AATD [110], and paediatric patients with AATD have been shown to have shorter telomeres with decreased telomerase activity compared with healthy controls [111].
AATD can result in a loss of proteostasis, with accumulation of the misfolded AAT protein and subsequent ER stress, but early studies suggest patients with AATD might also be at increased risk of other protein-misfolding conditions, such as amyloidosis. Transthyretin (TTR)-related familial amyloid polyneuropathy (ATTR) results from aggregation and extracellular disposition of misfolded TTR variants, and AAT is an important chaperone molecule for TTR, reducing aggregation [112]. Furthermore, changes in histone deacetylase activity (HDAC; involved in protein folding and proteostasis) have been described with age [113], and HDAC modulators have been shown to increase AAT expression in PiZZ cells, in vitro [114].
AATD is associated with an increased burden of ROS [115], and some evidence supports mitochondrial dysfunction [116]. AATD is associated with changes to the immune system, with some features of immunosenescence. Focusing again on neutrophils, evidence of enhanced neutrophil degranulation and proteinase activity in AATD is clear and consistent [117], and early studies suggest a reduction in the phagocytosis of opsonised bacteria, especially haemophilus influenzae, which appears to be detrimental in AATD [118]. However, studies suggest the accuracy of neutrophil migration is increased in AATD, especially in comparison with non-AATD COPD [119], highlighting that not all changes to the immune system in AATD map to an accelerated aging phenotype.
Silent information regulators of transcription (SIRT) are NAD + -dependant deacetylases associated with reduction–oxidation reactions and highly implicated in accelerated aging. SIRT1 inhibits cellular senescence, oxidative stress and autophagy via Foxhead Box 03 (FOXO3) deacetylation [120,121,122]; smokers have reduced SIRT1 levels and ‘smoking’ mice are protected against emphysema when given an activator of SIRT1 (SRT1720) [123]. SIRT1 also has a role in the stress response of cells, with downregulation of SIRT1 shown to increase the heat shock response [124], and in AATD there is evidence of increased levels of protein degradation with high levels of albumin protein fragments and heat shock protein (HSPA8) [125].
5.2 Organ Dysfunction and Aging in AATD
Although lung function measurements of 30-year-olds with AATD (diagnosed since birth) are within the normal range [126], clear deficits are noted in mid-life, and up to 80% of patients experience a faster decline in FEV1 than age alone would predict [127]. Although highly variable, the mean rate of decline has been described as being 49.9 ml/year in patients with AATD aged 40–60 years [128], higher than described in non-AATD COPD, which (although also highly variable) has been cited as an average of 33 ml/year [129].
Liver disease is also associated with aging, and the same processes of DNA damage, oxidative stress, telomere dysfunction, apoptosis resistance, cell cycle arrest, epigenetic changes and SASP release have been described in liver cirrhosis, non-alcoholic fatty liver disease and chronic hepatic infections [130]. For example, increased hepatocyte nuclear area and hepatocyte expression of p21, both markers of senescence, are associated with increased fibrosis stage and poor outcomes in non-alcohol-related fatty liver disease [131]. No papers on senescent markers in AATD-associated liver disease have yet been published, but such studies are underway.
5.3 Comorbidities and Aging in AATD
AATD is associated with significant age-related comorbidity, but—like the lung and liver disease—this is apparent at a younger age and with an increased prevalence compared with non-AATD controls. For example, CVD is more common in patients with AATD when increased cardiovascular risk is assessed noninvasively using arterial stiffness [132]. In non-AATD COPD, arterial stiffness relates to lung disease, including FEV1, FEV1/forced vital capacity (FVC) ratio [133] and emphysema on computed tomography scans [134]. Arterial stiffness is greater in patients with AATD than in age-matched healthy controls [135], and a small study of 19 patients with AATD found a relationship between increased arterial stiffness, age and FEV1 % predicted [57]. CVD is associated with inflammatory patterns similar to those seen in AATD, including higher concentrations of systemic TNFα (which predicts future cardiovascular events [136, 137]). There is also evidence of neutrophilic inflammation in CVD (especially pertinent in AATD), including associations with impaired microvascular perfusion, left ventricular dilation and adverse CVD outcomes [138]. We have already discussed the potential relationship between diabetes and AATD (Sect. 3).
The burden of osteoporosis is increased in AATD. Murine models suggest that AAT inhibits members of the TNFR superfamily in a dose-dependent manner, and this includes RANKL (receptor activator of NF-κB ligand) which, when activated, leads to osteoclast-associated mineral resorption and osteoporosis [139], perhaps providing some insight into the heightened burden of this disease in AATD. Sarcopenia is defined as a reduction in muscle mass and muscle function and is more prevalent with age [140]. AATD may favor the development of sarcopenia as exercise induces an increase in glycolytic type 2A myofibers, which are more fatigue prone and have higher energy consumption than the type 1 myofibers seen with exercise conditioning in non-AATD COPD [141]. The potential relationship between aging and AATD is summarized in Fig. 2.

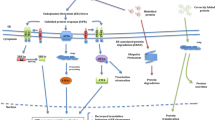
Mechanisms of damage in AATD and aging and potential therapeutic strategies. In AATD, a number of biological processes contribute to disease presentation, some of which are also seen in accelerated aging, but they can be targeted using appropriate therapeutics. a The genetic mutation leads to b misfolded AAT protein, which gets retained in the endoplasmic reticulum. Gene therapies may increase wild-type AAT expression, RNA interference strategies may reduce expression of the mutant AAT protein and chaperone molecules may assist with the removal of the misfolded protein. c In PiZZ AATD, polymer formation is common and is implicated in liver disease, but both chaperone molecules and drugs that increase autophagy may reduce polymer burden. Alternatively, small molecules may prevent misfolded protein formation. d Both the reduced systemic AAT and the presence of polymers are pro-inflammatory (shown as lightning strikes) and increase TNFα concentrations and bioavailability, as well as other pro-inflammatory cytokines and ROS. Specific anti-inflammatory therapies (e.g. targeting IL-8, leukotriene B4 or TNFα or their cogent receptors) may reduce this burden. e The inflammation leads to neutrophil activation, migration and degranulation, but proteinase and ROS release could be targeted using proteinase inhibitors and antioxidants. f Low AAT levels and increased proteinase (scissors) and ROS activity lead to tissue degradation, cleavage of immunoglobulins and inflammation, all of which are implicated in g the disease manifestations of AATD, which include (from figure top to bottom) liver disease, panniculitis, lung disease, but also chronic inflammatory comorbidities such as heart disease and diabetes. With aging, (1) genetic instability, telomere attrition, epigenetic modifications and loss of proteostasis (from left, clockwise) lead to (2) cell cycle arrest, immunosenescence, the release of the SASP and mitochondrial dysfunction (from left, clockwise). (3) This is proinflammatory (4), leading to neutrophil recruitment into tissues, proteinase release and oxidant damage, all implicated in disease. Current drugs to target aging processes, including those targeting telomeres and telomerase, reduce epigenetic modifications but also increase the removal of misfolded proteins, enhance autophagy, reduce the burden and function of immunosenescent cells and target inflammation, proteinases and ROS. AAT alpha-1 antitrypsin, AATD AAT deficiency, IL interleukin, PiZZ homozygous for Z allele, ROS reactive oxygen species, SASP senescence-associated secretory phenotype, TNF tumour necrosis factor
6 Current and Putative Treatments for AATD
There is great interest in expanding both the number of effective treatments in AATD and the ease of drug delivery for patients. Intravenous augmentation therapy has been the only specific treatment for AATD for decades. However, the current regimen only slows rather than prevents the progression of lung disease. For this reason, trials are now assessing better formulations, different doses and different routes of administration. Liver disease has been targeted with chemical chaperones to enhance the removal of mutated AAT from cells, RNA interference to prevent or reduce mutated AAT production, and viral vectors to enhance wild-type AAT production and secretion. As well as this, a number of studies are investigating repurposed drugs that have potential therapeutic effects in AATD. Table 1 provides a list of current AATD treatments either recently evaluated or under evaluation. Preclinical studies are also evaluating putative targets in lung disease, if not in AATD, and these are outlined in Table 2.
AAT augmentation therapy was first approved by the US FDA in 1987 for emphysema associated with severe AATD, but not all countries have licensed its use, including the UK, where augmentation therapy is only available for individual cases of panniculitis. Augmentation therapy modulates some manifestations of disease associated with a loss of AAT function, at least in some, but not all patients, including FEV1 decline and emphysema progression [142]. It is unclear whether augmentation therapy might affect other diseases associated with AAT function (with the exception of panniculitis, where the treatment is very effective), although the prevalence of CVD was lower in a population of patients with AATD receiving augmentation [143] and, as stated, AAT trials in diabetes are underway. However, there is significant redundancy in biological systems, and a loss of AAT may be mitigated via alternative cellular pathways, which could limit the impact or need for AAT augmentation in other putative AAT-related mechanisms of disease.
Although, classically, AAT augmentation is not thought to affect the progression of liver disease in AATD, some murine models suggest AAT can affect the progression of non-AATD-related liver failure [144]. Currently liver transplantation remains the only curative treatment for AATD-associated liver disease. While it can be successful, the limited availability of organs and the increasing burden of multimorbidity in elderly patients prevent widespread use. Therefore, therapies that reduce the burden of misfolded AAT in the liver are needed. One approach is to increase AAT transport from the ER with chemical chaperones. Phenylbutyrate has been used as a chaperone in several other diseases and increased AAT secretion in cell lines and animal models [145]; unfortunately, this did not translate to increased systemic AAT concentrations in AATD [146].
Harnessing autophagy (the process of orderly degradation and recycling of dysfunctional cellular components) is also of great interest in AATD-associated liver disease. Rapamycin and carbamazepine are medications used in different diseases but have been shown to affect autophagic pathways. Rapamycin is a mechanistic TOR (mTOR) inhibitor used for immunosuppression, but it has also been shown to enhance autophagy in murine models, with increased autophagic vacuoles, decreased AAT PiZ protein and improved hepatic fibrosis [147]. Carbamazepine (an anti-epileptic drug) has been shown to enhance the degradation of mutant AAT protein, through both autophagic and proteosomal mechanisms in murine models [148], and is currently in phase II/III clinical trials for patients, with trial reporting expected in 2020 (NCT01379469). The choleretic ursodeoxycholic acid (UDCA) shows similar promise.
There are also strategies to prevent the polymerisation of mutant AAT or increase the secretion of normal AAT in deficient patients through peptide, gene or mRNA therapy. The Z allele results from single base pair substitution, replacing lysine342 with glutamate, which induces the conformational change that allows unstable β-sheets to polymerise, generating insoluble aggregates. The underlying process of protein conformational change is amenable to targeting with monoclonal antibodies and small molecules, and to these ends drug development programmes have been established. Finally, stem cell and genetic therapies are also under development, but none to date have been approved in humans [149], although clinical trials are ongoing.
Currently, no therapies have been approved to address accelerated aging in general or specifically in AATD, but potential targets have been identified, and this is a fast-moving area for therapeutic discovery. Targets include the clearance of senescent cells, telomerase reactivation, epigenetic drugs and reducing both inflammation and ROS. A landmark study, recently published, suggested that transplanting senescent cells into young mice caused age-like physical dysfunction, and clearance of senescent cells with dasatinib plus quercetin improved physical function in mice and reduced pro-inflammatory cytokines in explanted human adipose tissue [150]. In oncology, there is interest in inhibiting telomerase to reduce the replicative burden of cancer cells, and concerns rose about increasing cancer burden if telomerase activity was enhanced. However, a recent murine study reported an increase in lifespan and healthspan when telomerase activity was enhanced, without increasing the cancer burden [151]. There are a high number of epigenetic targets to reduce accelerated aging, including SIRT1, calorific restriction to decrease mTOR signalling [152], and some repurposed drugs. These drugs include quetiapine [153] and especially simvastatin [154], which has been shown to improve age-associated neutrophil dysfunction [81] and is now being tested as a putative adjuvant therapy to enhance neutrophil functions in respiratory infections [155]. Whether any of these therapies enter clinical practice to help prevent an accelerated aging phenotype remains to be seen, but it is likely that our prescribing practices for many chronic diseases will substantially change over the next few decades.
7 Conclusion
Our understanding of the functions of AAT is evolving, and—with this—the complexity of AATD is increasingly evident. AATD reflects not only a pathological loss of function (reduced circulating AAT) but also a gain of function (the impact of mutant AAT accumulation and polymerisation, especially in hepatocytes) alongside a dysfunctional immune response and the presence of chronic inflammation, all within an aging host. These features vary between patients, and it is likely that other genetic and environmental factors will also influence AATD disease manifestation and progression. All in all, treating AATD is likely to be challenging. However, advancements in drugs and delivery systems are likely to provide us with multiple future options for treatment.
There is great interest in targeting biological processes associated with aging, but it remains unclear whether or how much accelerated aging contributes to the pathology seen in AATD. However, sound reasons exist for considering whether AATD is, in its manifestations, a disease of accelerated aging. Inflammation is a central feature of both AATD and accelerated aging. Pro-inflammatory signals are both a consequence of AATD and also drive AATD progression. These signals are a consequence of the primary hallmarks of aging and a cause of its development. Inflammation is a feature of many chronic non-communicable diseases such as CVD, T2DM and osteoporosis, all seen with increasing prevalence in AATD. Perhaps inflammation is the ‘one’ (from Emerson’s quote) that all diseases run into with age.
We have succeeded in reducing cigarette smoking in patients diagnosed with AATD, and augmentation therapy has slowed lung function decline for many patients and may delay the time to death [156]. Furthermore, trials of therapies to reduce the burden of misfolded mutant AAT protein are underway and offer hope for patients with AATD-associated liver disease. While this is undoubtedly progress, we still have much to learn about the pathophysiology of AATD. Understanding the impact of age and shared pathogenic mechanisms across age-associated diseases may provide new therapeutic strategies.
References
Eke PI, Thornton-Evans GO, Wei L, Borgnakke WS, Dye BA, Genco RJ. Periodontitis in US Adults: national Health and Nutrition Examination Survey 2009-2014. J Am Dent Assoc. 2018;149(7):576-88.e6.
Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Flaxman A, et al. The global burden of ischemic heart disease in 1990 and 2010: the Global Burden of Disease 2010 study. Circulation. 2014;129(14):1493–501.
Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, et al. Blue Journal Conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med. 2015;191(3):261–9.
Hobbins S, Chapple IL, Sapey E, Stockley RA. Is periodontitis a comorbidity of COPD or can associations be explained by shared risk factors/behaviors? Int J Chron Obstruct Pulmon Dis. 2017;12:1339–49.
Sevenoaks MJ, Stockley RA. Chronic Obstructive Pulmonary Disease, inflammation and co-morbidity–a common inflammatory phenotype? Respir Res. 2006;7(1):70.
Barnes PJ. Mechanisms of development of multimorbidity in the elderly. Eur Respir J. 2015;45(3):790.
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159(4):709–13.
Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, Grignolio A, et al. The continuum of aging and age-related diseases: common mechanisms but different rates. Front Med. 2018;5:61.
Kennedy BK, Pennypacker JK. Drugs that modulate aging: the promising yet difficult path ahead. Transl Res. 2014;163(5):456–65.
Hill AT, Bayley DL, Campbell EJ, Hill SL, Stockley RA. Airways inflammation in chronic bronchitis: the effects of smoking and alpha1-antitrypsin deficiency. Eur Respir J. 2000;15(5):886.
Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol. 2017;79(1):517–39.
Jacobsson KI. Studies on the determination of fibrinogen in human blood plasma. II. Studies on the trypsin and plasmin inhibitors in human blood serum. Scand J Clin Lab Invest. 1955;7:3–102.
Clemmensen SN, Jacobsen LC, Rørvig S, Askaa B, Christenson K, Iversen M, et al. Alpha-1-antitrypsin is produced by human neutrophil granulocytes and their precursors and liberated during granule exocytosis. Eur J Haematol. 2011;86(6):517–30.
Perlmutter DH, Cole FS, Kilbridge P, Rossing TH, Colten HR. Expression of the alpha 1-proteinase inhibitor gene in human monocytes and macrophages. Proc Natl Acad Sci USA. 1985;82(3):795–9.
Lou J, Triponez F, Oberholzer J, Wang H, Yu D, Buhler L, et al. Expression of alpha-1 proteinase inhibitor in human islet microvascular endothelial cells. Diabetes. 1999;48(9):1773.
Perlmutter DH, Daniels JD, Auerbach HS, De Schryver-Kecskemeti K, Winter HS, Alpers DH. The alpha 1-antitrypsin gene is expressed in a human intestinal epithelial cell line. J Biol Chem. 1989;264(16):9485–90.
Campbell EJ, Campbell MA, Boukedes SS, Owen CA. Quantum proteolysis by neutrophils; implications for pulmonary emphysema in alpha 1 anti trypsin deficiency. J Clin Invest. 1999;104:337–44.
Bucurenci N, Blake DR, Chidwick K, Winyard PG. Inhibition of neutrophil superoxide production by human plasma α1-antitrypsin. FEBS Lett. 1992;300(1):21–4.
Alfawaz B, Bergin DA, McElvaney NG, Reeves EP. Alpha-1 antitrypsin regulates neutrophil reactive oxygen species production via inhibition of key players of the respiratory burst oxidase system. BMC Proc. 2013;7(Suppl 1):P7.
Janciauskiene S, Tumpara S, Wiese M, Wrenger S, Vijayan V, Gueler F, et al. Alpha1-antitrypsin binds hemin and prevents oxidative activation of human neutrophils: putative pathophysiological significance. J Leukoc Biol. 2017;102(4):1127–41.
Lomas DA, Stone SR, Llewellyn-Jones C, Keogan MT, Wang ZM, Rubin H. The control of neutrophil chemotaxis by inhibitors of cathepsin G and chymotrypsin. J Biol Chem. 1996;270:23437–43.
Churg A, Wang X, Wang RD, Meixner SC, Pryzdial ELG, Wright JL. α1-antitrypsin suppresses TNF-α and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144–51.
Nita I, Hollander C, Westin U, Janciauskiene S-M. Prolastin, a pharmaceutical preparation of purified human alpha1-antitrypsin, blocks endotoxin-mediated cytokine release. Respir Res. 2005;6(1):12.
Cantin AM, Woods DE. Aerosolized prolastin suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Am J Respir Crit Care Med. 1999;160(4):1130–5.
Zhou X, Liu Z, Zhang J, Adelsberger JW, Yang J, Burton GF. Alpha-1-antitrypsin interacts with gp41 to block HIV-1 entry into CD4+ T lymphocytes. BMC Microbiol. 2016;16(1):172.
Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A, et al. alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA. 2008;105(42):16236–41.
Lu Y, Tang M, Wasserfall C, Kou Z, Campbell-Thompson M, Gardemann T, et al. α 1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum Gene Ther. 2006;17(6):625–34.
Zhang B, Lu Y, Campbell-Thompson M, Spencer T, Wasserfall C, Atkinson M, et al. α1-antitrypsin protects β-cells from apoptosis. Diabetes. 2007;56(5):1316.
Weir GC, Ehlers MR, Harris KM, Kanaparthi S, Long A, Phippard D, et al. Alpha-1 antitrypsin treatment of new-onset type 1 diabetes: an open-label, phase I clinical trial (RETAIN) to assess safety and pharmacokinetics. Pediatr Diab. 2018;19(5):945–54.
Gold M, Dolga AM, Koepke J, Mengel D, Culmsee C, Dodel R, et al. α1-antitrypsin modulates microglial-mediated neuroinflammation and protects microglial cells from amyloid-β-induced toxicity. J Neuroinflamm. 2014;11:165.
Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in α1-antitrypsin deficiency: molecular and cellular insights. Eur Respir J. 2009;34(2):475.
Parmar JS, Mahadeva R, Reed BJ, Farahi N, Cadwallader KA, Keogan MT, et al. Polymers of α1-antitrypsin are chemotactic for human neutrophils. Am J Respir Cell Mol Biol. 2002;26(6):723–30.
Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified alpha 1 antitrypsin standard to establish ranges for the common normal and deficient alpha 1 antitrypsin phenotypes. Chest. 1991;100(3):703–8.
Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294(24):1316–21.
O’Brien ML, Buist NRM, Murphey WH. Neonatal screening for alpha-1antitrypsin deficiency. J Pediatr. 1978;92(6):1006–10.
de Serres FJ. Worldwide racial and ethnic distribution of alpha 1 antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122(5):1818–29.
Blanco I, Bueno P, Diego I, Pérez-Holanda S, Casas-Maldonado F, Esquinas C, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chronic Obstr Pulm Dis. 2017;12:561–9.
Blanco I, Bueno P, Diego I, Pérez-Holanda S, Lara B, Casas-Maldonado F, et al. Alpha-1 antitrypsin Pi*SZ genotype: estimated prevalence and number of SZ subjects worldwide. Int J Chronic Obstr Pulm Dis. 2017;12:1683–94.
Luisetti M, Seersholm N. Alpha1-antitrypsin deficiency. 1: epidemiology of alpha1-antitrypsin deficiency. Thorax. 2004;59(2):164–9.
Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397–415.
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900.
Miravitlles M, Dirksen A, Ferrarotti I, Koblizek V, Lange P, Mahadeva R, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α1-antitrypsin deficiency. Eur Respir J. 2017;50(5):1700610.
Soriano JB, Lucas SJ, Jones R, Miravitlles M, Carter V, Small I, et al. Trends of testing for and diagnosis of alpha-1 antitrypsin deficiency in the UK: more testing is needed. Eur Respir J. 2018:1800360.
Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med. 1994;61:461–7.
Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of α-antitrypsin deficiency: a continuing problem. Chest. 2005;128(4):1989–94.
Laurell CB, Eriksson S. The electrophoretic α;1-globulin pattern of serum in α;1-antitrypsin deficiency. Scand J Clin Lab Invest. 1963;15(2):132–40.
Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in α1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med. 2004;170(11):1172–8.
Stockley RA, Edgar RG, Pillai A, Turner AM. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chronic Obstr Pulm Dis. 2016;11:1745–56.
Stockley RA, Miravitlles M, Vogelmeier C. Alpha One International R. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149.
Townsend SA, Edgar RG, Ellis PR, Kantas D, Newsome PN, Turner AM. Systematic review: the natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment Pharmacol Ther. 2018;47(7):877–85.
Townsend S, Newsome P, Turner AM. Presentation and prognosis of liver disease in alpha-1 antitrypsin deficiency. Exp Rev Gastroenterol Hepatol. 2018;12(8):745–7.
McElcaney NG, Stoller JK, Buist AS, Prakash UBS, Brantly ML, Schluchter MD, et al. Baseline characteristics of enrollees in the National Heart, Lung and blood institute registry of alpha 1-antitrypsin deficiency. Chest. 1997;111(2):394–403.
Parr DG, Guest PG, Reynolds JH, Dowson LJ, Stockley RA. Prevalence and impact of bronchiectasis in α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2007;176(12):1215–21.
Esnault VL, Testa A, Audrain M, Roge C, Hamidou M, Barrier JH, et al. Alpha 1-antitrypsin genetic polymorphism in ANCA-positive systemic vasculitis. Kidney Int. 1993;43:1329–32.
Segelmark M, Elzouk IAN, Wieslander J, Eriksson S. The PiZ gene of alpha 1-antitrypsin as a determinant of outcome in PR3-ANCA-positive vasculitis. Kidney Int. 1995;48:844–50.
Stone H, Pye A, Stockley RA. Disease associations in alpha-1-antitrypsin deficiency. Respir Med. 2014;108(2):338–43.
Duckers JM, Shale DJ, Stockley RA, Gale NS, Evans BAJ, Cockcroft JR, et al. Cardiovascular and musculskeletal co-morbidities in patients with alpha 1 antitrypsin deficiency. Respir Res. 2010;11(1):173.
Sandström CS, Ohlsson B, Melander O, Westin U, Mahadeva R, Janciauskiene S. An association between type 2 diabetes and α1-antitrypsin deficiency. Diabet Med. 2008;25(11):1370–3.
Greulich T, Nell C, Hohmann D, Grebe M, Janciauskiene S, Koczulla AR, et al. The prevalence of diagnosed α1-antitrypsin deficiency and its comorbidities: results from a large population-based database. Eur Respir J. 2017;49(1):1600154.
Smith SC Jr. Multiple risk factors for cardiovascular disease and diabetes mellitus. Am J Med. 2007;120(3):S3–11.
Strange C, Dickson R, Carter C, Carpenter MJ, Holladay B, Lundquist R, et al. Genetic testing for alpha 1 -antitrypsin deficiency. Genet Med. 2004;6(4):204–10.
Vestbo J, Søorensen T, Lange P, Brix A, Torre P, Viskum K. Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 1999;353(9167):1819–23.
Mittman C, Edelman NH, Norris AH, Shock NW. Relationship between chest wall and pulmonary compliance with age. J Appl Physiol. 1965;20:1211–6.
McClaran SR, Babcock MA, Pegelow DF, Reddan WG, Dempsey JA. Longitudinal effects of aging on lung function at rest and exercise in healthy active fit elderly adults. J Appl Physiol. 1995;78(5):1957–68.
Gillooly M, Lamb D. Airspace size in lungs of lifelong non-smokers: effect of age and sex. Thorax. 1993;48(1):39–43.
Enright PL, Kronmal RA, Higgins M, Schenker M, Haponik EF. Spirometry reference values for women and men 65 to 85 years of age: cardiovascular health study. Am Rev Respir Dis. 1993;147(1):125–33.
Stam H, Hrachovina V, Stijnen T, Versprille A. Diffusing capacity dependent on lung volume and age in normal subjects. J Appl Physiol. 1994;76(6):2356–63.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol Ser A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9.
Busse PJ, Zhang TF, Srivastava K, Schofield B, Li X-M. Effect of ageing on pulmonary inflammation, airway hyperresponsiveness and T and B cell responses in antigen-sensitized and -challenged mice. Clin Exp Allergy. 2007;37(9):1392–403.
Long D, Blake S, Song X-Y, Lark M, Loeser RF. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res Ther. 2008;10(1):R23.
Walker KA, Walston J, Gottesman RF, Kucharska-Newton A, Palta P, Windham BG. Midlife systemic inflammation is associated with frailty in later life: the ARIC study. J Gerontol A Biol Sci Med Sci. 2019;74(3):343–9. https://doi.org/10.1093/gerona/gly045.
Wenisch C, Patruta S, Daxbock F, Krause R, Horl W. Effect of age on human neutrophil function. J Leukoc Biol. 2000;67(1):40–5.
Fulop T, Larbi A, Douziech N, Fortin C, Guerard KP, Lesur O, et al. Signal transduction and functional changes in neutrophils with aging. Aging Cell. 2004;3(4):217–26.
Niwa Y, Kasama T, Miyachi Y, Kanoh T. Neutrophil chemotaxis, phagocytosis and parameters of reactive oxygen species in human aging: cross-sectional and longitudinal studies. Life Sci. 1989;44(22):1655–64.
Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123(2):239–48.
Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. In: Rommel C, Vanhaesebroeck B, Vogt PK, editors. Phosphoinositide 3-kinase in health and disease, vol. 1. Heidelberg: Springer; 2011. p. 183–202.
Butcher S, Chahel H, Lord JM. Ageing and the neutrophil: no appetite for killing? Immunology. 2000;100(4):411–6.
Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. 2001;70(6):881–6.
Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int. 2009;22(11):1041–50.
Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. 2014;13(4):690–8.
Sapey E, Patel JM, Greenwood HL, Walton GM, Hazeldine J, Sadhra C, et al. Pulmonary infections in the elderly lead to impaired neutrophil targeting, which is improved by simvastatin. Am J Respir Crit Care Med. 2017;196(10):1325–36.
Chatta GS, Andrews RG, Rodger E, Schrag M, Hammond WP, Dale DC. Hematopoietic progenitors and aging: alterations in granulocytic precursors and responsiveness to recombinant human G-CSF, GM-CSF, and IL-3. J Gerontol. 1993;48(5):M207–12.
Lord JM, Butcher S, Killampali V, Lascelles D, Salmon M. Neutrophil ageing and immunesenescence. Mech Ageing Dev. 2001;122(14):1521–35.
Fortin CF, Larbi A, Dupuis G, Lesur O, Fulop T Jr. GM-CSF activates the Jak/STAT pathway to rescue polymorphonuclear neutrophils from spontaneous apoptosis in young but not elderly individuals. Biogerontology. 2007;8(2):173–87.
Margolick JB, Ferrucci L. Accelerating aging research: how can we measure the rate of biologic aging? Exp Gerontol. 2015;64:78–80.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287.
Olovnikov AM. Telomeres, telomerase, and aging: origin of the theory. Exp Gerontol. 1996;31(4):443–8.
Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11(4):694–703.
Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev. 2011;10(2):205–15.
Zhang B, Fu D, Xu Q, Cong X, Wu C, Zhong X, et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat Commun. 2018;9(1):1723.
Fontana L, Partridge L, Longo VD. Extending healthy life span–from yeast to humans. Science (New York, NY). 2010;328(5976):321–6.
Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21(10):569–76.
Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2007;22(3):807–18.
Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, et al. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun. 2017;8:14063.
Stone H, McNab G, Wood AM, Stockley RA, Sapey E. Variability of sputum inflammatory mediators in COPD and alpha1-antitrypsin deficiency. Eur Respir J. 2012;40(3):561–9.
Bergin DA, Reeves EP, Meleady P, Henry M, McElvaney OJ, Carroll TP, et al. alpha-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120(12):4236–50.
O’Dwyer CA, O’Brien ME, Wormald MR, White MM, Banville N, Hurley K, et al. The BLT1 inhibitory function of α-1 antitrypsin augmentation therapy disrupts leukotriene B4 neutrophil signaling. J Immunol. 2015;195(8):3628.
Kirwan JP, Krishnan RK, Weaver JA, Del Aguila LF, Evans WJ. Human aging is associated with altered TNF-α production during hyperglycemia and hyperinsulinemia. Am J Physiol-Endocrinol Metabol. 2001;281(6):E1137–43.
Clark IA, Atwood CS. Is TNF a link between aging-related reproductive endocrine dyscrasia and Alzheimer’s disease? J Alzheimer’s Dis. 2011;27(4):691–9.
Bruunsgaard H, Skinhøj P, Pedersen AN, Schroll M, Pedersen BK. Ageing, tumour necrosis factor-alpha (TNF-alpha) and atherosclerosis. Clin Exp Immunol. 2000;121(2):255–60.
Lockett AD, Kimani S, Ddungu G, Wrenger S, Tuder RM, Janciauskiene SM, et al. α1-Antitrypsin modulates lung endothelial cell inflammatory responses to TNF-α. Am J Respir Cell Mol Biol. 2013;49(1):143–50.
Subramaniyam D, Virtala R, Pawłowski K, Clausen IG, Warkentin S, Stevens T, et al. TNF-α-induced self expression in human lung endothelial cells is inhibited by native and oxidized α1-antitrypsin. Int J Biochem Cell Biol. 2008;40(2):258–71.
Torres-Duran M, Lopez-Campos JL, Barrecheguren M, Miravitlles M, Martinez-Delgado B, Castillo S, et al. Alpha-1 antitrypsin deficiency: outstanding questions and future directions. Orphanet J Rare Dis. 2018;13(1):114.
Bergin DA, Reeves EP, Hurley K, Wolfe R, Jameel R, Fitzgerald S, et al. The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med. 2014;6(217):217ra1.
Maekawa T, Liu B, Nakai D, Yoshida K, Nakamura KI, Yasukawa M, et al. ATF7 mediates TNF-α–induced telomere shortening. Nucleic Acids Res. 2018;46(9):4487–504.
Eastman A, Potchen N, Carolan J, Malachowski A, Kryczek I, Kunkel S, et al. TNFα-induced epigenetic modifications support a DC1 program in dendritic cells during protective immunity to cryptococcal infection (MPF4P.732). J Immunol. 2015;194(1 Supplement):136.8.
Westbrook AM, Wei B, Hacke K, Xia M, Braun J, Schiestl RH. The role of tumour necrosis factor-α and tumour necrosis factor receptor signalling in inflammation-associated systemic genotoxicity. Mutagenesis. 2012;27(1):77–86.
Yan B, Wang H, Rabbani ZN, Zhao Y, Li W, Yuan Y, et al. Tumor necrosis factor-α is a potent endogenous mutagen that promotes cellular transformation. Can Res. 2006;66(24):11565.
Stanley SE, Merck SJ, Armanios M. Telomerase and the genetics of emphysema susceptibility. Implications for pathogenesis paradigms and patient care. Ann Am Thorac Soc. 2016;13(Suppl 5):S447–51.
Escribano A, Pastor S, Reula A, Castillo S, Vicente S, Sanz F, et al. Accelerated telomere attrition in children and teenagers with alpha1-antitrypsin deficiency. Eur Respir J. 2016;48(2):350–8.
Niemietz C, Fleischhauer L, Sandfort V, Guttmann S, Zibert A, Schmidt HHJ. Hepatocyte-like cells reveal novel role of SerpinA1 in transthyretin amyloidosis. J Cell Sci. 2018;131:jcs.219824. https://doi.org/10.1242/jcs.219824.
Willis-Martinez D, Richards HW, Timchenko NA, Medrano EE. Role of HDAC1 in senescence, aging, and cancer. Exp Gerontol. 2010;45(4):279–85.
Bouchecareilh M, Hutt DM, Szajner P, Flotte TR, Balch WE. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of α1-antitrypsin deficiency. J Biol Chem. 2012;287(45):38265–78.
Escribano A, Amor M, Pastor S, Castillo S, Sanz F, Codoñer-Franch P, et al. Decreased glutathione and low catalase activity contribute to oxidative stress in children with α-1 antitrypsin deficiency. Thorax. 2015;70(1):82.
Teckman JH, An J-K, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in α1-antitrypsin deficiency. Am J Physiol-Gastrointest Liver Physiol. 2004;286(5):G851–62.
Carter RI, Ungurs MJ, Mumford RA, Stockley RA. Aα-Val360: a marker of neutrophil elastase and COPD disease activity. Eur Respir J. 2012;41(1):31.
Walton GM, Belchamber KBR, Hughes SM, Barnes PJ, Stockley RA, Donnelly L, et al. Non-typeable haemophilus influenzae is associated with rapid lung function decline and poor macrophage and neutrophil phagocytosis in patients with alpha-1 anti-trypsin deficiency. D101 mechanistic and translational studies in COPD. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2017. p. A7363-A.
Sapey E, Stockley JA, Greenwood H, Ahmad A, Bayley D, Lord JM, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–86.
Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(8):861–70.
Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43(5):571–9.
Shi J, Yin N, Xuan LL, Yao CS, Meng AM, Hou Q. Vam3, a derivative of resveratrol, attenuates cigarette smoke-induced autophagy. Acta pharmacologica Sinica. 2012;33(7):888–96.
Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122(6):2032–45.
Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323(5917):1063–6.
Ohlmeier S, Nieminen P, Gao J, Kanerva T, Ronty M, Toljamo T, et al. Lung tissue proteomics identifies elevated transglutaminase 2 levels in stable chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1155–65.
Bernspång E, Wollmer P, Sveger T, Piitulainen E. Lung function in 30-year-old alpha-1-antitrypsin-deficient individuals. Respir Med. 2009;103(6):861–5.
Stockley JA, Stockley RA. Pulmonary physiology of chronic obstructive pulmonary disease, cystic fibrosis, and alpha-1 antitrypsin deficiency. Ann Am Thorac Soc. 2016;13(Suppl 2):S118–22.
Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in Alpha-1-Antitrypsin deficiency. Eur Respir J. 2009;1:1. https://doi.org/10.1183/09031936.00061208.
Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–92.
Hoare M, Das T, Alexander G. Ageing, telomeres, senescence, and liver injury. J Hepatol. 2010;53(5):950–61.
Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, et al. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol. 2013;58:549–56.
Nishijima T, Nakayama Y, Tsumura K, Yamashita N, Yoshimaru K, Ueda H, et al. Pulsatility of ascending aortic blood pressure waveform is associated with an increased risk of coronary heart disease. Am J Hypertens. 2001;14(5):469–73.
Sabit R, Bolton CE, Edwards PH, Pettit RJ, Evans WD, McEniery CM, et al. Arterial stiffness and osteoporosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(12):1259–65.
McAllister DA, Maclay JD, Mills NL, Mair G, Miller J, Anderson D, et al. Arterial stiffness is independently associated with emphysema severity in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176(12):1208–14.
Fisk M, Cheriyan J, Mohan D, McEniery CM, Forman J, Cockcroft JR, et al. Vascular inflammation and aortic stiffness: potential mechanisms of increased vascular risk in chronic obstructive pulmonary disease. Respir Res. 2018;19(1):100.
Sever PS, Poulter NR, Chang CL, Thom SA, Hughes AD, Welsh P, et al. Evaluation of C-reactive protein before and on-treatment as a predictor of benefit of atorvastatin: a cohort analysis from the Anglo-Scandinavian Cardiac Outcomes Trial lipid-lowering arm. J Am Coll Cardiol. 2013;62(8):717–29.
Ferrari R. The role of TNF in cardiovascular disease. Pharmacol Res. 1999;40(2):97–105.
Takahashi T, Hiasa Y, Ohara Y, Miyazaki S, Ogura R, Suzuki N, et al. Relationship of admission neutrophil count to microvascular injury, left ventricular dilation, and long-term outcome in patients treated with primary angioplasty for acute myocardial infarction. Circ J. 2008;72(6):867–72.
Akbar MA, Nardo D, Chen M-J, Elshikha AS, Ahamed R, Elsayed EM, et al. Alpha-1 antitrypsin inhibits RANKL-induced osteoclast formation and functions. Mol Med (Cambridge, Mass). 2017;23:57–69.
Doherty TJ. Invited review: aging and sarcopenia. J Appl Physiol (1985). 2003;95(4):1717–27.
Jarosch I, Gehlert S, Jacko D, Koczulla RA, Wencker M, Welte T, et al. Different training-induced skeletal muscle adaptations in COPD patients with and without alpha-1 antitrypsin deficiency. Respiration. 2016;92(5):339–47.
Edgar RG, Patel M, Bayliss S, Crossley D, Sapey E, Turner AM. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chronic Obst Pulm Dis. 2017;12:1295–308.
Fahndrich SW, Biertz F, Karch A, Kleibrink B, Koch A, Teschler H, et al. Comorbidity Patterns in Alpha-1-Antitrypsin Deficiency Depend on Natural History and Substitution Therapy. D103 alpha 1-antitrypsin deficiency: 50 years of progress. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2017. p. A7397-A.
Jedicke N, Struever N, Aggrawal N, Welte T, Manns MP, Malek NP, et al. Alpha-1-antitrypsin inhibits acute liver failure in mice. Hepatology. 2014;59(6):2299–308.
Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci USA. 2000;97(4):1796–801.
Fan J-Q, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal α–galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112.
Kaushal S, Annamali M, Blomenkamp K, Rudnick D, Halloran D, Brunt EM, et al. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med (Maywood, NJ). 2010;235(6):700–9.
Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229.
Connolly B, Isaacs C, Cheng L, Asrani KH, Subramanian RR. SERPINA1 mRNA as a treatment for alpha-1 antitrypsin deficiency. J Nucl Acids. 2018;2018:7.
Ivancich M, Schrank Z, Wojdyla L, Leviskas B, Kuckovic A, Sanjali A, et al. Treating cancer by targeting telomeres and telomerase. Antioxidants (Basel, Switzerland). 2017;6(1):15.
de Bernardes JB, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012;4(8):691–704.
Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166(4):822–39.
Lockwood LE, Youssef NA. Systematic review of epigenetic effects of pharmacological agents for bipolar disorders. Brain Sci. 2017;7(11):154.
Karlic H, Thaler R, Gerner C, Grunt T, Proestling K, Haider F, et al. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet. 2015;208(5):241–52.
Greenwood H, Patel J, Mahida R, Wang Q, Parekh D, Dancer RC, et al. Simvastatin to modify neutrophil function in older patients with septic pneumonia (SNOOPI): study protocol for a randomised placebo-controlled trial. Trials. 2014;15:332. https://doi.org/10.1186/1745-6215-15-332.
Rahaghi FF, Miravitlles M. Long-term clinical outcomes following treatment with alpha 1-proteinase inhibitor for COPD associated with alpha-1 antitrypsin deficiency: a look at the evidence. Respir Res. 2017;18(1):105.
Stocks JM, Brantly ML, Wang-Smith L, Campos MA, Chapman KR, Kueppers F, et al. Pharmacokinetic comparability of Prolastin(R)-C to Prolastin(R) in alpha(1)-antitrypsin deficiency: a randomized study. BMC Clin Pharmacol. 2010;10:13.
Campos MA, Kueppers F, Stocks JM, Strange C, Chen J, Griffin R, et al. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: a multicenter, randomized, double-blind, crossover study (SPARK). COPD. 2013;10(6):687–95.
Sorrells S, Camprubi S, Griffin R, Chen J, Ayguasanosa J. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir Med. 2015;109(4):490–9.
Seyama K, Nukiwa T, Sato T, Suzuki M, Konno S, Takahashi K, et al. Safety and pharmacokinetics of Alpha-1 MP (Prolastin((R))-C) in Japanese patients with alpha1-antitrypsin (AAT) deficiency. Respir Investig. 2019;57(1):89–96.
Sandhaus RA, Stocks J, Rouhani FN, Brantly M, Strauss P. Biochemical efficacy and safety of a new, ready-to-use, liquid alpha-1-proteinase inhibitor, GLASSIA (alpha1-proteinase inhibitor (human), intravenous). COPD. 2014;11(1):17–25.
Chapman KR, Burdon JG, Piitulainen E, Sandhaus RA, Seersholm N, Stocks JM, et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–8.
McElvaney NG, Burdon J, Holmes M, Glanville A, Wark PA, Thompson PJ, et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5(1):51–60.
Campos MA, Geraghty P, Holt G, Mendes E, Newby PR, Ma S, et al. The biological effects of double-dose alpha-1 antitrypsin augmentation therapy: a pilot study. Am J Respir Crit Care Med. 2019. https://doi.org/10.1164/rccm.201901-0010OC.
Barker AF, Campos MA, Brantly ML, Stocks JM, Sandhaus RA, Lee D, et al. Bioequivalence of a liquid formulation of alpha1-proteinase inhibitor compared with prolastin®-C (lyophilized alpha1-PI) in alpha1-antitrypsin deficiency. COPD. 2017;14(6):590–6.
Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antitrypsin in patients with alpha-1-antitrypsin deficiency: a preliminary study. J Pediatr Gastroenterol Nutr. 2004;39(1):34–7.
Turner AM, Stolk J, Bals R, Lickliter JD, Hamilton J, Christianson DR, et al. Hepatic-targeted RNA interference provides robust and persistent knockdown of alpha-1 antitrypsin levels in ZZ patients. J Hepatol. 2018;69(2):378–84.
Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing α1-antitrypsin: interim results. Hum Gene Ther. 2011;22(10):1239–47.
Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA. 2009;106(38):16363–8.
Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc. 2017;65(10):2297–301.
Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018;3(3):e93203. https://doi.org/10.1172/jci.insight.93203.
Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2010;25:341.
Füllgrabe J, Ghislat G, Cho D-H, Rubinsztein DC. Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci. 2016;129(16):3059.
Pastore N, Ballabio A, Brunetti-Pierri N. Autophagy master regulator TFEB induces clearance of toxic SERPINA1/alpha-1-antitrypsin polymers. Autophagy. 2013;9(7):1094–6.
Guo S, Booten SL, Aghajan M, Hung G, Zhao C, Blomenkamp K, et al. Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin-related liver disease in mice. J Clin Investig. 2014;124(1):251–61.
Li C, Xiao P, Gray SJ, Weinberg MS, Samulski RJ. Combination therapy utilizing shRNA knockdown and an optimized resistant transgene for rescue of diseases caused by misfolded proteins. Proc Natl Acad Sci USA. 2011;108(34):14258–63.
Mueller C, Tang Q, Gruntman A, Blomenkamp K, Teckman J, Song L, et al. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther. 2012;20(3):590–600.
Bjursell M, Porritt MJ, Ericson E, Taheri-Ghahfarokhi A, Clausen M, Magnusson L, et al. Therapeutic genome editing with CRISPR/Cas9 in a humanized mouse model ameliorates α1-antitrypsin deficiency phenotype. EBio Med. 2018;29:104–11.
Kaushal S, Annamali M, Blomenkamp K, Rudnick D, Halloran D, Brunt EM, et al. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med (Maywood). 2010;235(6):700–9.
Walton GM, Stockley JA, Griffiths D, Sadhra CS, Purvis T, Sapey E. Repurposing treatments to enhance innate immunity. Can statins improve neutrophil functions and clinical outcomes in COPD? J Clin Med. 2016;5(10):89. https://doi.org/10.3390/jcm5100089.
Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells. 2005;10(12):1113–25.
Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3(78):78ra32.
Patel JM, Thickett DR, Gao F, Sapey E. Statins for sepsis: distinguishing signal from the noise when designing clinical trials. Am J Respir Crit Care Med. 2013;188(7):874.
Balaguer C, Peralta A, Ríos Á, Iglesias A, Valera JL, Noguera A, et al. Effects of simvastatin in chronic obstructive pulmonary disease: results of a pilot, randomized, placebo-controlled clinical trial. Contemp Clin Trials Commun. 2016;2:91–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used in the preparation of this manuscript.
Conflict of interest
DC, ES and RAS have no conflicts of interest that are directly relevant to the content of this article.
Rights and permissions
About this article

Cite this article
Crossley, D., Stockley, R. & Sapey, E. Alpha-1 Antitrypsin Deficiency and Accelerated Aging: A New Model for an Old Disease?. Drugs Aging 36, 823–840 (2019). https://doi.org/10.1007/s40266-019-00684-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40266-019-00684-7