
Abstract
Tunlametinib (科露平®) is an oral, selective, mitogen-activated protein kinase kinase 1 and 2 (MEK 1/2) inhibitor being developed by Shanghai KeChow Pharma, Inc. for the treatment of solid tumours with RAS and RAF mutations, including melanoma, non-small cell cancer (NSCLC), colorectal cancer (CRC) and neurofibromatosis type 1 (NF1) plexiform neurofibromas. In March 2024, tunlametinib was granted conditional approval in China (based on surrogate endpoints) for use in patients with NRAS-mutated advanced melanoma who have failed anti-PD-1/PD-L1 treatment. This article summarizes the milestones in the development of tunlametinib leading to this first approval for the treatment of solid tumours with RAS and RAF mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.26103526. |
An oral selective MEK 1/2 inhibitor being developed by Shanghai KeChow Pharma, Inc for the treatment of solid tumours with RAS and RAF mutations, including melanoma, NSCLC, CRC and NF1 plexiform neurofibromas |
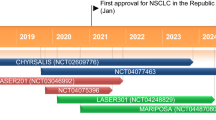
Received its first approval (conditional approval) on 15 March 2024 in China |
Approved for use in patients with NRAS-mutated advanced melanoma who have failed anti-PD-1/PD-L1 treatment |
1 Introduction
The mitogen-activated protein kinase (MAPK)/extracellular regulated protein kinases (ERK) signalling pathway (also known as the RAS/RAF/MEK/ERK signalling pathway) controls cell proliferation, survival and differentiation [1, 2]. Within the MAPK/ERK pathway, activated RAF phosphorylates and activates mitogen-activated protein kinase kinase 1 and 2 (MEK 1/2), resulting in downstream phosphorylation and activation of ERK1 and ERK2, which then triggers activation of targets associated with cell transcription, proliferation, differentiation and metabolism [1, 2]. Aberrant activation of the MAPK/ERK signalling pathway due to mutations in KRAS, NRAS and BRAF has been identified in > 20% of solid malignancies, including melanoma, non-small cell lung cancer (NSCLC), pancreatic cancer, and colorectal cancer (CRC) [3,4,5,6]; consequently, targeting the MAPK/ERK signalling pathway is a promising therapeutic approach [1, 2]. In the last decade, numerous MEK inhibitors have been investigated for use in tumours with RAS and RAF mutations, but poor exposure and drug toxicity issues have limited drug efficacy (mainly due to limited potency and/or an unfavourable pharmacokinetic profile) and few have been approved by regulatory agencies worldwide [1, 2]. Thus there is a need for MEK inhibitors that have good potency and improved efficacy and toxicity [2].

Tunlametinib (科露平®) is an oral, selective MEK1/2 inhibitor being developed by Shanghai KeChow Pharma, Inc. for the treatment of solid tumours with RAS and RAF mutations, including melanoma, non-small cell cancer (NSCLC), colorectal cancer (CRC) and neurofibromatosis type 1 (NF1) plexiform neurofibromas [7,8,9]. In March 2024, tunlametinib was granted conditional approval in China (based on surrogate endpoints) for use in adult patients with NRAS-mutated advanced melanoma who have failed anti-PD-1/PD-L1 treatment [7, 8]. Confirmation that the patient’s tumour sample carries NRAS mutations using a validated detection method is required before starting treatment with tunlametinib. An independent third-party designated by Shanghai KeChow Pharma, Inc should conduct an audit using the investigational companion diagnostic test to confirm that the patient is carrying the NRAS mutation and can continue to take the drug. The recommended dose of tunlametinib is 12 mg twice daily (approximately 12 h apart) with or without food. A missed dose can be taken provided if it is 8 h or more before the next dose; if the time to the next dose is < 8 h, the missed dose should not be taken [9]. Warnings and precautions related to tunlametinib treatment include decreased left ventricular ejection fraction (LVEF; baseline LEVF should be higher than the lower limit of normal and LVEF should be checked regularly during treatment), skin toxicity, ocular toxicity, interstitial lung disease, gastrointestinal reactions and increased blood creatine phosphokinase (CPK). Dose reductions, treatment interruption or treatment discontinuation may be required to manage decreased LVEF (including symptomatic congestive heart failure), ocular toxicity, pneumonia, rash, increased blood CPK and other grade 2–4 adverse reactions that are intolerable. Tunlametinib should be used with caution in patients with impaired liver function or with moderate/severe renal impairment. Pregnant women should not take tunlametinib and effective contraceptive measures should be taken by men and by women of childbearing potential during tunlametinib treatment and for 30 days after the last dose. Breastfeeding is not advised during tunlametinib treatment and within 30 days of the last dose [9].

2 Scientific Summary
2.1 Pharmacodynamics
Tunlametinib inhibits MEK 1/2 kinase activity, which blocks downstream ERK signalling pathways, inhibiting the ERK protein in tumour cells, inducing cell cycle arrest at the G0/G1 phase and inducing tumour cell apoptosis [9]. In cell-free enzyme assays, tunlametinib at 10 μmol/L showed a high selectivity for MEK1 (complete inhibition; no inhibition of numerous other kinases tested) and significant inhibitory activity (IC50 1.9 nM) against MEK1/MAP2K1(h) kinase [2]. Tunlametinib showed 18.5-fold greater inhibitory activity against MEK1/MAP2K1(h) kinase compared with binimetinib (IC50 12.1 vs 223.7 nM) [2]. In vitro, tunlametinib inhibited cell proliferation (IC50 0.67–59.89 nM) in RAS- or RAF-mutated melanoma, colon, lung cancer and myeloma cell lines, but had minimal effect (IC50 252.04 to > 1000 nM) on the proliferation of RAS-mutated LOX and RAF-mutated PANC-1 tumour cell lines, RAS/RAF wild-type tumour cell lines and normal cell lines [2, 9]. The inhibitory activity of tunlametinib in cell lines with RAS or RAF mutations was similar to that of trametinib (GSK212) and 10- to 100-fold greater than that of selumetinib (AZD6244) [2]. ERK phosphorylation levels of BRAF-mutated melanoma A375 cells decreased dose-dependently after treatment with tunlametinib 0.1–100 nmol/L and was almost completely blocked with tunlametinib 100 nmol/L. Tunlametinib dose-dependently increased the proportion of G0/G1 phase in A375 cells and induced apoptosis of COLO 205 cells in vitro [2]. In vivo, administration of tunlametinib 1-9 mg/kg once daily dose-dependently inhibited tumour growth and ERK phosphorylation in BRAF/KRAS-mutated melanoma and CRC or BRAF/KRAS wild type cell line-derived xenograft models, and tunlametinib 1 mg/kg once daily significantly (tumour growth inhibition > 79%; p < 0.05 vs vehicle) inhibited tumour growth in four BRAF-mutated CRC patient-derived xenograft models [2]. The combination of tunlametinib with BRAF, KRASG12C and SHP2 inhibitors or docetaxel showed a synergistic inhibitory effect on tumour cell proliferation in BRAF/KRAS-mutated melanoma, CRC or NSCLC cell lines in vitro and tumour growth in BRAF/KRAS-mutated cell line-derived xenograft models [2].
2.2 Pharmacokinetics
The pharmacokinetics of tunlametinib in patients with melanoma were generally dose-proportional after administration of single and multiple oral doses of tunlametinib 0.5–18 mg twice daily in a phase 1 trial (NCT03973151) [9, 10]. Tunlametinib was rapidly absorbed after oral administration, and peak concentrations were reached at a median 1 h. Steady state was achieved after 8 days’ administration of tunlametinib 12 mg twice daily; at steady state, Cmax was 122 ng/mL, Ctrough was 8.87 ng/mL and AUCτ was 360 ng·h/mL. The accumulation ratio was 1.66. Food has no clinically relevant effect on the absorption of tunlametinib [9]. Tunlametinib is highly bound (99.7%) to human plasma proteins. The mean Vd was ≈ 1940 L. Tunlametinib is primarily metabolized in the liver by CYP2C9 and to a lesser extent by CYP2C8 and CYP3A4. The mean CL/F was ≈ 51.9 L/h and the mean t1/2 was ≈ 26.2 h. Tunlametinib is excreted via faeces (59.6% of a total dose), urine (28.3%) and the lungs (2.8%) [9].
In vitro, tunlametinib is not an inhibitor of CYP3A4, CYP1A2, CYP2D6, CYP2C8 and CYP2B6. While tunlametinib is an inhibitor of CYP2C9 and CYP2C19, the effect is not clinically significant [9]. Tunlametinib is not a substrate of the transporters OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1 and MATE2-K, or of P-gp and BCRP in vitro and is not an inhibitor of OCT2 and MATE2-K. While tunlametinib is an inhibitor of OATP1B1, OATP1B3, OAT1, OAT3, MATE1, P-gp and BCRP in vitro, the effect is not clinically significant [9]. Clinical drug-drug interaction studies have not been formally conducted for tunlametinib; however, co-administration with inhibitors and inducers of CYP2C9 should be avoided [9].
2.3 Therapeutic Trials
Tunlametinib showed good efficacy in adult patients with advanced NRAS-mutated melanoma in a multicentre open label phase 2 trial (NCT05217303) conducted in China [11]. At data cut-off (19 February 2023; median follow-up 12.8 months), objective response rate (ORR) assessed by independent radiological review committee (IRRC) [primary endpoint] in patients receiving tunlametinib 12 mg twice daily (n = 95; full analysis set) was 35.8% [all partial response (PR)]. Median duration of response (DOR) was 6.1 months, median progression-free survival (PFS) was 4.2 months, disease control rate (DCR) was 72.6% and median overall survival (OS) was 13.7 months. In the subgroup of patients who had previously received anti-PD1/PD-L1 therapy (n = 64), ORR was 40.6%, median DOR was 6.3 months, median PFS was 4.1 months, DCR was 70.3% and median OS was 12.3 months [9, 11]. In patients who had not been treated with immunotherapy previously (n = 31), ORR was 25.8% [11]. Overall, tumor response and survival outcomes were sustained at a median follow-up of 24.4 months [12]. Eligible patients had histologically or cytologically confirmed unresectable stage III or IV melanoma, with documented NRAS mutations at baseline. In patients with central laboratory-confirmed NRAS mutations (n = 95), NRAS mutations were observed at Q61 (78.9%), G12 (15.8%) and G13 (5.3%). Median age at baseline was 58 years, most patients had stage IV melanoma (85.3%) and most (73.7%) had received prior systemic treatment (chemotherapy, immunotherapy, or both). Treatment was administered in 21-day cycles and continued until unacceptable toxicity, disease progression, consent withdrawal, death, or when clinical benefits were outweighed by the risks of treatment. If patients experienced treatment-related adverse events (TRAEs) requiring dose modification, dose reductions (one-level: 9 mg twice daily; two-level: 6 mg twice daily) were recommended. At data cut-off, 4 patients remained on treatment [11].
Features and properties of tunlametinib
Alternative names | 科露平®; HL-085 |
Class | Aniline compounds; antineoplastics; benzothiazoles; fluorobenzenes; iodobenzenes; small molecules |
Mechanism of action | MAP kinase kinase 1/2 inhibitor |
Route of administration | Oral |
Pharmacodynamics | High selectivity for MEK1 (complete inhibition); IC50 1.9 nM against MEK1/MAP2K1(h) kinase In vitro, ↓ tumour cell proliferation (IC50 0.67–59.89 nM), ↑ the proportion of G0/G1 phase, induced apoptosis in RAS- or RAF-mutated melanoma, colon, lung cancer and myeloma cell lines; minimal effect (IC50 > 1000 nM) on RAS or RAF wild-type tumour and normal cell lines ↓ ERK phosphorylation, tumour cell growth in vitro and in vivo Synergistic inhibition with BRAF, KRASG12C and SHP2 inhibitors or docetaxel on BRAF/KRAS-mutated tumour cell proliferation in vitro and in vivo |
Pharmacokinetics (steady state) | Mean Cmax 122 ng/mL, mean Ctrough 8.87 ng/mL, median Tmax 1 h, mean AUCτ 360 ng·h/mL, accumulation ratio 1.66, mean Vd ≈ 1940 L, mean CL/F ≈ 51.9 L/h, mean t1/2 ≈ 26.2 h |
Adverse events | |
Most frequent (any grade) | ↑ blood CPK, diarrhoea, facial oedema, peripheral oedema, increased AST, rash, ↑ blood lactate dehydrogenase, anaemia, ↑ ALT, asthenia, hypoalbuminemia, ↑ blood CPK MB, dermatitis acneiform |
Occasional (any grade) | left ventricular dysfunction, interstitial lung disease, gastrointestinal bleeding, ocular events |
Serious | Rash, hypoalbuminemia, pyrexia, peripheral oedema |
ATC codes | |
WHO ATC code | L01 (Antineoplastic Agents) |
EphMRA ATC code | L1 (Antineoplastics) |
Chemical name | 4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2- hydroxyethoxy)-1,3-benzothiazole-6-carboxamide |
In a phase 1 dose-escalation and dose expansion trial (NCT03973151) of tunlametinib in 42 adult patients with advanced NRAS-mutated melanoma conducted in China, PR was only seen in the tunlametinib 9, 12, 15 and 18 mg twice daily groups [13]. In the cohort of patients (n = 15) receiving tunlametinib 12 mg twice daily confirmed ORR was 26.7%, DCR was 86.7%, median DOR was 2.9 months and median PFS was 3.6 months patients. Eligible patients had histologically or cytologically confirmed unresectable stage III or IV melanoma, with documented NRAS mutations at baseline tested by centralized laboratories. Median age at baseline was 56 years and most patients had stage IV melanoma (n = 37). Tunlametinib 0.5–18 mg was administered twice daily in the dose-escalation phase and 12 mg twice daily in the dose-expansion phase [13].
In a phase 1 trial (NCT03781219) of tunlametinib plus vemurafenib in adult patients with advanced BRAF V600-mutated solid tumours conducted in China [14], at data cutoff (4 November 2022), ORR was 60.6%, median DOR was 11.3 months and median PFS was 11.7 months in the subgroup of patients with NSCLC (n = 33); in the subgroup of patients with mCRC (n = 24), ORR was 25.0%, median DOR was 5.5 months and median PFS was 6.2 months [14]. Eligible patients had progressed on or were intolerant to previous standard treatment. Patients were administered twice daily tunlametinib 0.5, 6, 9, 12 or 15 mg plus twice daily vemurafenib 960 mg in 21-day cycles in the dose-escalation phase, and twice daily tunlametinib 12 mg plus vemurafenib 960 mg, tunlametinib 12 mg plus vemurafenib 720 mg or tunlametinib 9 mg plus vemurafenib 720 mg in the dose-expansion phase [14].
Key clinical trials of tunlametinib (Shanghai KeChow Pharma, Inc.)
Drug(s) | Indication | Phase | Status | Location(s) | Identifier |
|---|---|---|---|---|---|
Tunlametinib, combination chemotherapy | Advanced NRAS-mutated melanoma | 3 | Enrolling | China | NCT06008106; HL-085-301 |
Tunlametinib | Advanced NRAS-mutated melanoma | 2 | Completed | China | NCT05217303; HL-085-101-II |
Tunlametinib, vemurafenib | Advanced BRAF V600E/K mutated melanoma | 2 | Ongoing | China | NCT05263453; HL-085-102-II-01 |
Tunlametinib | Advanced NRAS-mutated melanoma | 1/2 | Completed | China | NCT03973151; HL-085-101 |
Tunlametinib, vemurafenib | Metastatic colorectal cancer | 2 | Ongoing | China | NCT05233332, HL-085-201 |
Tunlametinib, vemurafenib | BRAF V600E-mutated metastatic colorectal cancer | 3 | Recruiting | China | NCT06008119; HL-085-304 |
Tunlametinib, vemurafenib | BRAF V600E-mutated non-small cell lung cancer | 2 | Recruiting | China | NCT05900219; HL-085-203 |
Tunlametinib | NF1 and inoperable plexiform neurofibromas | 2 | Ongoing | China | NCT05331105; HL-085-106-II |
Tunlametinib, vemurafenib | Advanced BRAF V600-mutated solid tumours | 1 | Ongoing | China | NCT03781219; HL-085-102 |
Tunlametinib | Advanced solid tumours | 1 | Completed | USA | NCT04683354; HL-085-US-102 |
2.4 Adverse Events
In the phase 2 trial (NCT05217303) of tunlametinib in adult patients with advanced NRAS-mutated melanoma, 100% of patients (n = 100) experienced a treatment-related adverse event (TRAE) of any grade; the incidence of TRAEs ≥ grade 3 was 68% [11, 13]. The most common adverse reactions (≥ 15%) included diarrhoea [74% (any grade); 4% (≥ grade 3)], facial oedema (64%; 0%), peripheral oedema (62%; 4%), rash (53%; 5%), anaemia (39%; 6%), asthenia (36%; 5%), hypoalbuminemia (34%; 0%), dermatitis acneiform (24%; 7%), vomiting (19%; 0%), hypokalaemia (19%; 4%), pruritus (18%; 0%), palmar-plantar erythrodysesthesia syndrome (17%; 0%), pyrexia (17%; 0%), hypocalcaemia (16%; 0%), increased weight (16%; 1%) and stomatitis (15%; 2%) [9]. The most common laboratory abnormalities (incidence ≥ 15%) that worsened from baseline included increased blood CPK (91%; 38%), increased AST (60%; 2%), increased blood lactate dehydrogenase (47%; 0%), increased ALT (40%; 0%), increased blood CPK MB (31%; 0%), increased γGT (16%; 0%) and increased blood myoglobin (16%; 0%) [9]. Serious TRAEs were reported in 26% of patients, the most common being rash (3%), hypoalbuminemia (3%), pyrexia (2%) and peripheral oedema (2%) [11]. TRAEs led to treatment interruption in 73% of patients, treatment interruption and dose reduction in 50% of patients, dose reduction in 37% of patients and treatment discontinuation in 9% of patients [9, 11].
In the phase 1 trial (NCT03781219) of tunlametinib plus vemurafenib in patients with advanced BRAF V600-mutated solid tumours, at cut-off date (n = 72 evaluable patients) the most common adverse events were CPK elevation, anaemia and rash [14].
2.5 Ongoing Clinical Trials
A phase 3 trial comparing tunlametinib with investigator-selected chemotherapy in patients with advanced NRAS-mutated melanoma who have previously received immunotherapy (NCT06008106) has commenced recruiting [15]. Other ongoing trials, all of which are being conducted in China, are: a phase 2 trial of tunlametinib plus vemurafenib in patients with advanced BRAF V600E/K-mutated melanoma (NCT05263453); a phase 2 trial of tunlametinib plus vemurafenib in patients with mCRC (NCT05233332); a phase 2 trial of tunlametinib in patients with NF1 and inoperable plexiform neurofibromas (NCT05331105); and a phase 1 trial of tunlametinib plus vemurafenib in patients with advanced BRAF V600-mutated solid tumours (NCT03781219). A phase 3 trial of tunlametinib plus vemurafenib in patients with BRAF V600E-mutated mCRC (NCT06008119) and a phase 2 trial of tunlametinib plus vemurafenib in patients with BRAF V600E-mutated NSCLC (NCT05900219) are recruiting patients.
References
Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules. 2017;22(10):1551.
Liu Y, Cheng Y, Huang G, et al. Preclinical characterization of tunlametinib, a novel, potent, and selective MEK inhibitor. Front Pharmacol. 2023;14:1271268.
Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res. 2020;80(14):2969–74.
Yaeger R, Corcoran RB. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 2019;9(3):329–41.
Garcia-Alvarez A, Ortiz C, Muñoz-Couselo E. Current perspectives and novel strategies of NRAS-mutant melanoma. Onco Targets Ther. 2021;14:3709–19.
Dean L. Vemurafenib therapy and BRAF and NRAS genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kattman BL, Malheiro AJ, editors. Medical genetics summaries. National Center for Biotechnology Information (US): Bethesda; 2012. p. 715–20.
National Medical Products Administration. State Food and Drug Administration conditional approval of tunlametinib capsules. 2024. https://www.nmpa.gov.cn/zhuanti/cxylqx/cxypxx/20240315082810127.html?type=pc&m. Accessed 12 Jun 2024.
Shanghai KeChow Pharma Inc. KeChow Pharma announces NMPA approval of tunlametinib (HL-085) as the first targeted therapy for patients with NRAS mutated advanced melanoma and previously treated with PD-1/PD-L1 [media release]. 15 Mar 2024. http://www.kechowpharma.com/.
Shanghai KeChow Pharma, Inc. Tunlametinib: Chinese prescribing information. 2024
Zhao Q, Wang T, Wang H, et al. Phase I pharmacokinetic study of an oral, small-molecule MEK inhibitor tunlametinib in patients with advanced NRAS mutant melanoma. Front Pharmacol. 2022;13:1039416.
Wei X, Zou Z, Zhang W, et al. A phase II study of efficacy and safety of the MEK inhibitor tunlametinib in patients with advanced NRAS-mutant melanoma. Eur J Cancer. 2024;202: 114008.
Wei X, Zou Z, Zhang W, et al. Overall survival and efficacy subgroup analysis of tunlametinib in patients with advanced NRAS-mutant melanoma: a multicenter, open-label, single-arm, phase 2 study. [abstract no. 9545 plus poster]. J Clin Oncol. 2024;42(Suppl 16)
Wang X, Luo Z, Chen J, et al. First-in-human phase I dose-escalation and dose-expansion trial of the selective MEK inhibitor HL-085 in patients with advanced melanoma harboring NRAS mutations. BMC Med. 2023. https://doi.org/10.1186/s12916-022-02669-7.
Shi YK, Zheng Y, Chen J, et al. Efficacy and safety of tunlametinib (HL-085) combined with vemurafenib in patients with advanced BRAF V600-mutated solid tumors: a multicenter, phase I study [abstract no. 1378P]. Ann Oncol. 2023;34(Suppl 2):790.
Wei X, Zhang J, Fang J, et al. A phase III, open-label, multicenter, randomized controlled trial of tunlametinib versus investigator-selected chemotherapy in patients with advanced NRAS-mutant melanoma who had previously received immunotherapy [abstract no. TPS9606]. J Clin Oncol. 2024;42:TPS9606.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Susan J. Keam is a contracted employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to this article and are responsible for its content.
Ethics Approval, Consent to Participate, Consent to Publish, Availability of Data and Material, Code Availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Keam, S.J. Tunlametinib: First Approval. Drugs 84, 1005–1010 (2024). https://doi.org/10.1007/s40265-024-02072-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-024-02072-x