
Abstract
The use of glucagon-like peptide-1 (GLP-1) receptor-based multi-agonists in the treatment of type 2 diabetes and obesity holds great promise for improving glycaemic control and weight management. Unimolecular dual and triple agonists targeting multiple gut hormone-related pathways are currently in clinical trials, with recent evidence supporting their efficacy. However, significant knowledge gaps remain regarding the biological mechanisms and potential adverse effects associated with these multi-target agents. The mechanisms underlying the therapeutic efficacy of GLP-1 receptor-based multi-agonists remain somewhat mysterious, and hidden threats may be associated with the use of gut hormone-based polyagonists. In this review, we provide a critical analysis of the benefits and risks associated with the use of these new drugs in the management of obesity and diabetes, while also exploring new potential applications of GLP-1-based pharmacology beyond the field of metabolic disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Metabolic diseases such as obesity and type 2 diabetes are growing global health challenges that require improved pharmacological solutions. |
A number of multi-agonist pharmacotherapies targeting different signalling pathways (glucagon like peptide 1 receptor [GLP-1R], glucose-dependent insulinotropic peptide [GIPR] and glucagon receptors [GCGR]) have shown promising preclinical and clinical efficacy as well as safety. |
Clinical data on chimeric peptide multi-agonists, such as GLP-1R/GIPR/GCGR, offer hope as a potent metabolic treatment strategy. However, the underlying biological mechanisms of their efficacy and potential hidden risks require further investigation. |
Beyond metabolic diseases, chimeric peptide multi-agonists based on GLP-1R agonism may hold promise for applications in improving bone health, addressing cognitive deficits and mitigating neurodegenerative diseases. |
1 Introduction
Just over 100 years ago, the life-saving discovery of insulin by Banting and colleagues [1] changed the lives of millions of people with type 1 diabetes (T1D). Today, we are witnessing a dramatic epidemic of obesity with increasing numbers of people affected by type 2 diabetes (T2D), and major discoveries since insulin are providing a new framework for long-term effective treatments.
Over the last century, the mechanisms of insulin secretion and action and new insights into the role of gastrointestinal (GI) hormones in body weight regulation have been largely elucidated, and the healing power of these hormones is now well accepted. However, challenges remain in achieving substantial weight loss and adequate glycaemic control in subjects with heterogeneous causes of disease onset and progression.
In the complex and urgent challenge posed by the obesity and metabolic disease epidemics, which require the development of pharmacological interventions with lifelong efficacy, the recent approval of tirzepatide for T2D represents a promising breakthrough. This peptide-based molecule, which targets both the glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) receptors, has attracted considerable attention for its multiple benefits in terms of weight loss, glycaemic control and improvement of the cardiometabolic profile, outperforming previous pharmacological treatments. The use of polyagonists based on pharmacological modulation of multiple gut hormone-related pathways, therefore, may have a transformative therapeutic impact on the global health threat of metabolic disorders [2,3,4,5,6,7,8]. However, the underlying mechanisms and potential limitations of this emerging pharmacological strategy are still under debate, and advances in this knowledge may pave the way for new therapeutic applications, such as obesity-related neurodegenerative diseases as well as several forms of addiction.
This review will provide an overview of the currently available treatment options for obesity and diabetes, with a particular focus on the often-mysterious mechanisms and hidden dangers associated with the use of gut hormone-based poly-agonists. We will offer an original view on the remaining challenges in preclinical research and pharmacological development of anti-obesity and anti-diabetes drugs, and provide a balanced analysis of whether and how emerging polyagonists may allow these challenges to be overcome.
2 Overview of Current Treatment Options for Obesity and Diabetes
Current treatment options for obesity and diabetes are varied. Lifestyle changes, including diet, physical activity and behavioural therapies to promote healthy habits, are often the first line of treatment and need to be implemented along with any additional therapeutic approach.
The modern environment of developed societies is full of obesogenic cues that drive maladaptive changes in eating behaviour towards energy-dense and palatable foods. Homeostatic biological mechanisms designed to maintain energy balance, i.e., the balance between energy intake and expenditure, are thrown out of control in response to these cues, resulting in exacerbated energy accumulation in the form of fat mass [9]. Maladaptive changes in energy balance may play a subtle but permissive role in the initial development of obesity in response to environmental cues and in conjunction with inherited genetic and epigenetic mechanisms. The interplay of biological and environmental cues is an even stronger driver of weight regain after initial weight loss [9]. Weight regain is often characterised by changes in biological responses favouring food intake and reduction in energy expenditure, undermining the efforts of individuals striving to maintain a healthy weight [10, 11]. The surge in appetite and food cravings especially, often observed following weight loss, presents a substantial challenge in the pursuit of long-term weight maintenance.
Metabolic surgery is a possible solution for maintaining a healthy body weight and metabolism in the long term. The surgical option is generally offered to people suffering from morbid obesity when, as it is often the case, lifestyle changes are not sufficient. The decision to have surgery is made on a case-by-case basis, considering the individual’s medical history, access to health care and psychological health.
Several procedures can be used to lose weight by surgically altering the digestive system, including gastric bypass, sleeve gastrectomy and adjustable gastric banding, which can help patients lose a significant amount of excess weight shortly after surgery (about 20% to 30% of initial body weight). Most patients maintain their relative weight loss for years after surgery, leading to improved long-term survival, a reduction in obesity-related comorbidities such as T2D and cardiovascular events like hypertension [12,13,14], and improved psychological health (increased self-confidence and self-esteem). A significant proportion of patients, estimated at 20–25%, have difficulty maintaining their weight over the long term after surgery, despite a significant improvement in their initial body weight and metabolic alterations [15,16,17]. In addition, some individuals experience a relapse of T2D within 5 years of surgery [18].
The mechanisms of weight loss induced by bariatric surgery are not yet fully understood. The entire architecture of the gut is altered and body weight loss may be due to mechanical changes in the GI tract resulting in reduced nutrient absorption and decreased appetite due to slower gastric emptying [19]. However, mechanical food restriction does not fully explain the sustained weight loss, as changes in the gut microbiome [20, 21], increased secretion of bile acids [22,23,24] and, most importantly, altered secretion of gut hormones [25, 26] also play a contributing role.
As with any surgical procedure, bariatric surgery can be costly depending on the health insurance system [27, 28] and also carries inherent risks, particularly in frail overweight individuals. For example, nutritional deficiencies can occur because the altered digestive system may not absorb nutrients as effectively, requiring lifelong supplementation with vitamins and minerals. Psychological support before and after surgery is as important as metabolic monitoring, as following strict dietary guidelines can be difficult, especially for those who are more prone to anxiety-related problems [29]. Finally, a key aspect of these operations is that they are largely irreversible.
Considering these inherent limitations, the risk-benefit ratio of metabolic surgery is certainly still positive, but an important question remains: is it possible to simulate the considerable benefits of surgery by pharmacological means and overcome the limitations mentioned above? Since the discovery of insulin [1] and leptin [30], a remarkable number of molecules have been tested in preclinical studies and clinical trials to improve the health of patients with T2D and obesity. Several pharmacological species have been developed over the past decades, generally with limited efficacy, high costs and in some cases significant side effects already reviewed [3, 4, 31,32,33]. Initially used for glycaemic control in diabetes, GLP-1 receptor (GLP1-R) agonists are now recognised as effective weight loss molecules. Targeting the GLP1-R is emerging as a necessary key component in achieving novel classes of molecules with the potential to induce benefits comparable to bariatric surgery, as will be discussed in the next session.
3 Past and Future of GLP1-R Mono-Agonists
3.1 Biology and Pharmacological Effects
The development of glucagon-like peptide-1 receptor (GLP-1R) agonists has flourished over the past 20 years, but why is there so much interest in targeting this peptide receptor?
Glucagon-like peptide-1 (GLP-1) is a 37-amino acid peptide hormone produced mainly (but not exclusively) by L-cells in the small intestine [34]. The peptide is cleaved from pre-proglucagon and processed by pro-convertase enzymes expressed not only in the gut, but also in the central nervous system (CNS) [35] and in the pancreas [36, 37]. This hormone is part of the incretin system, which regulates postprandial metabolism. By definition, incretin hormones are produced by enteroendocrine cells in response to nutrients in the gut lumen.
Meal-induced physiological GLP-1 secretion, as well as pharmacological administration of GLP-1R agonists, promote negative energy balance by reducing food intake [38,39,40,41], an effect that is maintained in patients with obesity [42]. Although the appetite-reducing efficacy of this pharmacological strategy is well-established, the impact on increasing energy expenditure remains less clear. Some animal studies report positive effects [43, 44], and others do not [45, 46]. The question of whether this class of molecule can promote weight loss by enhancing energy dissipation in humans remains to be definitively addressed.
Interestingly, GLP-1R agonists can also redirect food choice by altering food-related hedonic sensations in both rats [47] and humans [48,49,50]. The food-intake–related effects of GLP-1 may be mediated directly by neuronal circuits in the brain involved in feeding behaviour such as the hypothalamus and the Nucleus of Tractus Solitarii (NTS) [35, 38,39,40]. However, the systemic half-life of GLP-1 is quite short (about 2 minutes) due to rapid degradation by the enzyme dipeptidyl peptidase-4 (DDP4) [51] and how the peripherally produced hormone can reach distant GLP-1R-expressing tissues, such as the CNS, is still under debate.
Recent evidence suggests that endogenous GLP-1 biology may predominantly involve paracrine local effects from local synthesis of the hormone at the level of the gut [52] and the pancreas [36, 37]. Furthermore, the CNS effects of GLP-1 may depend on the local neuronal production of the hormone within the NTS [35, 53]. An important concept in unravelling the complex inter-organ circuits involved in the appetite-suppressing effects of GLP-1 [53] is the difference in pharmacokinetic properties between native GLP-1 and GLP1R agonists, which are resistant to DPP4-induced degradation and therefore have long-lasting activity and potentially differential brain penetration and distribution.
The prominent direct effects of GLP-1 biology on glucose metabolism constitute the key rational basis for the use of GLP-1R agonists for T2D treatment. Activation of the GLP-1R on pancreatic β-cells stimulates insulin synthesis in a glucose-dependent manner and β-cell survival in rodents, the latter effect being less clear in adult human β-cells [54, 55]. Reduced β-cell proliferation in humans can be explained by age-dependent signalling pathways [56].
Up to 70% of postprandial insulin secretion may be due to the incretin effect. Thus, any defect in the incretin axis leads to impaired insulin secretion, as seen in T2D [57].
At the level of the pancreatic islets, GLP-1-induced hormonal regulation involves a complex paracrine relationship between β-cells, α-cells and other islet endocrine cells [58, 59]. Besides, GLP-1–mediated improvements in glucose control can occur via inhibition of the production of the hyperglycaemic hormone glucagon [34]. Glucagon-like peptide-1 also lowers blood glucose in patients with T1D via its ability to slow down gastric emptying, indicating that not all of GLP-1 glycaemic effects are derived from its action on the β-cell or insulin action [60, 61].
Additional peripheral organs are implicated in GLP-1–mediated metabolic properties. Indeed, beside its anorexigenic action, GLP-1 reduces hepatic gluconeogenesis and steatosis, increases glucose uptake and lipolysis in white adipose tissue, and increases muscle insulin sensitivity, which are attractive properties for an anti-obesity drug. While these molecules have shown significant benefits on biochemical and histological markers of fatty liver and fibrosis in patients with non-alcoholic fatty liver disease (NAFLD), these outcomes are largely attributed to the impact of this class of compounds on weight loss and glycaemic control. Nevertheless, some studies have suggested potential direct effects on hepatocytes through local GLP-1R expression [62, 63]. However, the presence of these receptors in hepatocytes and their exact localisation in the liver remains a subject of debate [64].
The therapeutic potential of GLP-1 and its receptor were further revealed when exendin-4, a biologically active peptide isolated from the venom of the Gila monster lizard, was found to be resistant to DPP4 degradation [65]. This molecule formed the molecular basis of the first synthetic version of GLP-1 [66] developed and initially commercialized by Amylin Pharmaceutical, exenatide, which was approved as the first GLP-1R agonist for the treatment of T2D by subcutaneous (SC) injection [67]. Since then, several long-acting versions of GLP-1R agonists have been developed, either from the molecular structure of exendin-4 or from native GLP-1, as well as with chemical additions of linkers or fatty acid tails to increase half-life, making them more convenient for long-term use by patients (less frequent SC injections or oral route). Among these, dulaglutide, lixisenatide and liraglutide were all reported to cause absolute weight loss of between 2 and 6 kg at one year [32]. Liraglutide was the first GLP-1R agonist to be approved for the treatment of obesity in 2014, having been approved for the treatment of T2D four years earlier. While initial studies reported that T2D patients treated with 1.8 mg of SC liraglutide once daily lost 1.5 kg of body weight, people with obesity reported a maximum weight loss of almost 5.9 kg with an increased dose of 3 mg [68].
More recently (2017), the GLP-1R agonist semaglutide (Ozempic) was approved as a stand-alone treatment for T2D. The chemical structure of semaglutide differs from that of liraglutide, with a longer fatty acid chain and a spacer between the peptide and fatty acid. In addition, semaglutide is less susceptible to dipeptidyl peptidase IV (DPPIV) cleavage because the native alanine on the second residue has been replaced with an aminoisobutyric acid (AIB). Several clinical trials have shown that the molecule can induce significant weight loss in addition to glycaemic improvements, regardless of the route of administration [69], compared to other GLP-1R agonists. Ten randomised clinical trials from the SUSTAIN (for Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes) were initiated with the first primary endpoint of achieving better glycaemic control in T2D and safe use. Overall, T2D patients enrolled in SUSTAIN were overweight or obese with an initial body weight between 90 and 100 kg. Semaglutide (Ozempic) not only produced significantly greater reductions in HbA1c, but also up to three times greater weight loss (between 4 and 6 kg from baseline body weight) compared to any GLP-1R agonist or other antidiabetic drug tested before (insulin, metformin, SGLT2 inhibitors) [70,71,72,73,74,75,76,77,78]. The randomised, open-label, Phase 3b SUSTAIN7 study showed that weight loss with semaglutide 1.0 mg (SC) was − 5.8 to − 6.5 kg compared with − 3.0 kg with dulaglutide 1.5 mg [75] or − 1.9 kg with liraglutide [78]. In contrast to preclinical studies, semaglutide-induced weight loss in humans appears to be mainly mediated by reduced appetite and energy intake rather than increased energy expenditure. Semaglutide reduced energy intake by an average of approximately 25% over three meals [50], while liraglutide reduced energy intake by approximately 15% over one meal [79].
These exciting initial observations motivated the generation of several placebo-controlled trials (STEP trials) that definitively established the efficacy of semaglutide as a weight loss agent. To test the efficacy of semaglutide in patients with obesity, with or without T2D, the dose was increased to 2.4 mg, resulting in a significant reduction in body weight compared with placebo (− 9.6% to − 14.9% of body weight percentage change from baseline, compared to − 2.4 to − 3.4% difference with placebo), which had never been achieved previously with GLP-1R agonists [80, 81]. Nevertheless, treatment must last several months, if not years (with intensive behavioural interventions), to achieve the full benefits, as observed in the STEP4 and STEP5 trials [82, 83]. On the one hand, participants who stopped semaglutide after 20 weeks regained body weight, resulting in final overall weight loss of 5% after a total of 48 weeks, instead of 17.4% in those who continued to take semaglutide. On the other hand, long-term treatment with semaglutide 2.4 mg led to a substantial and sustained weight loss (− 15.2%) over two years compared to placebo. These exceptional results have led the US Food and Drug Administration (FDA) to approve semaglutide for the treatment of obesity with the Wegovy version in 2021.
It is important to remember that Ozempic and Wegovy are exactly the same molecule (semaglutide), but with different weekly SC doses for different diseases. While Ozempic maximum dose is 2 mg for patients with T2D, Wegovy maximum dose is 2.4 mg for patients with obesity (FDA access data reference ID: 5099892 Wegovy and reference ID: 5057152 Ozempic). This difference in dosing obviously affects the long-term bioavailability of the drug over time after the last injection. The superior weight loss efficacy of semaglutide compared to all other GLP-1R agonists tested may be closely related to modifications in the peptide backbone that ensures an optimal balance between a long plasma half-life and sufficiently high GLP-1R affinity in the presence of albumin [84]. The specific pharmacokinetic profile of semaglutide included a longer half-life (165 hours) compared to liraglutide injection (12–13 hours) [84,85,86].
Do other factors contribute to the superior efficacy of semaglutide despite its longer duration of action? In humans, liraglutide appears to have low brain penetrance, as suggested by the almost complete absence of liraglutide in the cerebrospinal fluid after 14 months of treatment with 1.8 mg of liraglutide [87]. Although both liraglutide and semaglutide rely on ependymal cells called tanycytes lining the third ventricle of the hypothalamus for brain penetration [88,89,90], semaglutide has been reported to have superior CNS penetration in rodents [90]. Further studies are needed to determine whether differences in the acyl moiety may explain the differential diffusion of the two compounds in the rodent brain.
Notably, GLP-1R agonists have also been considered as a therapeutic treatment for people with T1D who still have intact pancreatic β-cell mass. In a recent small case series, semaglutide (0.125 mg, weekly) allowed the elimination of pre-prandial insulin administration within three months in all enrolled patients [91]. This was associated with a reduction in glycated haemoglobin levels, and therefore better glycaemic control, in the year following initial diagnosis compared with a control cohort from four different studies receiving the same standard of care (education about T1D, insulin treatment with or without continuous glucose monitoring, and placebo). These preliminary human data merit further investigation, as the beneficial role of liraglutide in animal models of T1D has already been reported [92].
Besides the clear positive effects on glucose regulation and body weight control, GLP-1R agonists notably display additional favourable actions on cardiometabolic outcomes in both rodent and human preclinical studies [93, 94]. The SELECT trials showed that in a cohort of 17,500 patients with heart failure with preserved ejection fraction and obesity, those who received semaglutide (Wegovy 2.4 mg) had not only a greater weight loss compared to placebo, but also a 20% reduction in cardiovascular events and greater improvement in exercise function than those who received placebo [95,96,97]. Discontinuation of treatment with GLP-1R agonists was associated with an increased risk of major cardiovascular events, both in T2D patients with and without a history of cardiovascular events [98]. But what are the biological mechanisms underlying these effects on cardiovascular health? GLP-1R is present in atrial tissue in both humans and mice [99]. Studies in dogs [100], rats and more recently in diabetic db/db mice, which are known to develop cardiomyopathy, have shown that GLP-1R agonism has beneficial effects on the heart by reducing atrial fibrillation and reducing atrial remodelling [101].
Glucagon like peptide 1 receptor is expressed in cardiomyocytes as well as in endothelial and vascular smooth muscle cells. Therefore, the positive cardiovascular effects of GLP-1R agonists might be due to a direct action on GLP-1R-expressing endothelial cells [102, 103] and not to a reduction in body weight [104]. However, further elucidation of the precise effects of GLP-1R agonism on cardiovascular function (heart, blood vessels, kidney, etc.) in humans is still needed, which may help health care providers make informed treatment decisions and tailor therapy to individual patient needs.
The use of GLP-1R agonists has also been associated with improved baseline and postprandial lipid levels in numerous studies. A comprehensive meta-analysis of 35 trials confirmed that GLP-1R agonists can lead to reductions in LDL and total cholesterol, but did not show a significant increase in HDL cholesterol [105] by affecting hepatic lipid metabolism through mechanisms not yet elucidated [106].
3.2 Hidden Threats and Mysteries
Like any drugs, GLP-1R agonists can have side effects. Over the past decade, there have been some concerns about the potential long-term effects of using these treatments. Recently, concerns have been raised about potential psychiatric side effects reported with Ozempic (semaglutide), Saxenda (liraglutide) and Wegovy (semaglutide), and these are currently being investigated by the European Medicines Agency’s (EMA) Safety Committee [107]. More common adverse effects include pruritus and erythema at the injection site [108]. Moreover, semaglutide increases heart rate in rodents [99] and humans [109], as do all GLP-1R agonists. However, the beneficial effects of GLP-1R agonists on cardiovascular risk factors [74] and physiology outweigh the potential risk of the associated increase in heart rate [94].
As insulin secretagogues have been reported to be associated with acute pancreatitis and an increased risk of pancreatic cancer [110], there is concern that incretin-based drugs may contribute to pancreas-related complications over the long term. There have also been concerns about a possible increased risk of thyroid cancer [111]. Despite over two decades of GLP-1R agonist use, the evidence linking the use of these drugs to the development of thyroid tumours in humans is, at present, exceedingly limited and closely monitored [112]. It is important to note that conflicting results have emerged from preclinical investigations and epidemiological studies, adding to the complexity of the situation [113]. Glucagon like peptide 1 receptor activation induced proliferative effects in GLP-1R-expressing thyroid C-cells in rodents. However, it is less clear whether the human thyroid expresses the GLP-1R because of uncertainties about GLP-1R antibodies in histological analysis [113].
In addition, clinical data suggest rare cases of gallbladder disease, such as cholelithiasis, with some GLP-1R agonists [114], but this was not observed in the large cardiovascular outcomes study with extensive follow-up of these agents. Furthermore, recent meta-analyses show no significant effect on these outcomes despite a numerically higher incidence of pancreatitis [115, 116]. Yet, patients should be advised to report severe or persistent abdominal pain and despite the lack of proven causality, GLP-1R agonists are not recommended in individuals with a history of pancreatitis or pre-existing thyroid lesions. The likelihood of hypoglycaemia with GLP-1R agonists is low, except when these medications are combined with other treatments that lower blood glucose, such as insulin or sulfonylureas. In such cases, it is advisable to discontinue or reduce the dose of insulin or sulfonylurea when starting treatment with GLP-1R agonists.
The most common GI adverse events associated with GLP-1 receptor agonists are diarrhoea, mild nausea (25–60%) and vomiting (5–15%), which lessen over time [117].
A study from 2023 has revealed that the use of GLP-1R agonists for weight loss, when compared with the use of bupropion-naltrexone, is associated with an increased risk of pancreatitis, gastroparesis, and bowel obstruction [118]. However, there was no observed increase in the risk of biliary disease. The GI side effects make it difficult to increase doses, thus limiting their therapeutic impact. How to improve the GI tolerability of these agents remains an important puzzle, the resolution of which could significantly improve pharmacological compliance. One primary approach to mitigating GI side effects involves tailoring treatment through dose escalation. An emerging strategy involves also modifying drug delivery to specific tissues, particularly the gut and/or the CNS. Recent experimentation with a conjugated GLP-1R agonist, which exhibits reduced brain penetrance and does not induce emesis or anorexia [119], holds promise as a prospective solution. Alternatively, using GLP-1R agonists that target multiple metabolic pathways simultaneously holds the potential to achieve more comprehensive and substantial efficacy while addressing GI side effects.
Yet another mystery associated with the utilization of GLP-1R agonists revolves around the factors contributing to the relatively low treatment adherence. Clinical trials have revealed treatment discontinuation rates ranging from 5% to 15% [120]. The most common reasons for discontinuation cited by physicians include inadequate blood glucose control, as well as nausea/vomiting and GI side effects [120]. In contrast to these trial results, real-world analyses of adults with T2D starting treatment with GLP-1R agonists have shown even worse adherence [121]. A 2023 analysis of a Danish nationwide registry found that approximately half of first-time users of this class of pharmacological agents discontinued therapy within a span of five years [122]. Given the compelling evidence for the cardioprotective and reno-protective benefits of GLP-1R agonists, further insight into the reasons for discontinuation and the development of preventive initiatives, particularly for patients recently discharged from hospital, are warranted.
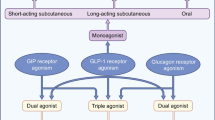
4 It Takes Two to Tango… or More? Polyagonists of GLP-1R and Other Receptors
With the exception of GLP-1R agonists, single hormone pharmacotherapies for the treatment of obesity have limited efficacy, typically resulting in moderate or no weight loss compared with placebo treatments. To improve outcomes, a combination of drugs at tolerable doses has been proposed (developed in Sect. 2.3), aiming for a synergistic improvement in metabolism beyond what each hormone can achieve individually. Given that metabolic surgery is associated with the reorientation of the levels of multiple hormones, it is reasonable to assume that the combined targeting of multiple hormonal pathways has the potential to mimic the substantial pharmacological effects of this procedure. However, co-injected agonists of different hormone receptors may differ in their half-life and duration of action due to differences in rates of absorption, distribution, metabolism and clearance. In addition, injecting multiple pharmacological species requires full clinical development of each molecular component, dramatically increasing production costs and delaying translation from bench to bedside [7, 8].
Combinations of several molecules in one single molecule have therefore been developed to potentially overcome these challenges. In an original version, these GLP-1-based molecules were created as peptide chimera, also known as co-agonists, in which the key biological components of each peptide hormone agonist (namely glucagon- and GIP-receptor agonists, abbreviated GCGR and GIPR, respectively) were fused into a unique chimera of similar chemical size to each individual agonist, as detailed below.
4.1 GLP-1R-GCGR Co-Agonists
Glucagon has long been restricted to its counterregulatory role in maintaining plasma glucose concentrations during fasting, exercise and hypoglycaemia by increasing hepatic glucose output to the circulation. The hormone can affect glucose homeostasis by promoting hepatic conversion of glycogen to glucose (glycogenolysis), stimulating de novo glucose synthesis (gluconeogenesis) and inhibiting glucose breakdown (glycolysis) and glycogen formation (glycogenesis). The rapid counterregulatory effects of this hormone under fasting or hypoglycaemic conditions mainly involve hepatic glycogenolysis and thus rapid hormone-controlled access to latent glucose stored in the liver via modulation of locally expressed GCGR [58, 123]. The GCGR agonists, therefore, were first developed to address insulin-induced iatrogenic hypoglycaemia during T1D. In this specific case, GCGR agonists seem appropriate because T1D is often linked with counter-regulatory failure of endogenous glucagon secretion in response to hypoglycaemia.
Because of its hyperglycaemic effect in response to fasting or hypoglycaemia, the concept of using GCGR activation as a therapeutic approach for patients with hyperglycaemic T2D initially seemed implausible. The opposite strategy of GCGR antagonism has been explored in both preclinical research and clinical trials for diabetes management. This approach has shown positive results in glycaemic regulation, although it is not without safety concerns, particularly with regard to alterations in hepatic lipid metabolism [124].
However, fasting hyperglucagonaemia in T2D, may be both a pathophysiological feature of glucose metabolism and a beneficial metabolic adaptation in hepatic lipid and amino acid metabolism [58, 125]. This second hypothesis has laid the foundation for exploring whether the activation of GCGR signalling might yield therapeutic advantages for metabolic diseases.
For instance, both native glucagon and GCGR agonists can limit hepatic steatosis, have a synergistic effect with GLP-1 on insulin secretion, a potential synergistic effect on reduced food intake [126] and increased energy expenditure [127,128,129,130].
Notably, glucagon also has insulinotropic actions: after binding to GCGR on β-cells, it increases insulin secretion and lowers glycaemia in fed postprandial conditions, i.e., only when β-cells are normally active [131]. This important observation must be put into perspective with the studies suggesting that α-cellderived glucagon may serve as an additional, albeit less potent, ligand for the β-cell GLP-1R [132], thereby extending the role of α-cells beyond that of a counterregulatory cell type and making them an ideal partner for GLP-1.
Given that GLP-1R agonism-mediated effects appear to be largely independent of energy expenditure, combined GCGR co-activation has the potential to extend the beneficial effects of GLP-1R pharmacology. So, from a mechanistic point of view, the biological mechanisms underlying the efficacy of GCGR agonism deserve further investigation, as the mechanisms of glucagon-induced increased energy expenditure [127,128,129,130] and the anorectic effect of glucagon [133,134,135] are not yet fully understood, but may occur in part via a liver-CNS axis [135] and also directly via the GCGR-expressing hypothalamic area [136].
The first GCGR agonists induced a negative energy balance by both promoting weight loss, partly indirectly in mice by stimulating hepatic Fibroblast Growth Factor 21 (FGF21) secretion (hormone will be described in paragraph 5.1.) and central FGF21 receptor actions [130, 137]; and increasing energy expenditure by increasing basal metabolic rate and inducing thermogenesis in brown adipose tissue (BAT), as shown in humans and in animal models [138,139,140]. However, it should be noted that the role of BAT thermogenesis in glucagon-mediated effects is still controversial [141].
The main challenge in the development of polyagonists targeting the GCGR is to balance the beneficial effects of glucagon on body weight and lipid metabolism with the potential hyperglycaemic effects of this hormone. The first GLP-1R-GCGR co-agonist used in rodents resulted in significant weight loss and adiposity reduction due to reduced food intake and increased energy expenditure [142].
Human co-infusion studies have confirmed the promise of the combined effects of GLP-1 and glucagon on both energy balance and glucose homeostasis [126, 143]. Cotadutide, the first GLP-1R-GCGR co-agonist in clinical development by Astra-Zeneca, improved glycaemic control and weight loss in participants with overweight/obesity and T2D over 54 weeks of treatment [144]. Ad hoc analyses showed improvements in liver parameters and support further evaluation of cotadutide in NASH in non-cirrhotic NASH patients with fibrosis (PROXYMO-ADV; NCT05364931). Mazdutide (also known as IBI362 or LY3305677), a novel once-weekly GLP-1R-GCGR co-agonist [145, 146], achieved 24-week weight loss of up to − 15.4% at 9 mg in overweight and obese patients (NCT04904913 and NCT05628311). Notably, a recent Phase 1 study evaluating the effects of the GLP-1R-GCGR co-agonist NNC9204-1177 (NN1177) in overweight or obese adults showed several unexpected treatment-related safety signals, including increased heart rate, decreased reticulocyte count, increased markers of inflammation, increased aspartate and alanine aminotransferase, impaired glucose tolerance and decreased blood levels of some amino acids, leading to a decision to discontinue clinical development [147].
Thus, while the potential to enhance the weight loss efficacy of the established GLP-1R agonist class through GCGR co-agonism is promising, recent evidence underscores the importance of ensuring the safety of these hybrid molecules. The key to optimising such co-targeting strategy may lie in understanding the implications of the relative pharmacological activity at each receptor. The ideal balance may also depend on the specific indication, be it weight loss or glycaemic control [148]. For example, to attenuate hepatic glucose production stimulated by GCGR agonism, it may be necessary to maintain a sufficiently high level of GLP-1 receptor agonism. Alternatively, given that the glycaemic benefits of glucagon have been observed in the fed state but not in the fasting state [131], further investigation of mealtime dosing schedules may provide valuable insights into optimising co-agonists for weight management and related therapeutic outcomes.
4.2 GLP-1R-GIPR Co-Agonists
Glucose-dependent insulinotropic polypeptide (GIP) is a 42-amino acid peptide first described in the early 1970s [149]. This hormone is released by K-cells at the level of the duodenum and the upper jejunum in response to food intake (anabolic conditions), particularly carbohydrates and fats [150]. As an incretin hormone, along with GLP-1, one of the main functions of GIP is to allow better handling of glucose after a meal. Glucose-dependent insulinotropic polypeptide also enhances lipogenesis and insulin sensitivity in adipose tissue, which is mainly observed in in vitro studies using the native hormone and GIPR inhibitors [151, 152]. The question remains whether GIP-induced glucose and lipid uptake is mediated directly or indirectly via insulin secretion in vivo. Whether GIP has CNS-mediated effects that promote a reduction in food intake remains controversial. Indeed, the hypophagic effects of the native hormone seem to be more pronounced in animal studies than in humans [153], for reasons that are still unknown [154, 155], because GIPR mRNA has been reported to be expressed, in both species, in several hypothalamic areas involved in the regulation of feeding behaviour [154].
Like GLP-1, the secretion of GIP is glucose dependent: these incretin hormones are relatively benign drug candidates because they limit the risk of hypoglycaemia sometimes seen with other antidiabetic drugs. However, patients with T2D have a reduced responsiveness to GIP [156], contributing to impaired glucose regulation, which initially limited preclinical studies of this hormone although more recent results with GLP-1R and GIPR co-agonism suggest that GIP enhances the beneficial effects of GLP-1. The attenuated insulinotropic effect of GIP in diabetes, together with the impaired incretin effect, is a reversible phenomenon. Short-term treatment with dexamethasone, which induces hepatic insulin resistance, not only leads to increased GIP concentrations but also to a transient impairment of the insulinotropic effect of GIP [157]. Conversely, treatment with sulfonylurea, xenin-25 (a neurotensin-related peptide secreted by intestinal K-cells) or sitagliptin (a DPP-4 inhibitor) has been shown to increase the insulin response to GIP in patients with impaired glucose tolerance [157], suggesting that functional GIP receptors are present on the β-cells of T2D patients. In support of this latter view, a recent study using a physiological mathematical model of the β-cell, which can effectively capture heterogeneous clinical protocols under a standardised paradigm, has shown that incretin-mediated enhancement of insulin secretion is reduced in T2D [158]. Importantly, this reduction is not absolute and the lack of effect of pharmacological doses of GIP is attributed to saturation of the GIP effect rather than insensitivity to the hormone [158].
Tirzepatide is a GIP and GLP-1 receptor co-agonist that shares amino acid sequences with both GLP-1 and GIP [2, 159]. A spectacular example of successful co-agonist generation was the recent FDA approval of the GLP-1R-GIPR co-agonist tirzepatide (Mounjaro) for the treatment of T2D, based on data from nine clinical trials. The SURPASS programme included several clinical trials evaluating the safety and efficacy of tirzepatide in patients with T2D. These trials were designed to assess various aspects of the molecule on glycaemic control, weight loss and cardiovascular outcomes. Tirzepatide was tested at three different weekly doses against various anti-diabetic agents (semaglutide, insulin degludec, insulin glargine) for 40 to 104 weeks in patients with diabetes who had persistently elevated HbA1C levels despite standard therapy with insulin and/or additional medications (metformin or SGLT2 inhibitors) [160,161,162,163,164]. It should be noted that obesity was prevalent in the population studied (BMI>30 kg/m2 at enrolment). Tirzepatide treatment consistently resulted in significantly greater HbA1c and weight reductions (up to − 10.9 kg compared to placebo at the 15-mg dose in the most recently published SURPASS 5 [164]) with − 9.1% relative weight loss. The significant weight loss observed in patients treated with tirzepatide compared to placebo led to the investigation of tirzepatide as a potential treatment for obesity in clinical trials (SURMOUNT). The Phase 3, double-blind, randomised, placebo-controlled SURMOUNT-1 and 2 studies (NCT04184622, NCT04657003) showed impressive weight loss in obese patients, with the highest dose of tirzepatide achieving − 20.9% and − 14.7% weight loss, respectively, making it the most effective drug so far generated against body weight loss [165] (Lilly press release https://investor.lilly.com/news-releases/news-release-details/lillys-tirzepatide-achieved-157-weight-loss-adults-obesity-or).
4.3 GLP-1R-GIPR-GCGR Tri-Agonists
The previous experience with chimeric co-agonists provided the basis for the development of long-acting chimeric unimolecular GLP-1 tri-agonists, which combine the beneficial effects of GLP-1, GIP and glucagon, with the idea of achieving even greater effects on weight loss and gluco-metabolic improvements.
The first GLP-1R-GIPR-GCGR tri-agonist dose-dependently reduced body weight by up to 25% over ∼4 weeks in diet-induced obese mice, outperforming previous reports for mono-agonists and receptor-balanced GLP-1R-GIPR co-agonists [166,167,168], but the clinical validation of these relevant preclinical findings remains to be confirmed.
SAR441255 is a unimolecular GLP-1R/GIPR/GCGR tri-agonist for once-daily subcutaneous injection in humans. In preclinical studies in diet-induced obese monkeys, SAR441255 reduced body weight by 12% after 7 weeks, confirming efficacy in advanced mammalian species. Following single doses in lean healthy humans, SAR441255 showed positive acute glucoregulatory effects during a mixed meal tolerance test and an acceptable safety profile in terms of GI tolerability and cardiovascular haemodynamics [169] (NCT04521738). While these initial human data in a relatively small number of healthy subjects are promising, further trials in larger populations of people suffering from overweight with T2D and obesity are required to confirm the therapeutic potential of GLP-1R-GIPR-GCGR co-targeting in comparison with existing selective GLP-1R, dual GLP-1R-GCGR, and dual GLP-1R-GIPR agonists.
Recently, another tri-agonist, retatrutide (LY3437943) was shown to reduce body weight and improve glycaemic control in obese mice. Body weight loss was due to the GCGR-mediated component of the molecule leading to increased energy expenditure, and by the superior hypophagic effect achieved with combined GIPR and GLP-1R activation compared to monotherapy [170]. In a Phase 2 clinical trial involving non-diabetic adult patients with a BMI of ≥30, or a BMI of 27 <30 plus at least one weight-related condition, body weight loss of − 24.2% was observed at the highest dose group (12 mg) compared to − 2.1% in the placebo group following treatment with LY3437943. Among the patient population in clinical trials, heterogeneity of responses is always observed, but the authors reported that up to 83% of patients (12 mg dose) achieved a 15% weight loss after 48 weeks [171], with tolerable reported GI side effects.
Finally, other beneficial improvements in obesity comorbidity factors, such as a reduction in non-alcoholic steatohepatitis (NASH), have been reported with HM15211, another tri-agonist developed by Hanmi Pharmaceuticals. These effects may be based on the hepatic actions of glucagon (ADA abstract Choi IY 2017). Results from the randomised, double-blind, placebo-controlled Phase 2 clinical trial are expected by the end of 2025 [172].
4.4 Hidden Threats and Mysteries
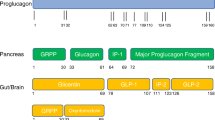
Although several biological and pharmacological mechanisms of gut-based hormone peptides for the treatment of metabolic diseases have emerged (Fig. 1), many mysteries still remain. An intriguing puzzle persists as researchers grapple with the diverse outcomes observed when modulating the activity of GIPR through various means, including pharmacological agents and transgenic models. The challenge ahead is to reconcile and make sense of the following perplexing observations.

Summary of the biological actions of glucose-dependent insulinotropic peptide (GIP), glucagon-like peptide 1 (GLP-1) and glucagon. Figure realized with Biorender
High-fat diet (HFD)-fed mice and non-human primates treated with GIPR antagonists, alone or in combination with GLP1RAs, showed resistance to weight gain and improvement in the glycaemic profile [173,174,175,176]. Similarly, GIPR knock-out mice are resistant to diet-induced obesity and protected from insulin resistance [177]. A recent study examining the phenotype of animals with CNS 'specific' ablation of GIPR via CRE-mediated recombination under the nestin promoter clearly demonstrates that CNS GIPR signalling plays an important role in the regulation of energy metabolism [155], although these data do not completely exclude some degree of off-target ablation of the receptor in certain cells/tissues outside the CNS that express low levels of the nestin gene, such as adipose tissue [155].
Based on these preclinical observations suggesting a protective role of GIPR inhibition in obesity, a humanised GIPR antagonist antibody linked to a GLP-1R agonist (AMG-133) was developed with the idea of providing beneficial metabolic effects. In a Phase 1 study, treatment with AMG-133 resulted in mean weight loss between 7.2% and 14.5% after 12 weeks in people with obesity (without diabetes) [178]. The main side effects were nausea and vomiting, which resolved after 48 h – a Phase 2 trial with this molecule is ongoing (NCT05669599). Of note, GIPR knock-out mice have higher blood glucose levels and impaired initial insulin response after oral glucose challenge [179], and as mentioned above, GIPR activation together with GLP-1R modulation clearly provides additional benefits on metabolism and body weight control. Thus, how can potentially beneficial effects of both GIPR agonism and GIPR antagonism be reconciled? This inconsistency in GIPR biology raises the question of whether constitutive deletion of incretin hormone receptors in transgenic animal models introduces bias and hinders translation or prediction of responses to pharmacological modulation of these receptors. For example, while it is widely accepted that GLP-1R agonism has anti-obesity and anti-diabetic effects, a puzzling inconsistency arises when evaluating the phenotype of GLP-1R-deficient transgenic animals, as these animals are protected from high-fat diet-induced weight gain and insulin resistance [180]. Thus, only limited interpretations can be drawn from findings in mouse models of whole-body genetic deletion of the incretin receptor from early embryonic life. Nevertheless, several alternative hypotheses could explain the lack of consensus on the physiological functions of the GIPR and, consequently, the mode of action of tirzepatide. Drawing from in vitro findings, tirzepatide may act as an unbalanced and biased agonist towards GLP-1R and GIPR. This implies that tirzepatide induces a signalling bias towards the cAMP pathway versus β-arrestin recruitment following GLP-1R binding. Reduced β-arrestin signalling limits GLP-1R internalization, whereas cAMP signalling increases insulin secretion compared to native GLP-1 [159], both of which may explain the observed efficacy of tirzepatide in clinical trials.
In addition, chronic GIPR agonism may desensitise GIPR activity in different tissues, as recently observed in primary adipocytes in vitro and adipose tissue in vivo [181], raising the possibility that effective strategies to promote weight loss and metabolic improvement should target optimal GIPR inhibition rather than activation. This consideration raises the question of whether GIPR antagonism, when combined with GLP-1R and GCGR co-activation, could offer a promising therapeutic pathway. However, there is conflicting information regarding the potential benefits of pharmacological GIPR inhibition in combination with GLP-1R agonism. Preclinical models have shown that both GIPR agonism and antagonism, when combined with GLP-1R agonism, induce weight loss by reducing food intake and fat mass. Targeting both receptors with a GIPR antagonist antibody and a GLP-1R agonist has demonstrated positive effects on body weight and various metabolic parameters in both mice and monkeys [175]. Nevertheless, not all preclinical studies have consistently shown the benefits of this combination strategy [182]. Moreover, in a small cohort of patients with T2D and overweight/obesity, co-infusion of a GIPR antagonist and a GLP-1R agonist had no discernible effect on appetite, food intake, resting energy expenditure, or adipose tissue triglyceride content [183]. Therefore, it is premature to conclusively assert that pharmacological GIPR antagonism in combination with GCGR and GLP-1R agonism can serve as an effective strategy to achieve similar or even greater pharmacological effects as observed with tirzepatide.
An additional puzzle is the apparent species-specific difference in the mechanism of action of GLP-1R-based tri-agonists. In diet-induced obese mice, treatment with GLP-1R-GIPR-GCGR tri-agonists results in a substantial increase in energy expenditure [167], which may not be as significant in humans, thus limiting efficacy in body weight loss. The GCGR component of the molecule is thought to contribute to the improved energy expenditure, but the mechanism by which this occurs remains to be fully elucidated. The GCGR-mediated modulation of BAT activity is a possible mechanism [138,139,140], although consensus is still lacking [141]. Whether BAT activation in humans contributes to the tri-agonist–mediated effects of weight loss remains controversial, which is a relevant question considering that the impact of BAT-mediated thermogenesis on whole-body energy dissipation may be negligible in humans compared to rodents [184]. Furthermore, while GIPR activation appears to be less important in stimulating insulin secretion in mouse islets, human islets may be more dependent on GIPR signalling to allow tirzepatide-mediated effects on insulin secretion [185]. One may consider interspecies differences in the response to the same ligand [186]. Antagonising GIPR activity in vitro consistently reduces the insulin response to tirzepatide in human islets [185]. Of note, tirzepatide also increases glucagon and somatostatin secretion in human islets [185]. This last observation raises the question of whether these beneficial effects observed in clinical trials may be mediated indirectly by other islet hormone secretions, such as indirect endogenous glucagon receptor agonism. Future development of poly-agonists with multiple targets and their subsequent possible indirect effects [185] will add complexity to this phenomenon.
Finally, an essential, unanswered question pertains to whether the GCGR-related component within the tri-agonist may induce undesired effects on cardiovascular function. Glucagon is widely thought to exert cardio-stimulatory effects via GCGR expressed on the myocardium [187], and recent reports have shown that tri-agonists can induce a transient stimulatory effect on heart rate [188], similar to observations made in preclinical studies of GLP-1R agonists. This potential risk holds particular significance in light of the consistent findings from the aforementioned large-scale cardiovascular outcome trials, which have demonstrated the cardioprotective attributes of GLP-1R agonism. Preclinical data suggest that tachycardia associated with GLP-1R agonists tends to diminish over time or can be mitigated through dose escalation [189]. Consequently, it is plausible that dose escalation protocols, a standard procedure for the administration of available GLP-1R agonists to alleviate GI side effects, may overcome potential adverse cardiovascular effects resulting from GCGR agonists. However, this hypothesis necessitates empirical validation within a clinical context.
5 New GLP-1-Based Pharmacological Trends on the Horizon
5.1 Novel Co-Treatment Strategies
Combining different hormones with GLP1 in physical mixtures may open new avenues for potentially beneficial synergistic metabolic effects. Examples include the use of GLP-1 agonists in combination with salmon calcitonin [190], peptide YY [191], cholecystokinin [192, 193], GIP [194], insulin [195], glucagon [143], amylin [196] or a melanocortin 4 receptor agonist [197].
Preclinical studies have shown promising effects of FGF21 in increasing insulin sensitivity [198], raising high hopes for the treatment of T2D. Due to its expression in several metabolically active organs (liver, muscle, pancreas, adipose tissue among others) and its interaction with many different tissues, FGF21 is considered to be a hormone rather than a growth factor. Interestingly, a recent study suggests that liraglutide may regulate body weight, at least in part, via FGF21 [199]. However, FGF21 has the same drawback as GLP-1 in that it has a relatively short circulating half-life, making it difficult to use in therapy in its native form. Efruxifermin, formerly known as AKR-001 from the Akero Therapeutics laboratory, is a fusion protein of FGF21 with an immunoglobulin Fc, which increases its half-life. Efruxifermin has been shown to be safe and well tolerated in T2D patients with non-alcoholic steatohepatitis (NASH), with improvements in markers of hepatitis after 3–4 months of treatment [200, 201]. When combined with GLP-1 in part of the SYMMETRY clinical trial (NCT05039450), efruxifermin showed enhanced benefits in reducing liver fat and key markers of fibrosis and liver injury [202].
There is also growing interest in the potential therapeutic use of Growth Differentiation Factor 15 (GDF15; also known as MIC-1) analogues for addressing obesity. Growth Differentiation Factor 15, a stress-regulated hormone, has recently emerged as a molecule that can suppress appetite [203, 204]. Promising preclinical studies conducted on rodents have illustrated GDF15 ability to effectively reduce both body weight and adiposity. However, it is worth noting that recent findings indicate that while GDF15 can reduce fat intake, it may not be essential for weight loss after bariatric surgery in mice [205]. In humans, an increase in serum GDF15 levels has been correlated with weight loss following activities such as exercise, metformin use, and Roux-en-Y bariatric surgery [206]. In 2023, a human study showed that high GDF15 expression was associated with lower hepatic fat content and sign of higher non-shivering thermogenesis in skeletal muscle (higher expression of sarcolipin) [207]. Used in a recent Phase 1 study conducted by Eli Lilly [208], LY346325, a long-acting GDF15 analogue, has shown efficacy in rodents and primates. This study, with a very small number of obese patients (n = 6 in each dose group), did not directly measure adiposity and showed no evidence of substantial body weight loss. However, enrolled patients reported a reduction in appetite and food intake. These observations present an exciting opportunity to embark on larger-scale clinical trials that aim to gain a more comprehensive understanding of how GDF15 functions in the context of human biology and pharmacology. Notably, there is growing anticipation surrounding the potential synergistic effects of combining GDF15 with GLP-1R agonists, as highlighted in a preclinical study indicating that GDF15 induces synergistic weight loss when administered in conjunction with liraglutide and semaglutide [209].
5.2 GLP-1R-Based Unimolecular Conjugates
Following the beneficial effects of combining GLP-1 with co-injection of adipogenic hormones, there has been increasing interest in generating unimolecular species that chemically link GLP-1 biology with FGF21- or GDF15-mediated actions. GLP1-FGF21 conjugates have been tested and reported to dose-dependently reduce body weight and improve blood glucose levels in obese db/db mice [210, 211]. More recently, QL1005, a dual agonist designed by fusing GLP-1 and GDF15 analogues through a peptide linker and conjugating it to a fatty acid for prolonged action, showed promising metabolic results in obese non-human primates with dose-dependent reductions in body weight, food intake, insulin and glucose with limited incidence of GI side effects [212].
G protein-coupled receptors (GPCRs) are suitable cellular targets for tissue-specific delivery of nuclear hormones. As GLP-1Rs are expressed in several peripheral tissues involved in metabolic control, GLP-1R-mediated nuclear hormone targeting has attracted considerable attention in the field. Several stable conjugates linking GLP-1R agonists to nuclear hormones have been developed to enhance pharmacological potency and potentially target the compound to specific tissues expressing the GLP-1R. The key pharmacological principle guiding this strategy was to generate a molecule in which the chemical linker is stable outside the cell and is only cleaved intracellularly after the molecules have been internalised to release the nuclear hormone.
One of the first experiences with peptide fusion was the conjugation of GLP-1 to 17-β-estradiol, which reduced food intake and body weight in several rodent models of metabolic syndrome, showing superior efficacy compared to the use of GLP-1 alone [46]. Importantly, targeting estradiol to GLP-1R-expressing tissues prevented the typical side effects of oestrogen, such as reproductive endocrine toxicity and oncogenicity, in both male and female mice. This conjugate also activates the insulin signalling pathway in β-cells [213], enhances anti-apoptotic pathways in human and mouse pancreatic islets [214], restores their function and leads to diabetes remission [46].
Other nuclear receptor agonists, such as thyroid hormone T3, have been combined with glucagon to selectively target the liver, leading to weight loss and correction of dyslipidaemia in several mouse models of obesity [215]. Conjugation of a GLP-1R agonist with dexamethasone, a synthetic agonist of the glucocorticoid receptor (GR), resulted in significant weight loss in obese mice with greater efficacy than GLP-1 alone [45] and attenuated hypothalamic and systemic obesity-induced inflammation. Glucagon-like peptide-1–directed dexamethasone delivery by-passes the adverse effects of glucocorticoids on glucose handling, bone integrity and hypothalamic-pituitary-adrenal axis activity in obese mice [45].
Conjugation of GLP-1 to dexamethasone via a bioactive linker alters the biodistribution of the synthetic glucocorticoid by limiting its action to tissues where the GLP-1R is located, resulting in fewer adverse effects on bone, liver and adrenal glands than would otherwise be observed with untargeted dexamethasone, while exploiting the dual pharmacodynamics of this hormonal pairing [216]. Thus, this preclinical evidence provides a first proof-of-concept for the use of a cell-selective immunosuppressive agent for the safe treatment of metabolic inflammation and obesity, and similar approaches may be used for precision targeting of obese patients with particularly altered inflammatory profiles. Finally, the insulinotropic effect of GLP-1R agonists has been shown to synergize with the insulin-sensitising effect of tesaglitazar, a PPARα/γ dual agonist. When covalently linked, this conjugate optimised glucose handling and the weight-lowering effect of a GLP-1R agonist alone in diet-induced obese male mice [216].
Therefore, the use of combination pharmacotherapy that includes other molecules in addition to gut hormones appears to be a promising approach to treating metabolic syndrome. It is important to note that most of these single molecule compounds have only been tested in preclinical models and further research is needed to determine whether their effects exceed those of clinically tested drugs. Obesity has a multifactorial basis and significant heterogeneity has been observed in the magnitude of response to anti-obesity drugs in general and pharmacological GLP-1R agonists in particular. Therefore, therapeutic approaches that target multiple converging molecular rearrangements in key metabolic organs may have a transformative impact on the global health threat of this disease.
5.3 Hidden Threats and Mysteries
The basic concept of a peptide-nuclear hormone conjugate is that the targeted peptide first binds to its specific receptor. The receptor is then internalised, carrying the nuclear hormone cargo with it. After the peptide-nuclear hormone conjugate enters the intracellular endolysosomal pathway, it is theorized that pH-dependent proteases play a role in cleaving and releasing the nuclear hormone from the peptide. This release allows nuclear activation and molecular reprogramming to take place.
Importantly, the linker must be stable enough to prevent the combined hormones from being released and affecting tissues in unwanted ways, such as estradiol on the reproductive system, which was already controlled in preclinical short-term studies [46]. It should also be mentioned that several steroid receptors, such as oestrogens, are known to have both membrane-mediated and nuclear-mediated signalling [217], which could have different metabolic effects [218]. The mechanisms of intracellular release of nuclear hormones are still poorly understood, and several questions remain: how exactly are nuclear hormones released intracellularly from the unimolecular conjugate, and can nuclear hormones with lipophilic properties cross membranes and diffuse out of the cell after intracellular release at clinically effective doses, leading to potential off-target effects after long-term pharmacological treatment? The question of whether and how the beneficial metabolic effects of these hormones are mediated via their membrane-bound receptors, as opposed to the intracellularly released pools, remains to be answered.
6 Conclusion and Perspectives
The use of GLP-1 receptor-based multi-agonists in T2D and obesity has shown great promise in improving glycaemic control and weight management (Table 1), but there are still important gaps in our knowledge of the biological mechanisms and potential adverse effects associated with the use of these multi-target molecules, which we have discussed in detail in this review.
As we search for new metabolic targets to enhance the efficacy of GLP-1R-based therapies to pharmacologically mimic metabolic surgery, we should not abandon the idea of assessing whether and how activation of GLP-1R signalling can be used to reprofile ‘historic’ anti-obesity targets, such as the cannabinoid receptor type 1 (CB1R).
In contrast to the psychiatric side effects of the first CB1R antagonist, rimonabant [219], which was brain penetrant, peripherally acting CB1R antagonists have now been developed and show safer preclinical profiles due to their reduced brain permeability [220]. In diet-induced obese mice, co-administration of a peripheral CB1R inhibitor (JD5037) with long-acting GLP-1R agonists achieves greater reductions in body weight and fat mass than monotherapies by promoting negative energy balance and also elicits significant improvements in systemic and hepatic insulin action, dyslipidaemia and hepatic steatosis [221]. Thus, chemical fusions of GLP-1R-based agonists with a growing number of peripheral CB1R inhibitors may have a transformative impact in obesity and diabetes pharmacotherapy, an exciting perspective given the emerging structural and functional plasticity of CB1R [222] and the emergence of novel generations of ‘fusion’ CB1R inhibitors [220].
Improving knowledge into the biology and pharmacology of gut–hormone-related drugs may also provide new avenues for expanding the therapeutic window of these agents beyond metabolic diseases. For instance, T2D frequently contributes to the onset of osteoporosis, a progressive bone disease characterized by loss of bone mass. Glucagon-like peptide-1 has been found to stimulate bone formation and suppress bone resorption [223, 224]. While the precise mechanisms and molecular pathways remain the subject of ongoing research, the accumulating evidence connecting GLP-1R agonists with enhanced bone health suggests that this drug class may offer valuable benefits for the increasing population of elderly individuals with T2D and osteoporosis, who are at a heightened risk of fractures.
Furthermore, GLP-1Rs are abundantly expressed in the CNS, particularly in regions that regulate mood, cognition and appetite [225]. The activation of these receptors by liraglutide has been shown to confer neuroprotection through various mechanisms, including enhancement of synaptic plasticity, reduction of oxidative stress and modulation of neuroinflammation, in both diabetic mice [226] and in animal models of neurodegenerative disease [227,228,229].
A dual GLP-1R and GIPR agonist (DA-JC1) has neuroprotective effects in a mouse model of Parkinson’s disease and it improves motor impairment [230]. The co-agonism of GLP-1R and GIPR restores visual and spatial memory deficits in a mouse model of mild traumatic brain injury [231], suggesting the potential cognitive benefits of this pharmacological approach. In addition, GLP-1 receptor agonists have been shown to promote neural stem cell proliferation and differentiation, which may have implications for cognitive function and mood regulation. Recently, a single-blind, randomised, controlled, crossover human functional magnetic resonance imaging study reported that adaptive learning is reduced when metabolic sensing is impaired in obesity, as indexed by reduced insulin sensitivity in participants [232]. Treatment with liraglutide normalises impaired learning of sensory associations in men and women with obesity and insulin resistance [232], providing insight into how metabolic signals can act as neuromodulators to adapt our behaviour to the internal state of our body and how GLP-1 receptor agonists work in the clinic. To take this further, it would be interesting to increase the number of participants (in the cited study there were only 44 people) [232] and to see if the duration of treatment with GLP-1 agonists could influence the outcome (here there were only acute liraglutide injections). Over the past decade, studies have reported that GLP-1R agonists lead to reductions in alcohol and substance abuse, effects that may be centrally mediated, although the precise mechanisms remain to be elucidated [233, 234].
One pressing concern is that many large pharmaceutical companies tend to offer these treatments at exorbitant prices, thereby limiting access to these drugs primarily to wealthier individuals. This is particularly problematic because studies consistently show that obesity disproportionately affects lower-income segments of the population. It is crucial to recognize that not all individuals with obesity are the same, which may help explain the significant variability in the ways that people respond to treatment and, in some cases, discontinue it.
To advance our understanding and optimize weight loss strategies, there is a need for a more comprehensive characterization of the heterogeneous pharmacological responses to weight loss and weight regain in obesity [235]. For instance, while it is evident that not all obese individuals respond effectively to GLP-1R–based pharmacology, the underlying mechanisms driving this variability remain unknown. Therefore, there should be a concerted effort to identify circulating biomarkers that can predict pharmacological responses.
In essence, the goal is two-fold: to make safe and effective treatments accessible to a broader spectrum of the population, especially those who need them the most, and to tailor these treatments more precisely by better understanding the diverse responses observed in patients with obesity. This holistic approach can lead to more equitable health care outcomes and improved strategies for managing metabolic diseases and beyond.
References
Banting FG. Early work on insulin. Science. 1937;85:594–6.
Petersen J, Strømgaard K, Frølund B, Clemmensen C. Designing poly-agonists for treatment of metabolic diseases: challenges and opportunities. Drugs. 2019;79:1187–97.
Perreault L, Skyler JS, Rosenstock J. Novel therapies with precision mechanisms for type 2 diabetes mellitus. Nat Rev Endocrinol. 2021;17:364–77.
Müller TD, Blüher M, Tschöp MH, DiMarchi RD. Anti-obesity drug discovery: advances and challenges. Nat Rev Drug Discov. 2022;21:201–23.
Campbell JE, Müller TD, Finan B, DiMarchi RD, Tschöp MH, D’Alessio DA. GIPR/GLP-1R dual agonist therapies for diabetes and weight loss-chemistry, physiology, and clinical applications. Cell Metab. 2023;35:1519–29.
Hammoud R, Drucker DJ. Beyond the pancreas: contrasting cardiometabolic actions of GIP and GLP1. Nat Rev Endocrinol. 2023;19:201–16.
Tschöp M, Nogueiras R, Ahrén B. Gut hormone-based pharmacology: novel formulations and future possibilities for metabolic disease therapy. Diabetologia. 2023;66:1796–808.
Nogueiras R, Nauck MA, Tschöp MH. Gut hormone co-agonists for the treatment of obesity: from bench to bedside. Nat Metab. 2023;5:933–44.
Maclean PS, Bergouignan A, Cornier M-A, Jackman MR. Biology’s response to dieting: the impetus for weight regain. Am J Physiol Regul Integr Comp Physiol. 2011;301:R581-600.
Greenway FL. Physiological adaptations to weight loss and factors favouring weight regain. Int J Obes (Lond). 2015;39:1188–96.
Sainsbury K, Evans EH, Pedersen S, Marques MM, Teixeira PJ, Lähteenmäki L, et al. Attribution of weight regain to emotional reasons amongst European adults with overweight and obesity who regained weight following a weight loss attempt. Eat Weight Disord. 2019;24:351–61.
Sjöström L, Narbro K, Sjöström CD, Karason K, Larsson B, Wedel H, et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med. 2007;357:741–52.
Adams TD, Gress RE, Smith SC, Halverson RC, Simper SC, Rosamond WD, et al. Long-term mortality after gastric bypass surgery. N Engl J Med. 2007;357:753–61.
Arterburn DE, Olsen MK, Smith VA, Livingston EH, Van Scoyoc L, Yancy WS, et al. Association between bariatric surgery and long-term survival. JAMA. 2015;313:62–70.
Brissman M, Beamish AJ, Olbers T, Marcus C. Prevalence of insufficient weight loss 5 years after Roux-en-Y gastric bypass: metabolic consequences and prediction estimates: a prospective registry study. BMJ Open. 2021;11: e046407.
Voorwinde V, Steenhuis IHM, Janssen IMC, Monpellier VM, van Stralen MM. Definitions of long-term weight regain and their associations with clinical outcomes. Obes Surg. 2020;30:527–36.
Noria SF, Shelby RD, Atkins KD, Nguyen NT, Gadde KM. Weight regain after bariatric surgery: scope of the problem, causes, prevention, and treatment. Curr Diab Rep. 2023;23:31–42.
Debédat J, Sokolovska N, Coupaye M, Panunzi S, Chakaroun R, Genser L, et al. Long-term relapse of type 2 diabetes after Roux-en-Y gastric bypass: prediction and clinical relevance. Diabetes Care. 2018;41:2086–95.
Janssen P, Vanden Berghe P, Verschueren S, Lehmann A, Depoortere I, Tack J. Review article: the role of gastric motility in the control of food intake: review: regulation of food intake by gastric motility. Aliment Pharmacol Ther. 2011;33:880–94.
Juárez-Fernández M, Román-Sagüillo S, Porras D, García-Mediavilla MV, Linares P, Ballesteros-Pomar MD, et al. Long-term effects of bariatric surgery on gut microbiota composition and faecal metabolome related to obesity remission. Nutrients. 2021;13:2519.
Guo Y, Huang Z-P, Liu C-Q, Qi L, Sheng Y, Zou D-J. Modulation of the gut microbiome: a systematic review of the effect of bariatric surgery. Eur J Endocrinol. 2018;178:43–56.
Ahmad NN, Pfalzer A, Kaplan LM. Roux-en-Y gastric bypass normalizes the blunted postprandial bile acid excursion associated with obesity. Int J Obes (Lond). 2013;37:1553–9.
Myronovych A, Kirby M, Ryan KK, Zhang W, Jha P, Setchell KD, et al. Vertical sleeve gastrectomy reduces hepatic steatosis while increasing serum bile acids in a weight-loss-independent manner. Obesity (Silver Spring). 2014;22:390–400.
Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509:183–8.
Garibay D, McGavigan AK, Lee SA, Ficorilli JV, Cox AL, Michael MD, et al. β-cell glucagon-like peptide-1 receptor contributes to improved glucose tolerance after vertical sleeve gastrectomy. Endocrinology. 2016;157:3405–9.
Albaugh VL, He Y, Münzberg H, Morrison CD, Yu S, Berthoud H-R. Regulation of body weight: lessons learned from bariatric surgery. Mol Metab. 2023;68: 101517.
Sundbom M, Franzén S, Ottosson J, Svensson A-M. Superior socioeconomic status in patients with type 2 diabetes having gastric bypass surgery: a case-control analysis of 10 642 individuals. BMJ Open Diabetes Res Care. 2020;8: e000989.
Alvarez R, Bonham AJ, Buda CM, Carlin AM, Ghaferi AA, Varban OA. Factors associated with long wait times for bariatric surgery. Ann Surg. 2019;270:1103–9.
Fulton S, Décarie-Spain L, Fioramonti X, Guiard B, Nakajima S. The menace of obesity to depression and anxiety prevalence. Trends Endocrinol Metab. 2022;33:18–35.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32.
Müller TD, Clemmensen C, Finan B, DiMarchi RD, Tschöp MH. Anti-obesity therapy: from rainbow pills to polyagonists. Pharmacol Rev. 2018;70:712–46.
Williams DM, Nawaz A, Evans M. Drug therapy in obesity: a review of current and emerging treatments. Diabetes Ther. 2020;11:1199–216.
Novikoff A, Müller TD. The molecular pharmacology of glucagon agonists in diabetes and obesity. Peptides. 2023;165: 171003.
Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72–130.
Brierley DI, Holt MK, Singh A, de Araujo A, McDougle M, Vergara M, et al. Central and peripheral GLP-1 systems independently suppress eating. Nat Metab. 2021;3:258–73.
Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B, et al. The role of β cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab. 2014;19:1050–7.
Holter MM, Saikia M, Cummings BP. Alpha-cell paracrine signaling in the regulation of beta-cell insulin secretion. Front Endocrinol (Lausanne). 2022;13: 934775.
Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J Clin Invest. 2014;124:2456–63.
Sisley S, Smith K, Sandoval DA, Seeley RJ. Differences in acute anorectic effects of long-acting GLP-1 receptor agonists in rats. Peptides. 2014;58:1–6.
Burmeister MA, Ayala JE, Smouse H, Landivar-Rocha A, Brown JD, Drucker DJ, et al. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes. 2017;66:372–84.
Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. a randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11–22.
Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515–20.
Lockie SH, Heppner KM, Chaudhary N, Chabenne JR, Morgan DA, Veyrat-Durebex C, et al. Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. Diabetes. 2012;61:2753–62.
Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes. 2014;63:3346–58.
Quarta C, Clemmensen C, Zhu Z, Yang B, Joseph SS, Lutter D, et al. Molecular integration of incretin and glucocorticoid action reverses immunometabolic dysfunction and obesity. Cell Metab. 2017;26:620-632.e6.
Finan B, Yang B, Ottaway N, Stemmer K, Müller TD, Yi C-X, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. 2012;18:1847–56.
Hansen G, Jelsing J, Vrang N. Effects of liraglutide and sibutramine on food intake, palatability, body weight and glucose tolerance in the gubra DIO-rats. Acta Pharmacol Sin. 2012;33:194–200.
Brindisi M-C, Brondel L, Meillon S, Barthet S, Grall S, Fenech C, et al. Proof of concept: effect of GLP-1 agonist on food hedonic responses and taste sensitivity in poor controlled type 2 diabetic patients. Diabetes Metab Syndr. 2019;13:2489–94.
Geisler CE, Antonellis MP, Trumbauer W, Martin JA, Coskun T, Samms RJ, et al. Tirzepatide suppresses palatable food intake by selectively reducing preference for fat in rodents. Diabetes Obes Metab. 2023;25:56–67.
Blundell J, Finlayson G, Axelsen M, Flint A, Gibbons C, Kvist T, et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes Metab. 2017;19:1242–51.
Windeløv JA, Wewer Albrechtsen NJ, Kuhre RE, Jepsen SL, Hornburg D, Pedersen J, et al. Why is it so difficult to measure glucagon-like peptide-1 in a mouse? Diabetologia. 2017;60:2066–75.
Zhang T, Perkins MH, Chang H, Han W, de Araujo IE. An inter-organ neural circuit for appetite suppression. Cell. 2022;185(14):2478–94.e28. https://doi.org/10.1016/j.cell.2022.05.007
Trapp S, Brierley DI. Brain GLP-1 and the regulation of food intake: GLP-1 action in the brain and its implications for GLP-1 receptor agonists in obesity treatment. Br J Pharmacol. 2022;179:557–70.
Tschen S-I, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58:1312–20.
Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human beta-cells. PLoS ONE. 2012;7: e35801.
Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, et al. Age-dependent human β cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J Clin Invest. 2017;127:3835–44.
Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52.
Capozzi ME, D’Alessio DA, Campbell JE. The past, present, and future physiology and pharmacology of glucagon. Cell Metab. 2022;34:1654–74.
Ørgaard A, Holst JJ. The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia. 2017;60:1731–9.
Gutniak M, Orskov C, Holst JJ, Ahrén B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326:1316–22.
Creutzfeldt WO, Kleine N, Willms B, Orskov C, Holst JJ, Nauck MA. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7–36) amide in type I diabetic patients. Diabetes Care. 1996;19:580–6.
Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, et al. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51:1584–92.
Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31:1285–97.
Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE−/− mice. Endocrinology. 2013;154:127–39.
Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267:7402–5.
Eng J. Exendin peptides. Mt Sinai J Med. 1992;59:147–9.
Furman BL. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon. 2012;59:464–71.
Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: The SCALE Maintenance randomized study. Int J Obes (Lond). 2015;39:187.
Pratley R, Amod A, Hoff ST, Kadowaki T, Lingvay I, Nauck M, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394:39–50.
Sorli C, Harashima S-I, Tsoukas GM, Unger J, Karsbøl JD, Hansen T, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:251–60.
Ahrén B, Masmiquel L, Kumar H, Sargin M, Karsbøl JD, Jacobsen SH, et al. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. Lancet Diabetes Endocrinol. 2017;5:341–54.
Ahmann AJ, Capehorn M, Charpentier G, Dotta F, Henkel E, Lingvay I, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label randomized, clinical trial. Diabetes Care. 2018;41:258–66.
Rodbard HW, Lingvay I, Reed J, de la Rosa R, Rose L, Sugimoto D, et al. Semaglutide added to basal insulin in type 2 diabetes (SUSTAIN 5): a randomized controlled trial. J Clin Endocrinol Metab. 2018;103:2291–301.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375:1834–44.
Pratley RE, Aroda VR, Lingvay I, Lüdemann J, Andreassen C, Navarria A, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6:275–86.
Lingvay I, Catarig A-M, Frias JP, Kumar H, Lausvig NL, le Roux CW, et al. Efficacy and safety of once-weekly semaglutide versus daily canagliflozin as add-on to metformin in patients with type 2 diabetes (SUSTAIN 8): a double-blind, phase 3b, randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7:834–44.
Zinman B, Bhosekar V, Busch R, Holst I, Ludvik B, Thielke D, et al. Semaglutide once weekly as add-on to SGLT-2 inhibitor therapy in type 2 diabetes (SUSTAIN 9): a randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019;7:356–67.
Capehorn MS, Catarig A-M, Furberg JK, Janez A, Price HC, Tadayon S, et al. Efficacy and safety of once-weekly semaglutide 1.0mg vs once-daily liraglutide 1.2mg as add-on to 1–3 oral antidiabetic drugs in subjects with type 2 diabetes (SUSTAIN 10). Diabetes Metab. 2020;46:100–9.
van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WHM. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int J Obes (Lond). 2014;38:784–93.
O’Neil PM, Birkenfeld AL, McGowan B, Mosenzon O, Pedersen SD, Wharton S, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392:637–49.
Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384:989–1002.
Rubino D, Abrahamsson N, Davies M, Hesse D, Greenway FL, Jensen C, et al. Effect of continued weekly subcutaneous semaglutide vs placebo on weight loss maintenance in adults with overweight or obesity: the STEP 4 randomized clinical trial. JAMA. 2021;325:1414–25.
Garvey WT, Batterham RL, Bhatta M, Buscemi S, Christensen LN, Frias JP, et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat Med. 2022;28:2083–91.
Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol (Lausanne). 2019;10:155.
Drucker DJ, Dritselis A, Kirkpatrick P. Liraglutide. Nat Rev Drug Discov. 2010;9:267–8.
Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–9.
Christensen M, Sparre-Ulrich AH, Hartmann B, Grevstad U, Rosenkilde MM, Holst JJ, et al. Transfer of liraglutide from blood to cerebrospinal fluid is minimal in patients with type 2 diabetes. Int J Obes (Lond). 2015;39:1651–4.
Bakker W, Imbernon M, Salinas CG, Moro Chao DH, Hassouna R, Morel C, et al. Acute changes in systemic glycemia gate access and action of GLP-1R agonist on brain structures controlling energy homeostasis. Cell Rep. 2022;41: 111698.
Imbernon M, Saponaro C, Helms HCC, Duquenne M, Fernandois D, Deligia E, et al. Tanycytes control hypothalamic liraglutide uptake and its anti-obesity actions. Cell Metab. 2022;34:1054-1063.e7.
Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5(e133429): 133429.
Dandona P, Chaudhuri A, Ghanim H. Semaglutide in early type 1 diabetes. N Engl J Med. 2023;389:958–9.
Villalba A, Rodriguez-Fernandez S, Perna-Barrull D, Ampudia R-M, Gomez-Muñoz L, Pujol-Autonell I, et al. Repurposed analog of GLP-1 ameliorates hyperglycemia in type 1 diabetic mice through pancreatic cell reprogramming. Front Endocrinol (Lausanne). 2020;11:258.
Nauck MA, Meier JJ, Cavender MA, Abd El Aziz M, Drucker DJ. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation. 2017;136:849–70.
Marx N, Husain M, Lehrke M, Verma S, Sattar N. GLP-1 Receptor agonists for the reduction of atherosclerotic cardiovascular risk in patients with type 2 diabetes. Circulation. 2022;146:1882–94.
Johnson B. Weight-loss drug lowers cardiovascular disease risk in new trial of Novo Nordisk’s Wegovy. Nat Med. 2023. https://doi.org/10.1038/d41591-023-00075-x
Borlaug BA, Kitzman DW, Davies MJ, Rasmussen S, Barros E, Butler J, et al. Semaglutide in HFpEF across obesity class and by body weight reduction: a prespecified analysis of the STEP-HFpEF trial. Nat Med. 2023;29:2358–65.
Kosiborod MN, Abildstrøm SZ, Borlaug BA, Butler J, Rasmussen S, Davies M, et al. Semaglutide in patients with heart failure with preserved ejection fraction and obesity. N Engl J Med. 2023;389(12):1069–84. https://doi.org/10.1056/NEJMoa2306963
Piccini S, Favacchio G, Panico C, Morenghi E, Folli F, Mazziotti G, et al. Time-dependent effect of GLP-1 receptor agonists on cardiovascular benefits: a real-world study. Cardiovasc Diabetol. 2023;22:69.
Baggio LL, Ussher JR, McLean BA, Cao X, Kabir MG, Mulvihill EE, et al. The autonomic nervous system and cardiac GLP-1 receptors control heart rate in mice. Mol Metab. 2017;6:1339–49.
Nakamura H, Niwano S, Niwano H, Fukaya H, Murakami M, Kishihara J, et al. Liraglutide suppresses atrial electrophysiological changes. Heart Vessels. 2019;34:1389–93.
Bohne LJ, Jansen HJ, Dorey TW, Daniel IM, Jamieson KL, Belke DD, et al. Glucagon-like peptide-1 protects against atrial fibrillation and atrial remodeling in type 2 diabetic mice. JACC Basic Transl Sci. 2023;8:922–36.
McLean BA, Wong CK, Kabir MG, Drucker DJ. Glucagon-like Peptide-1 receptor Tie2+ cells are essential for the cardioprotective actions of liraglutide in mice with experimental myocardial infarction. Mol Metab. 2022;66: 101641.
Ussher JR, Baggio LL, Campbell JE, Mulvihill EE, Kim M, Kabir MG, et al. Inactivation of the cardiomyocyte glucagon-like peptide-1 receptor (GLP-1R) unmasks cardiomyocyte-independent GLP-1R-mediated cardioprotection. Mol Metab. 2014;3:507–17.
Noyan-Ashraf MH, Shikatani EA, Schuiki I, Mukovozov I, Wu J, Li R-K, et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127:74–85.
Sun F, Wu S, Wang J, Guo S, Chai S, Yang Z, et al. Effect of glucagon-like peptide-1 receptor agonists on lipid profiles among type 2 diabetes: a systematic review and network meta-analysis. Clin Ther. 2015;37:225-241.e8.
Petrovic A, Igrec D, Rozac K, Bojanic K, Kuna L, Kolaric TO, et al. The Role of GLP1-RAs in direct modulation of lipid metabolism in hepatic tissue as determined using in vitro models of NAFLD. CIMB. 2023;45:4544–56.
https://www.ema.europa.eu/en/news/ema-statement-ongoing-review-glp-1-receptor-agonists. Accessed 11 Jul 2023
Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ. 2012;344: d7771.
Seufert J, Nauck M, Rosenstock J, Hansen T, Vrazic H, Vilsboll T. P2857Increase in pulse rate with semaglutide did not result in increased adverse cardiac events in subjects with type 2 diabetes in the SUSTAIN 6 cardiovascular outcomes trial. Eur Heart J [Internet]. 2018. https://doi.org/10.1093/eurheartj/ehy565.P2857/5080031.
Chen Y, Mushashi F, Son S, Bhatti P, Dummer T, Murphy RA. Diabetes medications and cancer risk associations: a systematic review and meta-analysis of evidence over the past 10 years. Sci Rep. 2023;13:11844.
Bezin J, Gouverneur A, Pénichon M, Mathieu C, Garrel R, Hillaire-Buys D, et al. GLP-1 Receptor Agonists and the Risk of Thyroid Cancer. Diabetes Care. 2023;46:384–90.
https://www.ema.europa.eu/en/news/meeting-highlights-pharmacovigilance-risk-assessment-committee-prac-23-26-october-2023. Accessed 27 Oct 2023
Butler PC, Elashoff M, Elashoff R, Gale EAM. A critical analysis of the clinical use of incretin-based therapies: are the GLP-1 therapies safe? Diabetes Care. 2013;36:2118–25.
Faillie J-L, Yu OH, Yin H, Hillaire-Buys D, Barkun A, Azoulay L. Association of Bile duct and gallbladder diseases with the use of incretin-based drugs in patients with type 2 diabetes mellitus. JAMA Intern Med. 2016;176:1474–81.
Kristensen SL, Rørth R, Jhund PS, Docherty KF, Sattar N, Preiss D, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019;7:776–85.
Sattar N, Lee MMY, Kristensen SL, Branch KRH, Del Prato S, Khurmi NS, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021;9:653–62.
Bettge K, Kahle M, Abd El Aziz MS, Meier JJ, Nauck MA. Occurrence of nausea, vomiting and diarrhoea reported as adverse events in clinical trials studying glucagon-like peptide-1 receptor agonists: a systematic analysis of published clinical trials. Diabetes Obes Metab. 2017;19:336–47.
Sodhi M, Rezaeianzadeh R, Kezouh A, Etminan M. Risk of gastrointestinal adverse events associated with glucagon-like peptide-1 receptor agonists for weight loss. JAMA [Internet]. 2023 [cited 2023 Nov 14]. https://jamanetwork.com/journals/jama/fullarticle/2810542. https://doi.org/10.1001/jama.2023.19574
Borner T, Workinger JL, Tinsley IC, Fortin SM, Stein LM, Chepurny OG, et al. Corrination of a GLP-1 Receptor Agonist for Glycemic Control without Emesis. Cell Rep. 2020;31: 107768.
Sikirica MV, Martin AA, Wood R, Leith A, Piercy J, Higgins V. Reasons for discontinuation of GLP1 receptor agonists: data from a real-world cross-sectional survey of physicians and their patients with type 2 diabetes. Diabetes Metab Syndr Obes. 2017;10:403–12.
Jung H, Tittel SR, Schloot NC, Heitmann E, Otto T, Lebrec J, et al. Clinical characteristics, treatment patterns, and persistence in individuals with type 2 diabetes initiating a glucagon-like peptide-1 receptor agonist: a retrospective analysis of the Diabetes Prospective Follow-Up Registry. Diabetes Obesity Metab. 2023;25:1813–22.
Malik ME, Falkentoft AC, Jensen J, Zahir D, Parveen S, Alhakak A, et al. Discontinuation and reinitiation of SGLT-2 inhibitors and GLP-1R agonists in patients with type 2 diabetes: a nationwide study from 2013 to 2021. Lancet Reg Health Eur. 2023;29: 100617.
Miller RA, Birnbaum MJ. Glucagon: acute actions on hepatic metabolism. Diabetologia. 2016;59:1376–81.
Pearson MJ, Unger RH, Holland WL. Clinical trials, triumphs, and tribulations of glucagon receptor antagonists. Diabetes Care. 2016;39:1075–7.
Hædersdal S, Andersen A, Knop FK, Vilsbøll T. Revisiting the role of glucagon in health, diabetes mellitus and other metabolic diseases. Nat Rev Endocrinol. 2023;19:321–35.
Cegla J, Troke RC, Jones B, Tharakan G, Kenkre J, McCullough KA, et al. Coinfusion of low-dose GLP-1 and glucagon in man results in a reduction in food intake. Diabetes. 2014;63:3711–20.
Finan B, Capozzi ME, Campbell JE. Repositioning glucagon action in the physiology and pharmacology of diabetes. Diabetes. 2020;69:532–41.
Atrens DM, Menéndez JA. Glucagon and the paraventricular hypothalamus: modulation of energy balance. Brain Res. 1993;630:245–51.
Salem V, Izzi-Engbeaya C, Coello C, Thomas DB, Chambers ES, Comninos AN, et al. Glucagon increases energy expenditure independently of brown adipose tissue activation in humans. Diabetes Obes Metab. 2016;18:72–81.
Habegger KM, Stemmer K, Cheng C, Müller TD, Heppner KM, Ottaway N, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62:1453–63.
Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, et al. Glucagon lowers glycemia when β-cells are active. JCI Insight. 2019;5(e129954): 129954.
Runge S, Wulff BS, Madsen K, Bräuner-Osborne H, Knudsen LB. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br J Pharmacol. 2003;138:787–94.
Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effect of glucagon on food intake and body weight in man. J Appl Physiol. 1957;11:419–21.
Penick SB, Hinkle LE. Depression of food intake induced in healthy subjects by glucagon. N Engl J Med. 1961;264:893–7.
Geary N, Le Sauter J, Noh U. Glucagon acts in the liver to control spontaneous meal size in rats. Am J Physiol. 1993;264:R116-122.
Inokuchi A, Oomura Y, Nishimura H. Effect of intracerebroventricularly infused glucagon on feeding behavior. Physiol Behav. 1984;33:397–400.
Nason SR, Antipenko J, Presedo N, Cunningham SE, Pierre TH, Kim T, et al. Glucagon receptor signaling regulates weight loss via central KLB receptor complexes. JCI Insight. 2021;6(e141323): 141323.
Billington CJ, Bartness TJ, Briggs J, Levine AS, Morley JE. Glucagon stimulation of brown adipose tissue growth and thermogenesis. Am J Physiol. 1987;252:R160-165.
Billington CJ, Briggs JE, Link JG, Levine AS. Glucagon in physiological concentrations stimulates brown fat thermogenesis in vivo. Am J Physiol. 1991;261:R501-507.
Nair KS. Hyperglucagonemia increases resting metabolic rate in man during insulin deficiency. J Clin Endocrinol Metab. 1987;64:896–901.
Kleinert M, Sachs S, Habegger KM, Hofmann SM, Müller TD. Glucagon Regulation of Energy Expenditure. Int J Mol Sci. 2019;20:5407.
Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749–57.
Tan TM, Field BCT, McCullough KA, Troke RC, Chambers ES, Salem V, et al. Coadministration of glucagon-like peptide-1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62:1131–8.
Nahra R, Wang T, Gadde KM, Oscarsson J, Stumvoll M, Jermutus L, et al. Effects of cotadutide on metabolic and hepatic parameters in adults with overweight or obesity and type 2 diabetes: a 54-week randomized phase 2b study. Diabetes Care. 2021;44:1433–42.
Jiang H, Pang S, Zhang Y, Yu T, Liu M, Deng H, et al. A phase 1b randomised controlled trial of a glucagon-like peptide-1 and glucagon receptor dual agonist IBI362 (LY3305677) in Chinese patients with type 2 diabetes. Nat Commun. 2022;13:3613.
Ji L, Gao L, Jiang H, Yang J, Yu L, Wen J, et al. Safety and efficacy of a GLP-1 and glucagon receptor dual agonist mazdutide (IBI362) 9 mg and 10 mg in Chinese adults with overweight or obesity: A randomised, placebo-controlled, multiple-ascending-dose phase 1b trial. EClinicalMedicine. 2022;54: 101691.
Friedrichsen MH, Endahl L, Kreiner FF, Goldwater R, Kankam M, Toubro S, et al. Results from three phase 1 trials of NNC9204-1177, a glucagon/GLP-1 receptor co-agonist: effects on weight loss and safety in adults with overweight or obesity. Mol Metab. 2023;78: 101801.
Capozzi ME, DiMarchi RD, Tschöp MH, Finan B, Campbell JE. Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocr Rev. 2018;39:719–38.
Brown JC, Dryburgh JR. A gastric inhibitory polypeptide. II. The complete amino acid sequence. Can J Biochem. 1971;49:867–72.
Yamane S, Harada N, Inagaki N. Mechanisms of fat-induced gastric inhibitory polypeptide/glucose-dependent insulinotropic polypeptide secretion from K cells. J Diabetes Investig. 2016;7(Suppl 1):20–6.
Getty-Kaushik L, Song DH, Boylan MO, Corkey BE, Wolfe MM. Glucose-dependent insulinotropic polypeptide modulates adipocyte lipolysis and reesterification. Obesity (Silver Spring). 2006;14:1124–31.
Mohammad S, Ramos LS, Buck J, Levin LR, Rubino F, McGraw TE. Gastric Inhibitory peptide controls adipose insulin sensitivity via activation of cAMP-response element-binding protein and p110β isoform of phosphatidylinositol 3-kinase. J Biol Chem. 2011;286:43062–70.
Asmar M, Tangaa W, Madsbad S, Hare K, Astrup A, Flint A, et al. On the role of glucose-dependent insulintropic polypeptide in postprandial metabolism in humans. Am J Physiol Endocrinol Metab. 2010;298:E614-621.
Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 2019;30:987-996.e6.
Zhang Q, Delessa CT, Augustin R, Bakhti M, Colldén G, Drucker DJ, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 2021;33:833-844.e5.
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–7.
Christensen MB, Gasbjerg LS, Heimbürger SM, Stensen S, Vilsbøll T, Knop FK. GIP’s involvement in the pathophysiology of type 2 diabetes. Peptides. 2020;125: 170178.
Grespan E, Giorgino T, Natali A, Ferrannini E, Mari A. Different mechanisms of GIP and GLP-1 action explain their different therapeutic efficacy in type 2 diabetes. Metabolism. 2021;114: 154415.
Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. 2020;5(e140532): 140532.
Rosenstock J, Hollander P, Chevalier S, Iranmanesh A, SERENADE Study Group. SERENADE: the study evaluating rimonabant efficacy in drug-naive diabetic patients: effects of monotherapy with rimonabant, the first selective CB1 receptor antagonist, on glycemic control, body weight, and lipid profile in drug-naive type 2 diabetes. Diabetes Care. 2008;31:2169–76.
Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. The Lancet. 2021;398:1811–24.
Ludvik B, Giorgino F, Jodar E, Frias JP, Lando LF, Brown K, et al. 78-LB: efficacy and safety of tirzepatide, a dual GIP/GLP-1 receptor agonist, compared with insulin degludec in patients with type 2 diabetes (SURPASS-3). Diabetes. 2021;70:78-LB.
Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385:503–15.
Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327:534.
Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022;387:205–16.
Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21:27–36.
Knerr PJ, Mowery SA, Douros JD, Premdjee B, Hjøllund KR, He Y, et al. Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice. Mol Metab. 2022;63: 101533.
Jall S, Sachs S, Clemmensen C, Finan B, Neff F, DiMarchi RD, et al. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol Metab. 2017;6:440–6.
Bossart M, Wagner M, Elvert R, Evers A, Hübschle T, Kloeckener T, et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 2022;34:59-74.e10.
Coskun T, Urva S, Roell WC, Qu H, Loghin C, Moyers JS, et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab. 2022;34:1234-1247.e9.
Jastreboff AM, Kaplan LM, Frías JP, Wu Q, Du Y, Gurbuz S, et al. Triple-hormone-receptor agonist retatrutide for obesity—a phase 2 trial. N Engl J Med. 2023;389:514–26.
Abdelmalek MF, Suzuki A, Sanchez W, Lawitz E, Filozof C, Cho H, et al. A phase 2, adaptive randomized, double-blind, placebo-controlled, multicenter, 52-week study of HM15211 in patients with biopsy-confirmed non-alcoholic steatohepatitis—study design and rationale of HM-TRIA-201 study. Contemp Clin Trials. 2023;130: 107176.
Killion EA, Wang J, Yie J, Shi SD-H, Bates D, Min X, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. 2018;10:eaat3392.
McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR. GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am J Physiol Endocrinol Metab. 2007;293:E1746-1755.
Lu S-C, Chen M, Atangan L, Killion EA, Komorowski R, Cheng Y, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. 2021;2: 100263.
Yang B, Gelfanov VM, El K, Chen A, Rohlfs R, DuBois B, et al. Discovery of a potent GIPR peptide antagonist that is effective in rodent and human systems. Mol Metab. 2022;66: 101638.
Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002;8:738–42.
https://www.amgen.com/newsroom/press-releases/2022/12/amgen-presents-new-amg-133-phase-1-clinical-data-at-wcirdc-2022. Accessed 1 Dec 2022
Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y, et al. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:14843–7.
Ayala JE, Bracy DP, James FD, Burmeister MA, Wasserman DH, Drucker DJ. Glucagon-like peptide-1 receptor knockout mice are protected from high-fat diet-induced insulin resistance. Endocrinology. 2010;151:4678–87.
Killion EA, Chen M, Falsey JR, Sivits G, Hager T, Atangan L, et al. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat Commun. 2020;11:4981.
Irwin N, McClean PL, Hunter K, Flatt PR. Metabolic effects of sustained activation of the GLP-1 receptor alone and in combination with background GIP receptor antagonism in high fat-fed mice. Diabetes Obes Metab. 2009;11:603–10.
Stensen S, Krogh LL, Sparre-Ulrich AH, Dela F, Hartmann B, Vilsbøll T, et al. Acute concomitant glucose-dependent insulinotropic polypeptide receptor antagonism during glucagon-like peptide 1 receptor agonism does not affect appetite, resting energy expenditure or food intake in patients with type 2 diabetes and overweight/obesity. Diabetes Obesity Metabolism. 2022;24:1882–7.
Carpentier AC, Blondin DP, Virtanen KA, Richard D, Haman F, Turcotte ÉE. Brown adipose tissue energy metabolism in humans. Front Endocrinol. 2018;9:447.
El K, Douros JD, Willard FS, Novikoff A, Sargsyan A, Perez-Tilve D, et al. The incretin co-agonist tirzepatide requires GIPR for hormone secretion from human islets. Nat Metab. 2023;5:945–54.
Sparre-Ulrich AH, Hansen LS, Svendsen B, Christensen M, Knop FK, Hartmann B, et al. Species-specific action of (Pro3)GIP1a full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. Br J Pharmacol. 2016;173:27–38.
Petersen KM, Bøgevig S, Holst JJ, Knop FK, Christensen MB. Hemodynamic effects of glucagon: a literature review. J Clin Endocrinol Metab. 2018;103:1804–12.
Urva S, Coskun T, Loh MT, Du Y, Thomas MK, Gurbuz S, et al. LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. The Lancet. 2022;400:1869–81.
Goodwill AG, Mather KJ, Conteh AM, Sassoon DJ, Noblet JN, Tune JD. Cardiovascular and hemodynamic effects of glucagon-like peptide-1. Rev Endocr Metab Disord. 2014;15:209–17.
Bello NT, Kemm MH, Ofeldt EM, Moran TH. Dose combinations of exendin-4 and salmon calcitonin produce additive and synergistic reductions in food intake in nonhuman primates. Am J Physiol Regul Integr Comp Physiol. 2010;299:R945-952.
Neary NM, Small CJ, Druce MR, Park AJ, Ellis SM, Semjonous NM, et al. Peptide YY3-36 and glucagon-like peptide-17-36 inhibit food intake additively. Endocrinology. 2005;146:5120–7.
Trevaskis JL, Sun C, Athanacio J, D’Souza L, Samant M, Tatarkiewicz K, et al. Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet-induced obese and leptin-deficient rodents. Diabetes Obes Metab. 2015;17:61–73.
Gutzwiller J-P, Degen L, Matzinger D, Prestin S, Beglinger C. Interaction between GLP-1 and CCK-33 in inhibiting food intake and appetite in men. Am J Physiol Regul Integr Comp Physiol. 2004;287:R562-567.
Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5:209ra151.
Balena R, Hensley IE, Miller S, Barnett AH. Combination therapy with GLP-1 receptor agonists and basal insulin: a systematic review of the literature. Diabetes Obes Metab. 2013;15:485–502.
Enebo LB, Berthelsen KK, Kankam M, Lund MT, Rubino DM, Satylganova A, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2·4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet. 2021;397:1736–48.
Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, et al. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015;7:288–98.
Kliewer SA, Mangelsdorf DJ. A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. 2019;29:246–53.
Le TDV, Fathi P, Watters AB, Ellis BJ, Besing G-LK, Bozadjieva-Kramer N, et al. Fibroblast growth factor-21 is required for weight loss induced by the glucagon-like peptide-1 receptor agonist liraglutide in male mice fed high carbohydrate diets. Mol Metab. 2023;72:101718.
Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. 2021;27:1262–71.
Harrison SA, Ruane PJ, Freilich B, Neff G, Patil R, Behling C, et al. A randomized, double-blind, placebo-controlled phase IIa trial of efruxifermin for patients with compensated NASH cirrhosis. JHEP Rep. 2023;5: 100563.
Carvalho T. Efruxifermin combined with a GLP-1 receptor agonist reduces liver fat in NASH. Nat Med. 2023;29:1881.
Lockhart SM, Saudek V, O’Rahilly S. GDF15: a Hormone Conveying Somatic Distress to the Brain. Endocr Rev. 2020;41:bnaa007.
Yang L, Chang C-C, Sun Z, Madsen D, Zhu H, Padkjær SB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med. 2017;23:1158–66.
Frikke-Schmidt H, Hultman K, Galaske JW, Jørgensen SB, Myers MG, Seeley RJ. GDF15 acts synergistically with liraglutide but is not necessary for the weight loss induced by bariatric surgery in mice. Mol Metab. 2019;21:13–21.
Breit SN, Brown DA, Tsai VW-W. The GDF15-GFRAL pathway in health and metabolic disease: friend or foe? Annu Rev Physiol. 2021;83:127–51.
Wang D, Townsend LK, DesOrmeaux GJ, Frangos SM, Batchuluun B, Dumont L, et al. GDF15 promotes weight loss by enhancing energy expenditure in muscle. Nature. 2023;619:143–50.
Benichou O, Coskun T, Gonciarz MD, Garhyan P, Adams AC, Du Y, et al. Discovery, development, and clinical proof of mechanism of LY3463251, a long-acting GDF15 receptor agonist. Cell Metab. 2023;35:274-286.e10.
Ghidewon M, Wald HS, McKnight AD, De Jonghe BC, Breen DM, Alhadeff AL, et al. Growth differentiation factor 15 (GDF15) and semaglutide inhibit food intake and body weight through largely distinct, additive mechanisms. Diabetes Obes Metab. 2022;24:1010–20.
Pan Q, Lin S, Li Y, Liu L, Li X, Gao X, et al. A novel GLP-1 and FGF21 dual agonist has therapeutic potential for diabetes and non-alcoholic steatohepatitis. EBioMedicine. 2021;63: 103202.
Gilroy CA, Capozzi ME, Varanko AK, Tong J, D’Alessio DA, Campbell JE, et al. Sustained release of a GLP-1 and FGF21 dual agonist from an injectable depot protects mice from obesity and hyperglycemia. Sci Adv. 2020;6:eaaz9890.
Zhang Y, Zhao X, Dong X, Zhang Y, Zou H, Jin Y, et al. Activity-balanced GLP-1/GDF15 dual agonist reduces body weight and metabolic disorder in mice and non-human primates. Cell Metab. 2023;35:287-298.e4.
Sachs S, Bastidas-Ponce A, Tritschler S, Bakhti M, Böttcher A, Sánchez-Garrido MA, et al. Targeted pharmacological therapy restores β-cell function for diabetes remission. Nat Metab. 2020;2:192–209.
Fuselier T, Mota P, Qadir de Sa MMF, Xu B, Allard C, Meyers MM, et al. Efficacy of glucagon-like peptide-1 and estrogen dual agonist in pancreatic islets protection and pre-clinical models of insulin-deficient diabetes. Cell Rep Med. 2022;3:100598.
Finan B, Clemmensen C, Zhu Z, Stemmer K, Gauthier K, Müller L, et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell. 2016;167:843-857.e14.
Quarta C, Stemmer K, Novikoff A, Yang B, Klingelhuber F, Harger A, et al. GLP-1-mediated delivery of tesaglitazar improves obesity and glucose metabolism in male mice. Nat Metab. 2022;4:1071–83.
Mauvais-Jarvis F, Lange CA, Levin ER. Membrane-initiated estrogen, androgen, and progesterone receptor signaling in health and disease. Endocr Rev. 2022;43:720–42.
Allard C, Morford JJ, Xu B, Salwen B, Xu W, Desmoulins L, et al. Loss of nuclear and membrane estrogen receptor-α differentially impairs insulin secretion and action in male and female mice. Diabetes. 2019;68:490–501.
Topol EJ, Bousser M-G, Fox KA, Creager MA, Despres J-P, Easton JD, et al. Rimonabant for prevention of cardiovascular events (CRESCENDO): a randomised, multicentre, placebo-controlled trial. The Lancet. 2010;376:517–23.
Quarta C, Cota D. Anti-obesity therapy with peripheral CB1 blockers: from promise to safe(?) practice. Int J Obes (Lond). 2020. https://doi.org/10.1038/s41366-020-0577-8
Zizzari P, He R, Falk S, Bellocchio L, Allard C, Clark S, et al. CB1 and GLP-1 Receptors Cross Talk Provides New Therapies for Obesity. Diabetes. 2021;70:415–22.
Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017;547:468–71.
Mabilleau G, Pereira M, Chenu C. Novel skeletal effects of glucagon-like peptide-1 (GLP-1) receptor agonists. J Endocrinol. 2018;236:R29-42.
Xie B, Chen S, Xu Y, Han W, Hu R, Chen M, et al. The Impact of Glucagon-Like Peptide 1 Receptor Agonists on Bone Metabolism and Its Possible Mechanisms in Osteoporosis Treatment. Front Pharmacol. 2021;12: 697442.
Diz-Chaves Y, Herrera-Pérez S, González-Matías LC, Lamas JA, Mallo F. Glucagon-Like Peptide-1 (GLP-1) in the Integration of Neural and Endocrine Responses to Stress. Nutrients. 2020;12:3304.
Yan W, Pang M, Yu Y, Gou X, Si P, Zhawatibai A, et al. The neuroprotection of liraglutide on diabetic cognitive deficits is associated with improved hippocampal synapses and inhibited neuronal apoptosis. Life Sci. 2019;231: 116566.
Cao B, Zhang Y, Chen J, Wu P, Dong Y, Wang Y. Neuroprotective effects of liraglutide against inflammation through the AMPK/NF-κB pathway in a mouse model of Parkinson’s disease. Metab Brain Dis. 2022;37:451–62.
McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:6587–94.
Parthsarathy V, Hölscher C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS ONE. 2013;8: e58784.
Ji C, Xue G-F, Lijun C, Feng P, Li D, Li L, et al. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson’s disease by increasing expression of BNDF. Brain Res. 2016;1634:1–11.
Tamargo IA, Bader M, Li Y, Yu S-J, Wang Y, Talbot K, et al. Novel GLP-1R/GIPR co-agonist “twincretin” is neuroprotective in cell and rodent models of mild traumatic brain injury. Exp Neurol. 2017;288:176–86.
Hanssen R, Rigoux L, Kuzmanovic B, Iglesias S, Kretschmer AC, Schlamann M, et al. Liraglutide restores impaired associative learning in individuals with obesity. Nat Metab. 2023;5:1352–63.
Klausen MK, Thomsen M, Wortwein G, Fink-Jensen A. The role of glucagon-like peptide 1 (GLP-1) in addictive disorders. Br J Pharmacol. 2022;179:625–41.
Jerlhag E. The therapeutic potential of glucagon-like peptide-1 for persons with addictions based on findings from preclinical and clinical studies. Front Pharmacol. 2023;14:1063033.
Cifuentes L, Ghusn W, Feris F, Campos A, Sacoto D, De la Rosa A, et al. Phenotype tailored lifestyle intervention on weight loss and cardiometabolic risk factors in adults with obesity: a single-centre, non-randomised, proof-of-concept study. EClinicalMedicine. 2023;58: 101923.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors would like to acknowledge funding from INSERM (C.Q., D.C.), Agence Nationale de la Recherche (ANR-20-CE14-0046 to C.Q.; ANR-18-CE14-0029, ANR-21-CE14-0018, ANR-22-CE14-0016, to D.C.), University of Bordeaux’s IdEx ‘Investments for the Future’ program/GPR BRAIN_2030 (D.C.) and the Fondation pour la Recherche Médicale (EQU202303016291 to D.C.; FRM-ARF201809006962 to C.A). C.A. is supported by the Société Francophone du Diabète. C.Q. is also supported by the Société Francophone du Diabète, Société Française d’Endocrinologie, Société Française de Nutrition, Institut Benjamin Delessert and the Fyssen Foundation.
Conflicts of Interest
The authors have no competing interests to declare.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
The authors provide consent to publish.
Availability of Data and Materials
Not applicable.
Code Availability
Not applicable.
Author Contributions
CA and CQ conceptualized and wrote the manuscript. CQ and DC edited the manuscript.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Allard, C., Cota, D. & Quarta, C. Poly-Agonist Pharmacotherapies for Metabolic Diseases: Hopes and New Challenges. Drugs 84, 127–148 (2024). https://doi.org/10.1007/s40265-023-01982-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-023-01982-6