
Abstract
Spondyloarthritis (SpA) represents one of the most frequent extraintestinal manifestations of inflammatory bowel disease (IBD). Evidence of shared genetic and molecular pathways underlying both diseases is emerging, which has led to rational approaches when treating patients with concomitant diseases. Clinical efficacy of tumor necrosis factor (TNF) antagonists has been ascertained over the years, and they currently represent the cornerstone of treatment in patients with IBD and SpA, but the therapeutic armamentarium in these cases has been recently expanded. Evidence for vedolizumab is controversial, as it was associated both with improvement and development of arthralgias, while ustekinumab, the first anti-interleukin 12/23 (IL-12/23) approved for IBD, has demonstrated good efficacy, especially in peripheral arthritis, and more IL-23 inhibitors are being developed in IBD. Tofacitinib was the first Janus kinase (JAK) inhibitor to be approved in IBD, and as it demonstrated efficacy in treating ankylosing spondylitis, it may represent a good choice in axial arthritis, while more selective JAK inhibitors are yet to be approved. Unexpectedly, the first anti-IL17 that was studied in IBD (secukinumab) has shown not to be effective in treating IBD, and the role of anti-IL17 drugs in these diseases needs further investigation. Therefore, as availability of biologics and small molecules is increasing, their positioning in clinical practice is becoming more and more challenging, and multidisciplinary management needs to be implemented in both research and clinical settings in order to enhance early recognition of SpA in IBD patients, optimize treatment and ultimately improve the patients’ quality of life.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Inflammatory bowel disease (IBD) is a group of chronic idiopathic conditions, including Crohn’s disease (CD), ulcerative colitis (UC) and IBD-unclassified (IBD-U), which are characterized by a chronic relapsing–remitting pattern of inflammation of the gastrointestinal tract due to immune dysregulation. Though these conditions primarily affect the gastrointestinal tract, extraintestinal manifestations (EIMs), which are inflammatory manifestations located outside the gut, are common and have an impact both on morbidity and treatment strategies [1, 2].
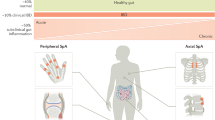
Musculoskeletal manifestations, especially those involving joints, are the most frequent EIMs [3, 4] and can be seen as a wide spectrum from unspecific joint pain without inflammatory features (arthralgia) to joint inflammation (arthritis) belonging to the spondyloarthritis (SpA) group. This category includes many arthropathies with similar clinical and imaging features, which encompasses ankylosing spondylitis (AS), reactive arthritis, psoriatic arthritis (PsA), IBD-related (or enteropathic) arthritis and undifferentiated SpA. Historically, these arthropathies have been known as seronegative due to the negativity of the rheumatoid factor and are associated with HLA-B27. More recently, these entities have been considered a continuum and according to the ASAS (Assessment of SpondyloArthritis Society) criteria as either axial SpA (axSpA) or peripheral SpA (pSpA) [5, 6]. One of the notable aspects of the new criteria has been the incorporation of the non-radiographic axial SpA (nr-AxSpA) concept and the inclusion of MRI for detection of sacroiliitis, which has led to earlier diagnosis and treatment. However, there are controversies regarding the lack of specificity of the new criteria, the high disagreement rates in imaging interpretation among non-dedicated radiologists and the highly variable progression rates from nr-AxSpA to radiographic SpA [7]. Similarly, the proposed reorganization of pSpA entities into groups based on clinical manifestations has been viewed by some experts as an advance and by others as detrimental—advances include the greater importance of enthesitis and dactylitis, and the inclusion of HLA-B27; drawbacks include the decision of not distinguishing between individual entities, which may confuse the interpretation of outcome measures and treatment responses. Moreover, it should be addressed that the diagnostic performance of both sets of classification criteria in the outpatient with low test probability of SpA is low and that these criteria could be misused. Possible overlap and change over time between degrees of axial and peripheral involvement may also pose challenges [8, 9].
AxSpA affects the spine and/or the sacroiliac joint and can be categorized into AS and sacroiliitis, whose prevalence in patients with IBD has been reported as high as 3% and 10%, respectively [10]. AS is characterized by persistent inflammatory low back pain and stiffness, which may ultimately lead to the formation of marginal syndesmophytes with a consequent fusion of vertebral bodies and ankylosis (i.e. bamboo spine). Sacroiliitis is usually bilateral, and it is characterized by inflammatory low back and buttock pain, even though a high prevalence of asymptomatic sacroiliitis has been observed, making it currently underdiagnosed. Sacroiliitis is usually HLA-B27 negative, possibly suggesting that AS and sacroiliitis may be two separate entities [11]. pSpA occurs in approximately 13% of patients with IBD [12] (10–20% among CD patients and 5–14% among UC patients) and is characterized by little or no joint destruction [12]. IBD and SpA are undoubtedly closely related, and IBD may also represent an extra-articular manifestation of SpA, being present in 4–14% of patients [13–15]. Additionally, gut inflammation has been observed in almost of 50% patients with SpA who underwent a colonoscopy [16, 17], even though a minority of patients eventually develops IBD [18].
This review focuses on the growing understanding of the underlying pathophysiology of IBD-associated SpA and the shared targets for pharmacological intervention, leading also to a brief summary of the most relevant clinical data on this topic.
2 Pathophysiology
Complex interactions between genetic, immunological, and microbial factors underlie the etiology of both IBD and SpA, with major implications for the gut-joint axis of inflammation. In an impressive prescient paper published in 1975, Zvaifler anticipated the currently hypothesized causative or correlative relationship between the gut and joint for SpA in IBD. Thanks to advances in technology for basic research and detailed tracking of well-defined patient cohorts, research in this field is producing data with unprecedented depth [19].
2.1 Genetics
At a genetic level, genealogic studies have shown shared heritability and familial clustering of IBD and AS [20]. However, even though HLA-B27 is tightly associated with AS, this link gets weaker when the IBD population with AS is considered and, in patients with IBD without AS, its prevalence does not even differ from that of the general population [21].
More recently, the era of genome-wide association studies (GWAS) has advanced the search for a shared genetic architecture between SpA and CD by identifying several risk loci for both diseases. The study performed by Danoy et al. [22] can be considered a watershed one as it showed that eight out of 53 genetic loci associated with CD in three separate GWAS were risk loci also for AS, namely IL23R (rs11465804), IL12B (rs10045431), CDKAL1 (rs6908425), LRRK2 (rs11175593), chr13q14 (rs3764147), chr1q32 (rs11584383), and STAT3 (rs6503695, rs744166). Interestingly, most of these loci are involved in gut homeostasis, unveiling the importance of the gut microbiome and mucosal immunity in both AS and CD. These results were confirmed by subsequent studies in which other risk loci implied in the gut barrier have been associated with both IBD and SpA [23–25].
2.2 Immunopathogenesis
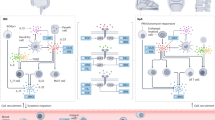
It is generally thought that these diseases share immunopathogenesis similarities and that inflammatory events in the gut precede and lead to inflammatory events in the joint. Growing evidence suggests that perturbation of gut barrier homeostasis leads to chronic intestinal inflammation. In healthy gut tissue, commensal microorganisms and dietary metabolites regulate Th3 cells (retinoic acid-related orphan receptor γt+ ILC3s, TC17 cells, and TH17 IL-23 R+ cells) in response to IL-23 stimulation. This results in IL-17 and IL-22 production that promotes epithelial cell–cell contact through tight junctions and mucus secretion by goblet cells, limiting the translocation of microorganisms through the epithelial barrier [26]. That a defective mucosal immunity is involved in the pathogenesis of AS has been suggested by Ciccia et al. [27], who observed that AxSpA patients with bacterial ileitis show increased expression of bacterial-induced zonulin, a protein that modulates the permeability of the aforementioned tight junctions.
A central role in the pathophysiology of IBD and SpA has been attributed to tumor necrosis factor (TNF), as the efficacy of TNF antagonists in these diseases is well known. TNF is a cytokine primarily secreted by macrophages, but also CD4+ lymphocytes, natural killer cells and other cells. Its primary function consists in regulating immune cells, with a mainly pro-inflammatory role, even though it is also involved in apoptosis, cachexia and tumorigenesis [28].
TNF is mainly produced as a transmembrane protein (mTNF) that can be cleaved by the TNF converting enzyme into a soluble form (sTNF) and its biological activity is exerted through its bond to specific receptors (TNFR1 and TNFR2) [29]. Through TNFR1 signaling, apoptosis of intestinal epithelial cells [30] and changes in the epithelial expression of tight junction proteins are induced [31], hence exacerbating the inflammation.
The role of TNF in inflammatory disorders has been investigated; one study showed that overexpression of TNF-α was associated with the development of chronic arthritis and colitis in mice [32], and TNF inhibition was found to attenuate mucosal inflammation in several experimental models of IBD-like colitis [33, 34]. Moreover, there is also evidence of an association of this cytokine with other immune-mediated diseases like psoriasis [35] and uveitis [36].
The first evidence of an association between SpA and TNF-α came from human sacroiliac joint specimens of patients with early AS, in which a high amount of TNF-α mRNA was found [37]. It is believed that TNF is the main cytokine involved in the early, inflammatory stages of AS, which is supported by the clinical efficacy reported with anti-TNFs [38]. TNF also induces osteoclastogenesis, which eventually leads to bone erosion—relatively mild in AS—while the role of TNF in the latest stages, with new bone formation, is less clear [38].
It is well established that dysbiosis in the gut microbiome is present in both diseases, but whether it occurs as a result of inflammation or precedes inflammatory changes, is still debated. Broadly defined, dysbiosis is the imbalance of gut microbiota associated with an unhealthy outcome, involving the loss of the beneficial commensal communities and/or the expansion of the pathogenic ones [39]. Patients with SpA have decreased fecal gut microbial diversity and increased abundance of Ruminococcus gnavus and the genus Dialister, which are positively correlated with disease activity [40, 41]. A possible role for dysbiosis in SpA has also been demonstrated in murine models, with the improvement of arthritis and inflammatory colitis features in germ-free HLA-B27-transgenic rats [42]. Similarly, the gut microbiota of IBD patients is characterized by low microbial diversity, and distinct dysbiotic profiles, resulting in lower saturated chain fatty acids (SCFA) concentrations compared with healthy individuals [43, 44]. However, it seems that the stability of the microbiome over time, rather than the degree of dysbiosis at a single time point, is predictive of gut inflammation, and microbiomes of IBD patients are more prone to fluctuations than those of healthy individuals [45].
Dysbiosis is thought to drive immune dysregulation through CXCR1+ CD59+ macrophage production of IL-23 as ILC3s IL23R + expansion is correlated with bacterial epithelial infiltration [46]. IL-23 is a heterodimeric cytokine composed of p19 and p40 subunits, the latter shared with IL-12. It has a prominent role in antigen-rich barrier surface modulation (gut barrier, skin barrier, etc.) through innate lymphoid cells and Th17 CD4+ T-cell production of IL-17 and IL-22 effector molecules [47]. IL-23 overexpression and Th3 IL-23 R+ cell expansion are the hallmarks of autoimmune diseases like IBD, SpA and psoriasis; and IL-23 characterizes the subclinical inflamed ileal gut of almost half of SpA patients [48].
In support of the gut–joint trafficking hypothesis, it has been postulated that Th3 IL-23 R+ cells, which express gut-specific trafficking receptors such as α4β7 integrin and the chemokine receptor CCR6, can recirculate from the gut and migrate to articular tissues, consequently mediating SpA inflammation [26, 49]. Photoconversion technology studies have demonstrated that cells labeled in the gut could be found in the enthesis or synovial tissue of arthritic mice models [50].
However, the mucosal vascular addressin cell adhesion molecule 1 (MADCAM-1), an α4β7 ligand, is found overexpressed only on endothelial cells from the gut and bone marrow, and not on the synovium, despite the presence of α4β7 on synovial T cells in SpA [26, 49]. Although conceptually fascinating, the lack of detectable gut inflammation in a sizable proportion of patients with SpA as well as the failure of some of the novel therapies in SpA and IBD does not support the causative hypothesis of gut-to-joint inflammation. A purely correlative scenario of gut–joint inflammation, attributable only to similarities in their immunopathogenesis, is still plausible. In this context, it should be noted that IL-23 also has a leading role in antigen-free barrier surfaces such as the joints, as IL-23-responsive cells appear to respond to high biomechanical stress [51]. Yet, the nature of the gut–joint link remains nebulous—profound knowledge of its underlying mechanisms is certainly desirable, as it has therapeutic implications for patients with IBD and SpA.
3 Targets for Pharmacological Interventions
3.1 Conventional Therapies and Immunosuppressants
Non-steroidal anti-inflammatory drugs (NSAIDs) are used in SpA as a first-line therapy; caution is generally recommended by many gastroenterologists in IBD patients, even the data on NSAID use and relapse of IBD are conflicting [51, 52].
Selective NSAIDs (e.g. celecoxib and etoricoxib) may be used, and short courses have shown not to induce clinical and endoscopic relapse both in patients with UC in remission and with CD [53, 54]. Therefore, selective NSAIDs may be used for short periods in patients with active SpA and quiescent IBD [55].
As a matter of fact, clinical practice suggests that systemic corticosteroid therapy, which is widely used as induction therapy in IBD patients, is also effective for the treatment of both AxSpA and pSpA [56], even though solid evidence is lacking. However, corticosteroids cannot be considered a maintenance treatment due to their well-known severe side effects.
Sulfasalazine and methotrexate are not recommended in patients with AxSpA [56–58] and their use in pSpA is provided only by indirect evidence in rheumatoid arthritis (RA) [59] and PsA, respectively. Sulfasalazine may be used in UC but not in CD, as two randomized controlled trials (RCTs) have shown that it was more effective than placebo for the induction and maintenance of remission in UC, but not in CD [60–63]. The current UC guideline suggests using sulfasalazine in patients who are in remission that have dominant peripheric arthritic symptoms [64]. Current data support the efficacy of methotrexate monotherapy for maintenance of clinical remission in CD [65], but not in UC [66, 67]. Therefore, it may be used as maintenance therapy in CD with concomitant peripheral arthritis, although definite data are lacking. Similarly, thiopurines (i.e. azathioprine and 6-mercaptopurine) are not recommended in this setting [56, 68].
3.2 Anti-Tumor Necrosis Factor (TNF)
Due to the involvement of TNF in the pathogenesis of autoimmune diseases, TNF antagonists have been successfully developed and applied in the clinical treatment of both IBD and SpA for 20 years [69–72].
All TNF antagonists target both sTNF and mTNF, but affinity differs greatly [73]. Accumulating evidence suggests that the efficacy of this class of drugs is due to neutralization of mTNF rather than sTNF: a study observed that the neutralization of soluble TNF alone was not effective in dampening inflammation in experimental colitis murine models [74]; moreover, a paradoxical increase of sTNF can be observed during successful treatment with infliximab or adalimumab [75]. These findings may explain why etanercept, an anti-TNF that preferentially binds to the soluble form, was not found to be effective in treating IBD and has even been associated with paradoxical new onset of IBD in patients with SpA, while infliximab and adalimumab, which preferentially bind to mTNF, are effective [76–78].
Clinical efficacy of TNF antagonists has been ascertained across many RCTs, and currently represent the cornerstone of treatment in patients with IBD and/or SpA [55, 68]. Infliximab [79], 80, adalimumab [81–83] and certolizumab [84] are effective in moderate to severe CD, while in UC, RCTs have shown infliximab [85], adalimumab [86], 87 and golimumab [88, 89] to be effective in inducing and maintaining clinical remission in patients with moderate to severe disease activity.
TNF antagonists also demonstrated efficacy in patients with SpA; infliximab [90], adalimumab [91] and golimumab [92] were all found to be effective in several RCTs in AS. As mentioned before, etanercept is also effective in this setting [72]. Although no RCT specifically addressed the efficacy of TNF antagonists in IBD-related SpA, their effectiveness has been clearly demonstrated by real-world experience [93].
However, a safety issue has emerged for anti-TNFs; due to the physiological role of TNF-α in defense against microbial pathogens and tumorigenesis, a thorough diagnostic workup is required to exclude any latent or manifest infection or malignancy before initiating treatment with anti-TNF [94], 95]. Moreover, longstanding therapy with anti-TNFs has been associated with the onset of psoriasiform skin eruptions, which can be considered paradoxical, as these agents are used to treat psoriasis. The study of Tillack et al. [96] showed the presence in these lesions of infiltrates of interferon-γ-secreting Th1 cells and IL-17A/IL-22-secreting Th17 cells. As a good response to ustekinumab, an anti-IL-12/IL-23 antibody was observed; the switch to this biologic agent may be an option in CD patients with psoriasiform skin lesions not responding to topical therapies.
3.3 Anti-Intestinal T-Cell Trafficking Drugs
Vedolizumab is a monoclonal antibody approved for IBD that targets α4β7 integrin to prevent the trafficking of immune cells to the gut [97, 98]. The efficacy of vedolizumab for the treatment of musculoskeletal manifestations in IBD is controversial.
On one hand, if a correlative relationship of the gut–joint axis is hypothesized, the gut-specificity of vedolizumab should make it ineffective for arthritis manifestations. As mentioned above, the lack of MADCAM-1 expression in synovial tissue despite the presence of α4β7 on synovial T cells in SpA provides a scientific rationale for the differential response to joint versus gut symptoms. On the other hand, the causative relationship of the gut–joint axis may explain the improvement in arthralgias in vedolizumab-treated IBD patients shown in other studies [99, 100]. Moreover, a recent post-hoc analysis of GEMINI trials in IBD reported a decreased likelihood of new or worsening arthralgia in CD and no increased incidence of these events in UC upon vedolizumab administration [101], suggesting that the control of gut inflammation may drive the response at least on arthralgias. Notably, the same study assessed that the worsening or the new onset arthralgias seen in vedolizumab-treated patients can be attributed to corticosteroid tapering/withdrawal instead of a paradoxical mechanism of action of vedolizumab.
Sphingosine 1-phosphate (S1P) modulation is another strategy to control leukocyte traffic by blocking T-cell egress from lymph nodes into the circulation. In contrast to α4β7 blockade, S1PR1 agonists may be able to downregulate both gut and joint inflammation that depends on circulating lymphocytes. Ozanimod and etrasimod have shown promising results in IBD [102–104]; etrasimod has also shown efficacy in a collagen-induced model of arthritis [105]. Recently, IMMH001 (SYL930), an S1PR1 and S1PR4 modulator, inhibited arthritis progression in Sprague-Dawley rats, diminishing pro-inflammatory cytokines and chemokines in damaged joints. Despite these encouraging results, there are no ongoing clinical studies in RA, nor specific data on IBD-associated SpA [106].
3.4 Anti-IL-17/23 Axis Drugs
Knowledge of Th3 cells has translated into the development of effective IL-23– and IL-17A–blocking antibodies for the treatment of SpA and IBD, although some discrepancies in efficacy between the two diseases need to be noted.
Ustekinumab is a fully humanized IgG1 monoclonal antibody that binds to the p40 subunit of both IL-23 and IL-12, subsequently blocking Th17 and Th1 immune responses mediated by IL-23 and IL-12, respectively. Ustekinumab, first approved for psoriasis and PsA, also showed efficacy in both CD and UC [107, 108]. Although the pivotal trials did not evaluate EIMs, a recent literature review showed ustekinumab effectiveness for arthralgia and PsA in 152 out of 254 IBD patients through three high-quality studies [109]. Also, an Italian real-world multicenter study has demonstrated that ustekinumab was able to obtain a response on articular symptoms in nearly half of the patients with CD and active SpA, even though patients with purely axial forms did not respond [110].
Risankizumab, mirikizumab, brazikumab and guselkumab specifically bind to the p19 subunit of IL-23, producing a selective inhibition of IL-23 [111]. It has been speculated that this more restricted targeting could increase overall safety, by sparing IL-12–mediated Th1 responses that are crucial for both malignancy surveillance and host immunity [112–114]. Among the IL-23p19 inhibitors, guselkumab has been approved for PsA and risankizumab for both PsA and CD by the US FDA.
The DISCOVER-1 and -2 pooled analyses in axial PsA patients showed that patients in guselkumab treatment groups had a significantly higher percentage of achieving BASDAI50 scores when compared with placebo [115, 116]. This result was quite encouraging since ustekinumab and risankizumab did not show efficacy in clinical trials of axSpA and hence IL-23 inhibitors were initially thought to be not effective on the axial skeleton. Phase IIa/IIb and III studies on these drugs are still ongoing in IBD [117–120] and promising results of phase III ADVANCE and MOTIVATE risankizumab trials have recently been published [121]. Interestingly, in three head-to-head phase II and phase III RCTs in psoriatic patients, risankizumab has resulted in improved treatment efficacy compared with ustekinumab [122, 123]. This enhanced efficacy was also demonstrated in the NAVIGATE trial with guselkumab being superior to ustekinumab in patients with moderate to severe plaque psoriasis [124].
Interestingly, all IL-23 inhibitors have a good safety profile with a low risk of serious and opportunistic infections. This clinical observation is concordant with colitis-resistant IL-23p19−/− mice that developed normal Th1 responses while being severely impaired in the development of IL-17-producing T cells [125].
IL-17A, IL-17F and IL17A/F heterodimers are prominent members of the IL-17 family of cytokines that via a common receptor subunit (IL-17RA) regulate both innate and adaptive immunity. The IL-17A–blocking secukinumab is currently approved in Europe for the treatment of axSpA, psoriasis and PsA. Considering its efficacy in these diseases and animal models of autoimmunity [126], Hueber et al. in 2012 conducted a multicentric phase IIa RCT of secukinumab in patients with moderate to severe CD. Unexpectedly, this trial was prematurely stopped as it showed a statistically significant difference in mean Crohn's Disease Activity Index (CDAI) in favor of placebo [127]. Crohn’s symptoms worsening was also observed by Targan et al. assessing the efficacy of brodalumab, an IL-17RA antibody currently approved for psoriasis with promising results on PsA [128, 129]. Multiple uncontrolled case series also support the new onset or worsening of pre-existing IBD in secukinumab-treated patients [130]. A possible explanation for this is that IL-23-independent IL-17 suppression leads to gut expansion of Candida albicans, and consequently to CD disease exacerbation [131].
On the other hand, the long-term safety results of clinical trials carried out with secukinumab for dermatological and rheumatic diseases describe a low incidence of exacerbations or new cases of IBD [132, 133]. Indeed, it should be noted that whether secukinumab unmasks subclinical IBD or induces de novo IBD is hard to prove, since psoriasis patients have already an increased risk of IBD.
Interestingly, studies in murine colitis models showed that IL-17F deficiency protected against colitis symptoms [134]. Indeed, IL-17F promotes inflammation by inhibiting the colonization of the gut by regulatory T-cell–promoting commensals. These findings indicate that specific IL-17F inhibitors, instead of anti–IL-17A ones, could be a potential option for treatment of IBD.
3.5 Janus Kinase (JAK) Inhibitors
Janus kinase (JAK) and signal transducer and activator of transcription (STAT) molecules are crucial transducers for various pro-inflammatory pathways implicated in both IBD and SpA. Respective of the several cytokines relying on JAK-STAT signaling, the inhibition of this pathway offers multiple possibilities to modulate the immune response, offering an advantage over selective cytokine inhibitors such as anti-TNFα, anti-α4β7, or anti-IL-12/23.
The JAK family is comprised of four members (JAK1, JAK2, JAK3 and TYK2) that pair to each other. For this reason, the different combinations of JAKs are associated with different cytokine receptors; thus, the inhibition of each type of JAK leads to inhibition of signaling of a specific subset of cytokines and different effects.
Tofacitinib, a pan-JAK inhibitor, was first approved for RA patients with incomplete response to conventional disease-modifying anti-rheumatic drugs [135–137]. Subsequently, tofacitinib has proven to be effective for PsA [138, 139], AS [140] and UC [141]. Disappointingly, similar efficacy data were not found in two phase IIb trials in CD patients, therefore the further development of tofacitinib in CD was stopped [142, 143]. The randomized, non-inferiority, phase IIIb/IV, ORAL Surveillance safety study compared the relative risk of major adverse cardiovascular events and malignancies of tofacitinib at two doses (5 mg or 10 mg twice daily) with anti-TNF therapy in RA patients aged >50 years with at least one cardiovascular risk factor [144]. Results showed that for the two co-primary endpoints, the non-inferiority criteria for both doses of tofacitinib compared with anti-TNFs were not met. Moreover, an increased risk of venous thromboembolism and mortality rates with the tofacitinib 10-mg dose relative to the tofacitinib 5-mg dose and anti-TNFs was found.
Despite conflicting opinions on the design and generalization of the study’s results, the ORAL study has led to changes in FDA recommendations for the use of JAK inhibitors, raising safety concerns for a higher dose of tofacitinib in a high-risk population [145, 146]. In addition, this study has evoked the question of whether more selective agents might result in fewer adverse effects. Filgotinib and upadacitinib are oral JAK1 selective inhibitors whose efficacy has been studied for both SpA and IBD.
Filgotinib has been evaluated as monotherapy or in combination with methotrexate in several RCTs in patients with RA [147–149], PsA [150] and AS [151]. The phase IIb/III SELECTION trial has demonstrated good efficacy and safety data of filgotinib in inducing and maintaining remission in patients with UC [152]; though the same exciting results, once again, were not shown for CD patients [153]. However, despite the disappointing rates for mucosal healing in these patients, the overall positive data resulted in a large phase III induction and maintenance trial for moderate to severe CD, a phase II trial for fistulizing CD and a phase II trial for small bowel CD. Efficacy of upadacitinib in PsA has also recently been reported, with the 30-mg upadacitinib group found to be superior to adalimumab [154, 155]. Upadacitinib 45 mg was also superior to placebo for induction of clinical remission in both UC and CD patients according to data presented from phase III trials [156–158]. In a recent network meta-analysis including biologics and small molecule drugs, Lasa et al. found that upadacitinib was the best performing agent for induction of clinical remission in UC patients, according to SUCRA (surface under the cumulative ranking curve) [159].
Moreover, ritlecitinib (PF-06651600), a selective JAK-3 inhibitor, is in a phase III clinical trial for UC and phase II for RA. Additional promising JAK inhibitors that are being investigated for both rheumatological diseases and IBD are ivarmacitinib (SHR0302), a JAK 1 inhibitor; PF-06700841, a TYK2/JAK1 inhibitor; deucravacitinib (BMS-986165), a TYK2 inhibitor and PF-06826647, a TYK2 inhibitor.
4 From Pathophysiology to Clinical Practice
Given the increasing shared immunosuppressive treatments, the management of patients with coexisting IBD and SpA represents a challenge for a single specialist. In fact, a multidisciplinary approach should be implemented to improve early recognition of SpA, especially if some red flags are identified (i.e. chronic low back pain, dactylitis, enthesitis and pain/swelling of peripheral joints) [160], in order to optimize treatment and decision making and ultimately improve the patients’ quality of life [161].
A possible practical clinical management of patients with both IBD and SpA is summarized in Table 1. However, in every clinical scenario, a rheumatological or gastroenterological consult is strongly recommended, especially in patients unresponsive to several lines of therapy and persistent clinical activity. Note that the quality of the evidence behind some of these recommendations is low, as few high-quality studies are available, especially regarding recently approved drugs. Finally, this brief table does not consider drugs that are not yet approved for CD or UC and may have an important role in the future, such as upadacitinib.
5 A Personalized Treatment Approach for IBD-Associated SpA: Future Perspectives
Understanding whether the pathophysiology of the gut–joint axis is of correlative or causative nature would have major clinical practice implications. If inflammation in the gut is indeed causative of joint inflammation, targeting key pathways in the gut should prevent joint manifestations. Conversely, if a simple correlation exists between gut and joint inflammation, they should be independently targeted. In this regard, almost every currently approved therapy for IBD has been designed for and experimented first in rheumatologic diseases and only at a later time in IBD; that this modus operandi is not ideal was proved by the unexpected failure of IL-17 blockade in IBD, which resulted in improving our knowledge on the gut barrier, but ultimately failed in providing new effective treatments despite the resources invested.
As availability of biologics and small molecules is increasing, their positioning in clinical practice is becoming more and more challenging. In fact, in real-world clinical practice, costs and bureaucratic difficulties in prescribing novel therapies, rather than their actual efficacy, play a huge role in choosing between one drug and another, an approach that, in addition to the common loss of response to treatments, leads to unsatisfactory results for a non-irrelevant percentage of patients and subsequently has an impact on their quality of life. An additional challenge is represented by the different response to these agents between CD and UC, whose underlying mechanisms are still basically unknown. In an ideal world, drug positioning would be personalized and based on objective findings (e.g. genetics, biomarkers, radiological or endoscopic findings, etc.) but so far clinical management has continued to rely on a ‘trial and error’ approach.
An appealing concept that could change our perspective as clinicians is represented by the association of two pharmacological agents (biologics and/or small molecules; i.e. dual targeted therapy), which is usually only considered as a ‘rescue’ therapy in cases of refractoriness. However, as new, more selective targeted therapies have been and will be developed, dual targeted therapy has the potential to become the new standard of care in patients with IBD with concomitant SpA resistant to one biological agent, if no additional safety concerns emerge.
Another crucial point is that EIMs are often overlooked in RCTs, and head-to-head comparisons of drug efficacy in patients with IBD and SpA are still lacking. Moreover, as highlighted in a recent literature review, the studies analyzing IBD-SpA cohorts are often clinical-based, single-center and cross-sectional in design with no validation of the recognized ASAS criteria [162].
In conclusion, it is undeniable that an individualized approach that involves different specialists within a multidisciplinary team is desirable and often necessary, and should be encouraged not only in the management of these patients but also in the design of clinical trials while hoping for breakthroughs provided by translational research in the field of immune-mediated diseases.
References
Magro F, Gionchetti P, Eliakim R, et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11(6):649–70. https://doi.org/10.1093/ecco-jcc/jjx008.
Gionchetti P, Dignass A, Danese S, et al. 3rd European evidence-based consensus on the diagnosis and management of crohn’s disease 2016: Part 2: surgical management and special situations. J Crohns Colitis. 2017;11(2):135–49. https://doi.org/10.1093/ecco-jcc/jjw169.
Bourikas LA, Papadakis KA. Musculoskeletal manifestations of inflammatory bowel disease. Inflamm Bowel Dis. 2009;15(12):1915–24. https://doi.org/10.1002/ibd.20942.
Algaba A, Guerra I, Ricart E, et al. Extraintestinal manifestations in patients with inflammatory bowel disease: study based on the ENEIDA registry. Dig Dis Sci. 2021;66(6):2014–23. https://doi.org/10.1007/s10620-020-06424-x.
Di Jiang C, Raine T. IBD considerations in spondyloarthritis. Ther Adv Musculoskelet Dis. 2020. https://doi.org/10.1177/1759720X20939410.
Sieper J, Rudwaleit M, Baraliakos X, et al. The Assessment of SpondyloArthritis international Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis. 2009;68:1–44. https://doi.org/10.1136/ard.2008.104018.
Ashrafi M, Ermann J, Weisman MH. Spondyloarthritis evolution: what is in your history? Curr Opin Rheumatol. 2020;32(4):321–9. https://doi.org/10.1097/BOR.0000000000000712.
Lipton S, Deodhar A. The new ASAS classification criteria for axial and peripheral spondyloarthritis: Promises and pitfalls. Int J Clin Rheumatol. 2012;7(6):675–82. https://doi.org/10.2217/ijr.12.61.
Robinson PC, van der Linden S, Khan MA, et al. Axial spondyloarthritis: concept, construct, classification and implications for therapy. Nat Rev Rheumatol. 2021;17:109–18. https://doi.org/10.1038/s41584-020-00552-4.
Karreman MC, Luime JJ, Hazes JMW, Weel AEAM. The prevalence and incidence of axial and peripheral spondyloarthritis in inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis. 2017;11(5):631–42. https://doi.org/10.1093/ecco-jcc/jjw199.
Salvarani C, Fries W. Clinical features and epidemiology of spondyloarthritides associated with inflammatory bowel disease. World J Gastroenterol. 2009;15(20):2449–55. https://doi.org/10.3748/wjg.15.2449.
Orchard TR, Wordsworth BP, Jewell DP. Peripheral arthropathies in inflammatory bowel disease: their articular distribution and natural history. Gut. 1998;42(3):387–91. https://doi.org/10.1136/gut.42.3.387.
Stolwijk C, van Tubergen A, Castillo-Ortiz JD, Boonen A. Prevalence of extra-articular manifestations in patients with ankylosing spondylitis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(1):65–73. https://doi.org/10.1136/annrheumdis-2013-203582.
de Winter JJ, van Mens LJ, van der Heijde D, Landewé R, Baeten DL. Prevalence of peripheral and extra-articular disease in ankylosing spondylitis versus non-radiographic axial spondyloarthritis: a meta-analysis. Arthritis Res Ther. 2016;18(1):196. https://doi.org/10.1186/s13075-016-1093-z.
Fragoulis GE, Liava C, Daoussis D, Akriviadis E, Garyfallos A, Dimitroulas T. Inflammatory bowel diseases and spondyloarthropathies: From pathogenesis to treatment. World J Gastroenterol. 2019;25(18):2162–76. https://doi.org/10.3748/wjg.v25.i18.2162.
Cypers H, Varkas G, Beeckman S, et al. Elevated calprotectin levels reveal bowel inflammation in spondyloarthritis. Ann Rheum Dis. 2016;75(7):1357–62. https://doi.org/10.1136/annrheumdis-2015-208025.
Leirisalo-Repo M, Turunen U, Stenman S, Helenius P, Seppälä K. High frequency of silent inflammatory bowel disease in spondylarthropathy. Arthritis Rheum. 1994;37(1):23–31. https://doi.org/10.1002/art.1780370105.
De Vos M, Mielants H, Cuvelier C, Elewaut A, Veys E. Long-term evolution of gut inflammation in patients with spondyloarthropathy. Gastroenterology. 1996;110(6):1696–703. https://doi.org/10.1053/gast.1996.v110.pm8964393.
Ashrafi M, Kuhn KA, Weisman MH. The arthritis connection to inflammatory bowel disease (IBD): why has it taken so long to understand it? RMD Open. 2021;7(1): e001558. https://doi.org/10.1136/rmdopen-2020-001558.
Thjodleifsson B, Geirsson AJ, Björnsson S, Bjarnason I. A common genetic background for inflammatory bowel disease and ankylosing spondylitis: a genealogic study in Iceland. Arthritis Rheum. 2007;56(8):2633–9. https://doi.org/10.1002/art.22812.
De Vos M. Joint involvement associated with inflammatory bowel disease. Dig Dis. 2009;27(4):511–5. https://doi.org/10.1159/000233290.
Danoy P, Pryce K, Hadler J, et al. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn’s disease. PLoS Genet. 2011. https://doi.org/10.1371/annotation/0ee7d13b-c55e-4be6-ab3e-8e8df5bb2c97.
Yang X, Li M, Wang L, Hu Z, Zhang Y, Yang Q. Association of KIF21B genetic polymorphisms with ankylosing spondylitis in a Chinese Han population of Shandong Province. Clin Rheumatol. 2015;34(10):1729–36. https://doi.org/10.1007/s10067-014-2761-5.
Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40(8):955–62. https://doi.org/10.1038/ng.175.
Ellinghaus D, Jostins L, Spain SL, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. 2016;48(5):510–8. https://doi.org/10.1038/ng.3528.
Gracey E, Vereecke L, McGovern D, et al. Revisiting the gut-joint axis: links between gut inflammation and spondyloarthritis. Nat Rev Rheumatol. 2020;16(8):415–33. https://doi.org/10.1038/s41584-020-0454-9.
Ciccia F, Guggino G, Rizzo A, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis. 2017;76(6):1123–32. https://doi.org/10.1136/annrheumdis-2016-210000.
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. https://doi.org/10.1016/s0092-8674(01)00237-9.
MacEwan DJ. TNF ligands and receptors–a matter of life and death. Br J Pharmacol. 2002;135(4):855–75. https://doi.org/10.1038/sj.bjp.0704549.
Schulzke JD, Bojarski C, Zeissig S, Heller F, Gitter AH, Fromm M. Disrupted barrier function through epithelial cell apoptosis. Ann N Y Acad Sci. 2006;1072:288–99. https://doi.org/10.1196/annals.1326.027.
Zeissig S, Bürgel N, Günzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56(1):61–72. https://doi.org/10.1136/gut.2006.094375.
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–98. https://doi.org/10.1016/s1074-7613(00)80038-2.
Neurath MF, Fuss I, Pasparakis M, et al. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur J Immunol. 1997;27(7):1743–50. https://doi.org/10.1002/eji.1830270722.
Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1(7):553–62. https://doi.org/10.1016/1074-7613(94)90045-0.
Ogawa E, Sato Y, Minagawa A, Okuyama R. Pathogenesis of psoriasis and development of treatment. J Dermatol. 2018;45(3):264–72. https://doi.org/10.1111/1346-8138.14139.
Wakefield D, Yates W, Amjadi S, McCluskey P. HLA-B27 anterior uveitis: immunology and immunopathology. Ocul Immunol Inflamm. 2016;24(4):450–9. https://doi.org/10.3109/09273948.2016.1158283.
Braun J, Bollow M, Neure L, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 1995;38(4):499–505. https://doi.org/10.1002/art.1780380407.
Lata M, Hettinghouse AS, Liu CJ. Targeting tumor necrosis factor receptors in ankylosing spondylitis. Ann N Y Acad Sci. 2019;1442(1):5–16. https://doi.org/10.1111/nyas.13933.
Zeng MY, Inohara N, Nuñez G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017;10(1):18–26. https://doi.org/10.1038/mi.2016.75.
Breban M, Tap J, Leboime A, et al. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann Rheum Dis. 2017;76:1614–22.
Tito RY, Cypers H, Joossens M, et al. Brief report: Dialister as a microbial marker of disease activity in spondyloarthritis. Arthritis Rheumatol. 2017;69:114–21.
Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359–64.
Andoh A, Kuzuoka H, Tsujikawa T, et al. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn’s disease. J Gastroenterol. 2012;47(12):1298–307. https://doi.org/10.1007/s00535-012-0605-0.
Huda-Faujan N, Abdulamir AS, Fatimah AB, et al. The impact of the level of the intestinal short chain Fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem J. 2010;4:53–8. https://doi.org/10.2174/1874091X01004010053.
Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;13(2):17004.
Ciccia F, Guggino G, Zeng M, et al. Proinflammatory CX3CR1+CD59+ tumor necrosis factor–like molecule 1A+Interleukin-23+ monocytes are expanded in patients with ankylosing spondylitis and modulate innate lymphoid cell 3 immune functions. Arthritis Rheum. 2018;70(12):2003–13.
Sherlock JP, Cua DJ. Interleukin-23 in perspective. Rheumatology. 2021;60(4):1–3. https://doi.org/10.1093/rheumatology/keab461.
Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009;60:955–65. https://doi.org/10.1002/art.24389.
Salmi M, Jalkanen S. Human leukocyte subpopulations from inflamed gut bind to joint vasculature using distinct sets of adhesion molecules. J Immunol. 2001;166(7):4650–7. https://doi.org/10.4049/jimmunol.166.7.4650.
Norman E, Lefferts A, Kuhn K. Gut-joint T cell trafficking in a model of bacteria-driven murine IBD-SpA [abstract]. Arthritis Rheumatol. 2018;70:1828.
Evans JM, McMahon AD, Murray FE, McDevitt DG, MacDonald TM. Non-steroidal anti-inflammatory drugs are associated with emergency admission to hospital for colitis due to inflammatory bowel disease. Gut. 1997;40(5):619–22. https://doi.org/10.1136/gut.40.5.619.
Bonner GF, Fakhri A, Vennamaneni SR. A long-term cohort study of nonsteroidal anti-inflammatory drug use and disease activity in outpatients with inflammatory bowel disease. Inflamm Bowel Dis. 2004;10(6):751–7. https://doi.org/10.1097/00054725-200411000-00009.
Sandborn WJ, Stenson WF, Brynskov J, et al. Safety of celecoxib in patients with ulcerative colitis in remission: a randomized, placebo-controlled, pilot study. Clin Gastroenterol Hepatol. 2006;4(2):203–11. https://doi.org/10.1016/j.cgh.2005.12.002.
El Miedany Y, Youssef S, Ahmed I, El Gaafary M. The gastrointestinal safety and effect on disease activity of etoricoxib, a selective cox-2 inhibitor in inflammatory bowel diseases. Am J Gastroenterol. 2006;101(2):311–7. https://doi.org/10.1111/j.1572-0241.2006.00384.x.
Pouillon L, Bossuyt P, Vanderstukken J, et al. Management of patients with inflammatory bowel disease and spondyloarthritis. Expert Rev Clin Pharmacol. 2017;10(12):1363–74. https://doi.org/10.1080/17512433.2017.1377609.
van der Heijde D, Ramiro S, Landewé R, et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis. 2017;76(6):978–91. https://doi.org/10.1136/annrheumdis-2016-210770.
Braun J, Zochling J, Baraliakos X, et al. Efficacy of sulfasalazine in patients with inflammatory back pain due to undifferentiated spondyloarthritis and early ankylosing spondylitis: a multicentre randomised controlled trial. Ann Rheum Dis. 2006;65(9):1147–53. https://doi.org/10.1136/ard.2006.052878.
Chen J, Lin S, Liu C. Sulfasalazine for ankylosing spondylitis. Cochrane Database Syst Rev. 2014;11:4800. https://doi.org/10.1002/14651858.CD004800.pub3.
Dougados M, Vam der Linden S, Leirisalo-Repo M, et al. Sulfasalazine in the treatment of spondylarthropathy. A randomized, multicenter, double-blind, placebo-controlled study. Arthritis Rheum. 1995;38:618–27. https://doi.org/10.1002/art.1780380507.
Baron JH, Connell AM, Lennard-Jones JE, et al. Sulphasalazine and salicylazosulphadimidine in ulcerative colitis. Lancet. 1962;1:1094–6.
Dick AP, Carpenter RB, Petrie A. Controlled trial of sulphasalazine in the treatment of ulcerative colitis. Br Med J. 1964;5:437–42.
Dissanayake AS, Truelove SC. A controlled therapeutic trial of long-term maintenance treatment of ulcerative colitis with sulphazalazine (Salazopyrin). Gut. 1973;14:923–6.
Misiewitz LJJ, Connell AM, Baron JH, et al. Controlled trial of sulfasalazine in maintenance therapy for ulcerative colitis. Lancet. 1965;1:185–8.
Ko CW, Singh S, Feuerstein JD, Falck-Ytter C, Falck-Ytter Y, Cross RK. American Gastroenterological Association Institute Clinical Guidelines Committee AGA Clinical Practice Guidelines on the Management of Mild-to-Moderate Ulcerative Colitis. Gastroenterology. 2019;156(3):748–64.
Feagan BG, Rochon J, Fedorak RN, Irvine EJ, Wild G, Sutherland L, Steinhart AH, Greenberg GR, Gillies R, Hopkins M, et al. Methotrexate for the treatment of Crohn’s disease The North American Crohn’s Study Group Investigators. N Engl J Med. 1995;332(5):292–7. https://doi.org/10.1056/NEJM199502023320503.
Herfarth H, Barnes EL, Valentine JF, et al. Methotrexate is not superior to placebo in maintaining steroid-free response or remission in ulcerative colitis. Gastroenterology. 2018;155(4):1098–108. https://doi.org/10.1053/j.gastro.2018.06.046.
Macaluso FS, Renna S, Cottone M, Orlando A. The METEOR trial: the burial of methotrexate in ulcerative colitis? Gastroenterology. 2016;151(1):211–2. https://doi.org/10.1053/j.gastro.2016.02.085.
Olivieri I, Cantini F, Castiglione F, et al. Italian Expert Panel on the management of patients with coexisting spondyloarthritis and inflammatory bowel disease. Autoimmun Rev. 2014;13(8):822–30. https://doi.org/10.1016/j.autrev.2014.04.003.
Brandt J, Haibel H, Reddig J, et al. Successful short term treatment of severe undifferentiated spondyloarthropathy with the anti-tumor necrosis factor-alpha monoclonal antibody infliximab. J Rheumatol. 2002;29:118–22.
Raun J, Baraliakos X, Brandt J, Listing J, Zink A, Alten R, Burmester G, Gromnica-Ihle E, Kellner H, Schneider M, Sörensen H, Zeidler H, Sieper J. Persistent clinical response to the anti-TNF-alpha antibody infliximab in patients with ankylosing spondylitis over 3 years. Rheumatology (Oxford). 2005;44:670–6. https://doi.org/10.1093/rheumatology/keh584.
Baeten D, Van den BF, Kruithof E, Mielants H, Veys EM. Infliximab in patients who have spondyloarthropathy: clinical efficacy, safety, and biological immunomodulation. Rheum Dis Clin North Am. 2003;29:463–79.
Brandt J, Khariouzov A, Listing J, Haibel H, Sorensen H, Rudwaleit M, Sieper J, Braun J. Successful short term treatment of patients with severe undifferentiated spondyloarthritis with the anti-tumor necrosis factor-alpha fusion receptor protein etanercept. J Rheumatol. 2004;31:531–8.
Billmeier U, Dieterich W, Neurath MF, Atreya R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol. 2016;22(42):9300–13. https://doi.org/10.3748/wjg.v22.i42.9300.
Perrier C, de Hertogh G, Cremer J, et al. Neutralization of membrane TNF, but not soluble TNF, is crucial for the treatment of experimental colitis. Inflamm Bowel Dis. 2013;19(2):246–53. https://doi.org/10.1002/ibd.23023.
Eder P, Korybalska K, Łykowska-Szuber L, et al. An increase in serum tumour necrosis factor-α during anti-tumour necrosis factor-α therapy for Crohn’s disease - A paradox or a predictive index? Dig Liver Dis. 2016;48(10):1168–71. https://doi.org/10.1016/j.dld.2016.06.038.
Braun J, van der Horst-Bruinsma IE, Huang F, Burgos-Vargas R, Vlahos B, Koenig AS, Freundlich B. Clinical efficacy and safety of etanercept versus sulfasalazine in patients with ankylosing spondylitis: a randomized, double-blind trial. Arthritis Rheum. 2011;63:1543–51.
Sandborn WJ, Hanauer SB, Katz S, et al. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121:1088–94.
Toussirot E, Houvenagel E, Goeb V, et al. Development of inflammatory bowel disease during anti-TNF-alpha therapy for inflammatory rheumatic disease: a nationwide series. Joint Bone Spine. 2012;79:457–63.
Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541–9. https://doi.org/10.1016/S0140-6736(02)08512-4.
Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337(15):1029–35. https://doi.org/10.1056/NEJM199710093371502.
Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology. 2006;130(2):323–591. https://doi.org/10.1053/j.gastro.2005.11.030.
Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology. 2007;132(1):52–65. https://doi.org/10.1053/j.gastro.2006.11.041.
Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med. 2007;146(12):829–38. https://doi.org/10.7326/0003-4819-146-12-200706190-00159.
Schreiber S, Rutgeerts P, Fedorak RN, et al. A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn’s disease. Gastroenterology. 2005;129(3):807–18. https://doi.org/10.1053/j.gastro.2005.06.064.
Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462–76. https://doi.org/10.1056/NEJMoa050516.
Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60(6):780–7. https://doi.org/10.1136/gut.2010.221127.
Sandborn WJ, van Assche G, Reinisch W, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142(2):257–65. https://doi.org/10.1053/j.gastro.2011.10.032.
Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):85-e15. https://doi.org/10.1053/j.gastro.2013.05.048.
Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):96-109.e1. https://doi.org/10.1053/j.gastro.2013.06.010.
Braun J, Brandt J, Listing J, et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet. 2002;359(9313):1187–93. https://doi.org/10.1016/s0140-6736(02)08215-6.
van der Heijde D, Kivitz A, Schiff MH, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2006;54(7):2136–46. https://doi.org/10.1002/art.21913.
Inman RD, Davis JC Jr, Heijde D, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum. 2008;58(11):3402–12. https://doi.org/10.1002/art.23969.
Vavricka SR, Gubler M, Gantenbein C, et al. Anti-TNF treatment for extraintestinal manifestations of inflammatory bowel disease in the Swiss IBD cohort study. Inflamm Bowel Dis. 2017;23(7):1174–81. https://doi.org/10.1097/MIB.0000000000001109.
Rahier JF. Prevention and management of infectious complications in IBD. Dig Dis. 2012;30(4):408–14. https://doi.org/10.1159/000338143.
Beaugerie L, Rahier JF, Kirchgesner J. Predicting, preventing, and managing treatment-related complications in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2020;18(6):1324-1335.e2. https://doi.org/10.1016/j.cgh.2020.02.009.
Tillack C, Ehmann LM, Friedrich M, et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-gamma-expressing Th1 cells and IL-17A/ IL-22-expressing Th17 cells and respond to anti-IL12/IL-23 antibody treatment. Gut. 2014;63:567–77.
Vermeire S, Loftus EV Jr, Colombel JF, et al. Long-term efficacy of vedolizumab for crohn’s disease. J Crohns Colitis. 2017;11(4):412–24.
Loftus EV Jr, Colombel JF, Feagan BG, et al. Long-term efficacy of vedolizumab for ulcerative colitis. J Crohns Colitis. 2017;11(4):400–11.
Tadbiri S, et al. Impact of vedolizumab therapy on extra-intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV-IBD cohort. Aliment Pharmacol Ther. 2018;47:485–93.
Macaluso FS, Orlando R, Fries W, Scolaro M, Magnano A, Pluchino D, Cappello M, Morreale GC, Siringo S, Privitera AC, Ferracane C, Belluardo N, Alberghina N, Ventimiglia M, Rizzuto G, Renna S, Cottone M, Orlando A. The real-world effectiveness of vedolizumab on intestinal and articular outcomes in inflammatory bowel diseases. Dig Liver Dis. 2018;50(7):675–81. https://doi.org/10.1016/j.dld.2018.02.013.
Feagan BG, Sandborn WJ, Colombel JF, et al. Incidence of arthritis/arthralgia in inflammatory bowel disease with long-term vedolizumab treatment: post hoc analyses of the GEMINI trials. J Crohns Colitis. 2019;13(1):50–7. https://doi.org/10.1093/ecco-jcc/jjy125.
Feagan BG, Sandborn WJ, Danese S, et al. Ozanimod induction therapy for patients with moderate to severe Crohn’s Disease: a single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol Hepatol. 2020;5:819–28.
Sandborn WJ, Feagan BG, Dhaens G, et al. True North Study Group. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2021;385(14):1280–91. https://doi.org/10.1056/NEJMoa2033617.
Vermeire S, Chiorean M, Panés J, et al. Long-term safety and efficacy of etrasimod for ulcerative colitis: results from the open-label extension of the OASIS study. J Crohns Colitis. 2021;15(6):950–9. https://doi.org/10.1093/ecco-jcc/jjab016.
Tsunemi S, Iwasaki T, Kitano S, Imado T, Miyazawa K, Sano H. Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clin Immunol. 2010;136(2):197–204.
Jin J, Ji M, Fu R, et al. Sphingosine-1-phosphate receptor subtype 1 (S1P1) modulator IMMH001 regulates adjuvant- and collagen-induced arthritis. Front Pharmacol. 2019;10:1085.
Feagan BG, Sandborn WJ, Gasink C, et al. UNITI–IM-UNITI Study Group. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2016;375(20):1946–60. https://doi.org/10.1056/NEJMoa1602773.
Sands BE, Sandborn WJ, Panaccione R, O’Brien CD, Zhang H, Johanns J, Adedokun OJ, Li K, Peyrin-Biroulet L, Van Assche G, Danese S, Targan S, Abreu MT, Hisamatsu T, Szapary P, Marano C. UNIFI Study Group Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201–14. https://doi.org/10.1056/NEJMoa1900750.
Guillo L, D’Amico F, Danese S, Peyrin-Biroulet L. Ustekinumab for extra-intestinal manifestations of inflammatory bowel disease: a systematic literature review. J Crohns Colitis. 2021;15(7):1236–43. https://doi.org/10.1093/ecco-jcc/jjaa260.
Macaluso FS, Fries W, Viola A, Costantino G, Muscianisi M, Cappello M, Guida L, Giuffrida E, Magnano A, Pluchino D, Ferracane C, Magrì G, Di Mitri R, Mocciaro F, Privitera AC, Camilleri S, Garufi S, Renna S, Casà A, Scrivo B, Ventimiglia M, Orlando A. Effectiveness of ustekinumab on crohn’s disease associated spondyloarthropathy: real-world data from the sicilian network for inflammatory bowel diseases (SN-IBD). Expert Opin Biol Ther. 2020;20(11):1381–4. https://doi.org/10.1080/14712598.2020.1830057.
Macaluso FS, Orlando A, Cottone M. Anti-interleukin-12 and anti-interleukin-23 agents in Crohn’s disease. Expert Opin Biol Ther. 2019;19(2):89–98.
Bowman EP, Chackerian AA, Cua DJ. Rationale and safety of anti-interleukin-23 and anti interleukin-17A therapy. Curr Opin Infect Dis. 2006;19(245–52):529.
Fieschi C, Casanova JL. The role of interleukin-12 in human infectious diseases: only a faint signature. Eur J Immunol. 2003;33(1461–4):531.
Meeran SM, Mantena SK, Meleth S, Elmets CA, Katiyar SK. Interleukin-12-deficient mice are 532 at greater risk of UV radiation-induced skin tumors and malignant transformation of papillomas to 533 carcinomas. Mol Cancer Ther. 2006;5:825–32.
Mease PJ, Rahman P, Gottlieb AB, Kollmeier AP, Hsia EC, Xu XL, Sheng S, Agarwal P, Zhou B, Zhuang Y, van der Heijde D, McInnes IB. DISCOVER-2 Study Group Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395(10230):1126–36. https://doi.org/10.1016/S0140-6736(20)30263-4.
Deodhar A, Helliwell PS, Boehncke WH, Kollmeier AP, Hsia EC, Subramanian RA, Xu XL, Sheng S, Agarwal P, Zhou B, Zhuang Y, Ritchlin CT. DISCOVER-1 Study Group Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395(10230):1115–25. https://doi.org/10.1016/S0140-6736(20)30265-8.
Danese S, Sandborn WJ, Feagan BG, et al. The effect of guselkumab induction therapy on early clinical outcome measures in patients with Moderately to Severely Active Crohn’s Disease: Results from the phase 2 GALAXI 1 study (abstract)
Hanžel J, D’Haens GR. Anti-interleukin-23 agents for the treatment of ulcerative colitis. Expert Opin Biol Ther. 2020;20(4):399–406. https://doi.org/10.1080/14712598.2020.1697227.
Sandborn WJ, Ferrante M, Bhandari BR, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with ulcerative colitis. Gastroenterology. 2020;158(3):537-549.e10. https://doi.org/10.1053/j.gastro.2019.08.043.
Sands BE, Peyrin-Biroulet L, Kierkus J, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with Crohn’s disease. Gastroenterology. 2022;162(2):495–508. https://doi.org/10.1053/j.gastro.2021.10.050.
Schreiber SW, Ferrante M, Panaccione R, Colombel JF, Hisamatsu T, et al. OP26 risankizumab induces early clinical remission and response in patients with moderate-to-severe Crohn’s disease: Results from the phase 3 ADVANCE and MOTIVATE studies. J Crohn’s Colitis. 2021;15(1):S026–7. https://doi.org/10.1093/ecco-jcc/jjab075.025.
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(1551–60):541.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–61.
Langley RG, Tsai TF, Flavin S, et al. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double-blind, phase III NAVIGATE trial. Br J Dermatol. 2018;178(1):114–23. https://doi.org/10.1111/bjd.15750.
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–40. https://doi.org/10.1084/jem.20041257.
Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2(52):52–72. https://doi.org/10.1126/scitranslmed.3001107.
Hueber W, Sands BE, Lewitzky S, et al. Secukinumab in Crohn’s Disease Study Group Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–700.
Targan SR, Feagan B, Vermeire S, et al. A randomized, double-blind, placebo-controlled phase 2 study of brodalumab in patients with moderate-to-severe crohn’s disease. Am J Gastroenterol. 2016;111(11):1599–607. https://doi.org/10.1038/ajg.2016.298.
Mease PJ, Helliwell PS, Hjuler KF, et al. Brodalumab in psoriatic arthritis: results from the randomised phase III AMVISION-1 and AMVISION-2 trials. Ann Rheum Dis. 2021;80:185–93.
Kukol W, Jose LA, Marino D. P055 Development of Crohn’s disease with use of secukinumab. Am J Gastroenterol. 2019. https://doi.org/10.14309/01.ajg.0000578292.95094.87.
Colombel JF, Sendid B, Jouault T, et al. Secukinumab failure in Crohn’s disease: the yeast connection? Gut. 2013;62:800–1.
Van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.e4.
Baeten D, Sieper J, Braun J, et al. MEASURE 1 Study Group. MEASURE 2 Study Group Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. 2015;373:2534–48.
Tang C, Kakuta S, Shimizu K, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat Immunol. 2018;19(7):755–65.
Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–86.
Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebocontrolled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507.
Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin-Mola E, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–61.
Mease P, Hall S, FitzGerald O, van der Heijde D, Merola JF, Avila-Zapata F, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med. 2017;377:1537–50.
Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med. 2017;377:1525–36.
Deodhar A, Sliwinska-Stanczyk P, Xu H, et al. Tofacitinib for the treatment of ankylosing spondylitis: a phase III, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2021;80:1004–13.
Sandborn WJ, Su C, Panes J. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;377(5):496–7. https://doi.org/10.1056/NEJMc1707500.
Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W. Study A3921043 Investigators A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12:1485–93.
Panés J, Sandborn WJ, Schreiber S, et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: results of two phase IIb randomised placebo-controlled trials. Gut. 2017;66:1049–59.
Ytterberg SR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–26.
U.S. Food and Drug Administration. FDA approves Boxed Warning about increased risk of blood clots and death with higher dose of arthritis and ulcerative colitis medicine tofacitinib (Xeljanz, Xeljanz XR). https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-blood-clots-and-death-higher-dose-arthritis-and (2021).
Winthrop KL, Cohen SB. Oral surveillance and JAK inhibitor safety: the theory of relativity. Nat Rev Rheumatol. 2022. https://doi.org/10.1038/s41584-022-00767-7.
Genovese MC, Kalunian K, Gottenberg JE, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA. 2019;322:315–25.
Kavanaugh A, Kremer J, Ponce L, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. 2017;76:1009–19.
Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis. 2017;76:998–1008.
Mease P, Coates LC, Helliwell PS, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): results from a randomised, placebocontrolled, phase 2 trial. Lancet. 2018;392:2367–77.
van der Heijde D, Baraliakos X, Gensler LS, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392:2378–87.
Feagan BG, Danese S, Loftus EV Jr. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet. 2021;397(10292):2372–84. https://doi.org/10.1016/S0140-6736(21)00666-8.
Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. 2017;389:266–75.
Mease PJ, Lertratanakul A, Anderson JK, Papp K, Van den Bosch F, Tsuji S, et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann Rheum Dis. 2020;2:2.
McInnes IB, Anderson JK, Magrey M, Merola JF, Liu Y, Kishimoto M, et al. Trial of upadacitinib and adalimumab for psoriatic arthritis. N Engl J Med. 2021;384:1227–39.
Vermeire, S et al. OP23 Efficacy and safety of upadacitinib as induction therapy in patients with moderately to severely active ulcerative colitis: results from phase 3 U-ACCOMPLISH study. ECCO presentation 2021
Danese S. et al. OP24 Efficacy and safety of upadacitinib induction therapy in patients with Moderately to Severely Active Ulcerative Colitis: Results from the phase 3 U-ACHIEVE study. ECCO presentation 2021
AbbVie. Data on File: ABVRRTI73568.
Lasa JS, Olivera PA, Danese S, Peyrin-Biroulet L. Efficacy and safety of biologics and small molecule drugs for patients with moderate-to-severe ulcerative colitis: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2022;7(2):161–70. https://doi.org/10.1016/S2468-1253(21)00377-0.
Felice C, Leccese P, Scudeller L, et al. Red flags for appropriate referral to the gastroenterologist and the rheumatologist of patients with inflammatory bowel disease and spondyloarthritis. Clin Exp Immunol. 2019;196(1):123–38. https://doi.org/10.1111/cei.13246.
Rizzello F, Olivieri I, Armuzzi A, et al. Multidisciplinary management of spondyloarthritis-related immune-mediated inflammatory disease. Adv Ther. 2018;35(4):545–62. https://doi.org/10.1007/s12325-018-0672-6.
Schwartzman M, Ermann J, Kuhn KA, Schwartzman S, Weisman MH. Spondyloarthritis in inflammatory bowel disease cohorts: systematic literature review and critical appraisal of study designs. RMD Open. 2022;8(1): e001777. https://doi.org/10.1136/rmdopen-2021-001777.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors received no specific funding for this work.
Conflicts of interest
FSM served as an advisory board member and/or received lecture grants from AbbVie, Biogen, Galapagos, Janssen, MSD, Pfizer, Samsung Bioepis, and Takeda Pharmaceuticals. AO served as an advisory board member for AbbVie, Galapagos, MSD, Janssen, Pfizer, Takeda Pharmaceuticals, and received lecture grants from AbbVie, MSD, Sofar, Chiesi, Janssen, Pfizer, and Takeda Pharmaceuticals. AO served as an advisory board member for AbbVie, MSD, Janssen, Pfizer, Takeda Pharmaceuticals, and received lecture grants from AbbVie, MSD, Sofar, Chiesi, Janssen, Pfizer, and Takeda Pharmaceuticals. SR served as an advisory board member for AbbVie, Janssen and MSD Pharmaceuticals, and received lecture grants from AbbVie, Janssen, MSD and Takeda Pharmaceuticals. FC, MG, EMB, NM, GR, AC declared no conflicting interests.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Author contributions
FC and MG drafted the article. FC, MG, EMB, NM, GR, AC were responsible for critical revision of the manuscript for important intellectual content. FSM and AO were responsible for study supervision. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Rights and permissions
About this article

Cite this article
Crispino, F., Grova, M., Bruno, E.M. et al. Spondyloarthropathy in Inflammatory Bowel Disease: From Pathophysiology to Pharmacological Targets. Drugs 82, 1151–1163 (2022). https://doi.org/10.1007/s40265-022-01750-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01750-y