
Abstract
Mantle cell lymphoma is a rare B-cell non-Hodgkin’s lymphoma that retains a sobering prognosis despite an extensive research effort. Mantle cell lymphoma remains incurable even with aggressive, and at times toxic, chemoimmunotherapy with early incorporation of autologous stem cell transplantation. Given this, attention has turned to the use of targeted therapies addressing dysregulation of B-cell signaling pathways. Drugs such as immunomodulatory agents, proteasome inhibitors, and Bruton’s tyrosine kinase inhibitors have shown success in the relapsed/refractory population, and there is ongoing investigation into the utilization of novel Bruton’s tyrosine kinase, B-cell leukemia/lymphoma-2, and spleen tyrosine kinase inhibitors alone or in combination in both the front-line and relapsed settings. Other areas of research in novel immunotherapies include investigations of bispecific T-cell engagers and antibody-drug conjugates. Most recently, chimeric antigen receptor T-cell therapy has been granted US Food and Drug Administration approval as a result of durable remissions even in high-risk patients who have classically done poorly with traditional chemoimmunotherapy. The intent of this article is to review the literature describing these selective therapies and discuss their current and future roles in the treatment of mantle cell lymphoma.
Similar content being viewed by others
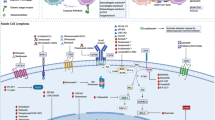

Avoid common mistakes on your manuscript.
Mantle cell lymphoma has remained incurable despite increasing attention to research on targeted therapies for use following upfront chemoimmunotherapy and autologous stem cell transplantation. |
Current options in the relapsed/refractory setting include Bruton’s tyrosine kinase inhibitors, proteasome inhibitors, and immunomodulatory agents. Many of these are being investigated in combination with other novel agents such as B-cell leukemia/lymphoma-2 inhibitors and immunotherapies, and there is growing evidence for the use of antibody-drug conjugates and bispecific T-cell engagers. |
Chimeric antigen receptor T cells represent an option for the management of aggressive disease, and have shown favorable results in patients whose disease displays high-risk characteristics such as blastoid histology and TP53 mutations. |
1 Introduction
Mantle cell lymphoma (MCL) is a rare B-cell malignancy comprising about 3–10% of all non-Hodgkin’s lymphomas (NHLs) [1]. The median age at diagnosis is 68 years and there is a male predominance with a ratio of 2.5:1 [2]. Mantle cell lymphoma classically involves the lymph nodes, but extranodal disease, particularly of the spleen, bone marrow, Waldeyer ring, and the gastrointestinal tract is common [1, 3]. This extra-nodal involvement accounts for the high frequency of advanced-stage disease at diagnosis, in fact, some guidelines recommend endoscopy to assess for occult extranodal disease of the gastrointestinal tract because of the rarity of a truly limited stage presentation [4]. Mantle cell lymphoma is a heterogenous disease as some patients can present with a leukemic non-nodal variant, which is typically more indolent and can be managed expectantly [5], while others exhibit a much more aggressive course with frequent relapses.
Classical MCL arises from mature B cells that have undergone an (11; 14) translocation with resulting overexpression of nuclear cyclin D1. The archetypal immunohistochemical profile is positive for BCL-2, CD5, and FMC7, while being negative for CD10, BCL-6, and CD23. Rarely, MCL can be negative for cyclin D1. In these cases, expression of SOX11, a transcription factor that has been implicated in the pathogenesis of the malignant cells, can help define a lymphoma as mantle cell with high specificity [6, 7].
There are morphological, proliferative, and mutational markers that affect prognosis. Lymphomas comprising cells with a large blastic appearance are characterized as pleomorphic or blastoid variants. Found in about 10% of MCL, these cytologic subtypes are associated with an inferior prognosis and poor response to conventional chemotherapy [8, 9]. This may be due to the overlap with increased Ki67 indices in these subtypes, which has been consistently shown to correlate with worse overall survival (OS) with increasing proliferation rates [10, 11]. The Mantle Cell Lymphoma International Prognostic Index was developed to help divide those with MCL into three separate groups with differing median OS based on age, Eastern Cooperative Oncology Group performance status, lactate dehydrogenase, and leukocyte count, and the Ki67 rate is also incorporated into the biologic index (Mantle Cell Lymphoma International Prognostic Index) [12]. In addition, those with a mutation in TP53 have also been shown to have an extremely poor prognosis despite intensive therapy [13].
At the time of initial diagnosis, some patients may be able to undergo close observation without an impact on OS. While a strict definition of which patients should be observed at initial presentation has yet to be developed, several retrospective studies have suggested that asymptomatic patients with a leukemic-only presentation, low nodal burden, low Ki67, or early-stage disease may be good candidates [14,15,16].
Overall survival for patients with MCL used to be 3–5 years [1]; however, as discussed below, recent clinical trial data show some patients now far exceed that expectation. Optimization of treatment for patients whose disease exhibits a more aggressive clinical course, particularly those with TP53 mutations or blastoid histology, and in those patients who disease quickly relapses following initial upfront therapy remains an unmet need in the field.
2 Initial Chemoimmunotherapy and the Role of Transplantation
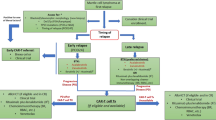
First-line therapy for MCL outside of a clinical trial should be determined by the patient’s ability to tolerate intensive chemoimmunotherapy and their eligibility for eventual autologous stem cell transplantation (ASCT) (Fig. 1). In young fit patients, there are data to support intensive induction with high-dose cytarabine-containing regimens as discussed below. Autologous stem cell transplantation in the first remission has been shown to increase progression-free survival (PFS), but not OS [17, 18]. Intensive chemoimmunotherapy regimens are available for younger patients and often incorporate the use of high-dose cytarabine. The Nordic Lymphoma Group assessed dose-intensified R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) alternating with rituximab and high-dose cytarabine followed by autologous transplantation, and showed a median OS of greater than 10 years [19]. R-CHOP alternating with R-DHAP (rituximab, dexamethasone, high-dose cytarabine, and cisplatin) followed by ASCT vs R-CHOP followed by ASCT was evaluated in the MCL-Younger trial. It identified a longer time to treatment failure with a median of 9.1 years vs 3.9 years in favor of the cytarabine-containing regimen, although there was no OS benefit [20]. R-Hyper-CVAD alternating with high-dose methotrexate and cytarabine without immediate consolidative ASCT was pioneered by the MD Anderson Group. Long-term follow-up of this regimen showed a median time to treatment failure of 4.6 years and a median OS that had not been reached at 10 years [21]. However, other evaluations of this regimen in multicenter studies have been fraught with high rates of toxicity and limitations in completing therapy, making older patients ineligible for this approach [22, 23].

Approach to mantle cell lymphoma (MCL) utilizing targeted agents. ASCT autologous stem cell transplantation, BR endamustine and rituximab, BTK Bruton’s tyrosine kinase, CAR-T chimeric antigen receptor T-cell, DOR duration of response, R2 lenalidomide and rituximab, R-CHOP rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone, VR-CAP bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone
There are patient groups who do not benefit from approaches containing high-dose cytarabine, even if they are eligible for this approach to induction. In an analysis of 183 patients treated with front-line chemotherapy containing high-dose cytarabine, a TP53 mutation was shown to portend a median OS of 1.7 years vs 12.7 years for those without a mutation [13]. Therefore, for these patients, we strongly consider clinical trial enrollment with novel therapies, such as the ongoing BOVEN trial with zanubrutinib, obinutuzumab, and venetoclax in untreated patients with TP53 mutations (NCT03824483).
In patients who are felt to be candidates for ASCT but who are unable to tolerate high-dose cytarabine, conventional therapy includes induction with BR (bendamustine and rituximab), R-CHOP, or VR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone). These regimens are typically well tolerated with good response rates, although the duration of response remains generally less than a few years [24,25,26]. Following ASCT, maintenance rituximab every 2 months for 3 years has been shown to improve PFS and OS [27].
Older patients who are not felt to be transplant candidates are generally given conventional chemoimmunotherapy with BR, VR-CAP, R-CHOP, or lenalidomide and rituximab as high-dose cytarabine-containing regimens can be toxic in the elderly. Maintenance rituximab can be considered until disease progression. There is evidence that maintenance rituximab following R-CHOP results in a 4-year OS rate of 87% compared with 63% with interferon-alfa [28]. There has been a lack of evidence regarding the survival benefit of maintenance rituximab following BR; however, a recent multicenter retrospective outcomes analysis did show an improvement in OS in those who did not undergo an autologous transplant and this remains an ongoing area of research [29, 30]. Further discussion of conventional chemoimmunotherapy and ASCT is beyond the scope of this review, and we turn our focus to the available evidence regarding targeted therapies. Table 1 summarizes the US Food and Drug Administration (FDA)-approved agents for relapsed/refractory MCL, and Table 2 highlights selected investigational agents in this setting discussed below.
3 Small Molecular Therapies
3.1 Bortezomib
Bortezomib, a proteasome inhibitor commonly used in multiple myeloma, has efficacy in MCL. It acts as an antineoplastic agent through a variety of mechanisms, including inducing apoptosis, limiting angiogenesis, NF-κB activity, and inhibiting cell-cycle progression. Notably, it also inhibits cyclin D1 expression in MCL [31]. In the PINNACLE study, 155 patients with relapsed or refractory (R/R) MCL were treated on days 1, 4, 8, and 11 of a 21-day cycle. There was a 33% response rate, with a median duration of response of 9.2 months in patients with one to three prior therapies. Adverse events included neuropathy (13%), fatigue (12%), and thrombocytopenia (11%). In an extended follow-up with a median of 26.4 months, median time to progression in patients who showed an initial response was 12.4 months, and in those who had a complete response (CR), the median OS was 36 months [32]. Bortezomib received FDA approval in 2006 for R/R MCL on the basis of these results, in addition to several other studies that showed efficacy and tolerability [31, 33, 34]. An expanded assessment of one study showed similar response rates and duration of PFS between patients with relapsed disease to those with refractory disease, suggesting that bortezomib may still be equally effective in patients who are resistant to other therapies [35]. Bortezomib can also be combined with rituximab or rituximab and bendamustine in the relapsed setting with improved efficacy without overlapping toxicity [36, 37].
Bortezomib also has activity in first-line therapy in combination with chemoimmunotherapy as previously discussed and was FDA approved for use in the front-line setting in 2014. VR-CAP, in which bortezomib replaces vincristine in the traditional R-CHOP regimen, was trialed vs R-CHOP in 487 transplant-ineligible patients. The study was performed prior to rituximab maintenance being established as beneficial following R-CHOP, thus no maintenance was given. VR-CAP displayed longer PFS (24.7 months vs 14.4 months), higher CR rate (53% vs 42%), and an improved 4-year OS rate (64% vs 54%). VR-CAP had higher rates of grade 3/4 neutropenia and thrombocytopenia, although it did not affect the number of completed cycles or rates of discontinuation [26].
3.2 Lenalidomide
Lenalidomide, an immunomodulatory agent with a variety of antineoplastic properties, was first assessed in MCL in the R/R population, during a time when bortezomib was the only agent FDA approved in that setting. A subgroup analysis of patients with MCL included in a trial of lenalidomide in aggressive NHL showed a 53% overall response rate (ORR) in 15 patients with a 20% CR rate. The most common grade 3 or 4 adverse events were thrombocytopenia and neutropenia [38]. The EMERGE study next assessed the efficacy of lenalidomide in 134 patients who were relapsed or refractory to bortezomib. There was an ORR of 28%, with a median duration of response of 16.6 months, and importantly a very rapid time to response with a median of 2.2 months. The rate of cytopenias was similar to prior studies. The FDA approval was granted in 2013 for patients who have progressed on two lines of therapy, including bortezomib, on the basis of this trial [39]. The SPRINT/MCL-002 trial compared lenalidomide with investigator’s choice monotherapy (rituximab, gemcitabine, fludarabine, chlorambucil, or cytarabine) in 254 transplant-ineligible patients who had relapsed at least once. Lenalidomide had a longer PFS (8.7 months vs 5.2 months) compared with alternative monotherapies [40]. Lenalidomide and rituximab is also well tolerated in the R/R setting with improved clinical efficacy compared with monotherapy; it showed a 57% ORR and duration of repsonse (DOR) of 18.9 months in one study and thus we utilize the combination more frequently than the monotherapy [41].
Given the success and tolerability in the R/R setting, attention was turned to utilizing lenalidomide in combination with rituximab in the upfront setting, particularly for those patients who were deemed ineligible for the standard intensive chemotherapy regimens typically used first line. Thirty-eight patients were treated with an ORR of 92%, with a CR rate of 64%. Fifty percent of patients experienced neutropenia, 29% had a rash, and 13% had thrombocytopenia. Importantly in this population, quality of life was also improved in response to treatment and a relative lack of toxic effects, based on the FACT-Lym questionnaire [42]. Five-year follow-up data showed that 75% of the patients completed more than 3 years of continuous treatment, with a 5-year estimated OS of 77% and PFS of 64%. During maintenance therapy, the most common adverse events remained hematologic [43].
3.3 BTK Inhibitors: Ibrutinib, Acalabrutinib, Zanubrutinib, and Novel Inhibitors
In MCL, there is deregulation of the B-cell pathway resulting in abnormal proliferation of B cells despite a lack of antigen binding. Bruton’s tyrosine kinase (BTK) is a protein downstream of the signal receptor that helps amplify the activation of B cells via NF-κB and other transcription factors [44]. Bruton’s tyrosine kinase inhibitors, of which ibrutinib is the first-in-class agent, block the kinase activity thereby leading to the death of B cells.
Ibrutinib was FDA approved for MCL based on a phase II study in R/R patients who had received a median of three prior therapies. A total of 111 patients, including high-risk patients such as those with high Mantle Cell Lymphoma International Prognostic Index scores or extra-nodal involvement, received 560 mg of ibrutinib. Sixty-eight percent of patients had a response, and benefits were seen across key subgroups associated with chemotherapy failure. The responses were quite durable, with a median duration of 17.5 months. The most common adverse events were diarrhea (50%), fatigue (41%), upper respiratory infection (URI) (23%), neutropenia (16%), and thrombocytopenia (11%). There were five instances of grade 3 bleeding events, and four patients had subdural hematomas, all of whom were receiving antiplatelet or anticoagulant therapy and developed in the setting of trauma. The response rate was much higher than typically seen with single-agent therapy in these heavily pre-treated patients, with favorable tolerability and safety [45]. Long term follow-up of this study showed a 24-month PFS of 31%, with a median duration of response of 17.5 months, and OS of 47%. It was also noted that the risk of infection, diarrhea, and bleeding generally improved after 6 months of therapy [46].
Ibrutinib was compared to temsirolimus, an mTOR inhibitor approved for single-agent use in R/R MCL used in Europe. Ibrutinib showed superior PFS (14.6 vs 6.2 months) and tolerability compared to temsirolimus [47]. In the final analysis with a median follow-up of 38.7 months, 24% of patients remained taking ibrutinib vs 0% in the temsirolimus group, and the ORR for ibrutinib was reported as 77% [48]. A pooled analysis of ibrutinib single-agent use in 370 patients with R/R MCL from three studies showed that half of all patients had a longer PFS than on the previous line of therapy, which is atypical for successive lines of chemoimmunotherapy [49]. In addition to superior cancer control, a health-related quality-of-life assessment of this study showed that those taking ibrutinib indicated better well-being on two patient-reported outcome instruments [50]. Mantle cell lymphoma that has an initial response to ibrutinib appears to be able to acquire resistance, and overcoming this resistance is an area of ongoing research [51].
Some drug-specific side effects of ibrutinib to note include atrial fibrillation (up to 10.3% at 2 years of therapy in one study) [52], bleeding (causing patients taking warfarin to be excluded from some trials) [53], and infections, notably pulmonary aspergillosis [54]. These side effects are felt to be due to off-target kinase inhibition, and therefore more selective BTK inhibitors are being developed in an attempt to limit these side effects [55].
Acalabrutinib was first utilized in MCL in a phase II trial using a dose of 100 mg twice daily in 124 patients with R/R disease. Eighty-one percent of patients responded, including 40% with a CR. Notably, there were no cases of atrial fibrillation and only one grade 3 gastrointestinal bleed; the most common adverse events were headache (38%), diarrhea (31%), and neutropenia (10%) [56]. Based on this study, acalabrutinib was also approved for use in R/R MCL by the FDA. The ibrutinib study [45] enrolled a more heavily pre-treated and high-risk population than the acalabrutinib study, which limits the ability to directly compare the two drugs. Long term follow-up of acalabrutinib in MCL confirmed efficacy with a median duration of response of 26 months, and did not show any instances of atrial fibrillation and only two additional episodes of grade 3 bleeding [57]. We await the results of the head-to-head trial between ibrutinib and acalabrutinib in CLL, which will provide additional insight into their adverse effects (NCT02477696).
Zanubrutinib, another selective BTK inhibitor, was evaluated in a phase II trial in MCL, with an ORR of 84% in 85 patients; CR rate was 67%. The most frequent adverse events were neutropenia (48% with 20% being grade 3 or higher), infection (34% with 9.3% grade 3 or higher), and rash (33%). There were no episodes of atrial fibrillation, and 1.2% with grade 3 bleeding [58] [59]. Zanubrutinib has been approved by the FDA for use at either 160 mg twice daily or 320 mg once daily. While there has not been a direct comparison in MCL, in a recent study of zanubrutinib vs ibrutinib in patients with Waldenstrom’s macroglobulinemia, the incidence of BTK inhibitor-related toxicities was lower with zanubrutinib than with ibrutinib [60]. We consider utilizing zanubrutinib or acalabrutinib as our BTK inhibitor of choice given the more limited side-effect profile of these medications compared with ibrutinib.
Two other notable BTK inhibitors have not yet been granted FDA approval. Orelabrutinib showed an ORR of 86% and 84% with 100 mg twice daily and 150 mg once daily, respectively, in a phase II trial with no reported episodes of grade 3/4 atrial fibrillation or bleeding [61]. LOXO-305 is a non-covalent, highly selective BTK inhibitor that was developed to limit resistance to BTK inhibition, and is undergoing evaluation particularly in those who have previously progressed on a covalent BTK inhibitor. In an abstract presented at the 2020 American Society of Hematology meeting, Wang et al. reported an ORR of 52% in heavily pre-treated patients with MCL, including 77% who had previously progressed taking a BTK inhibitor and several who progressed following chimeric antigen receptor T-cell (CAR-T) therapy. The only treatment-emergent adverse effects occurring in > 10% of patients were fatigue (16%) and diarrhea (15%) [62]. This may present an attractive future option to minimize side effects, particularly in patients who have progressed on a covalent BTK inhibitor.
4 Novel Combination Therapies
4.1 Ibrutinib Plus Rituximab in the R/R Setting
In the initial phase II study of ibrutinib [45], Wang et al. noted a 34% incidence of transient lymphocytosis, thought to be due to BTK inhibition leading to inability of the malignant B cells to adhere to tissues with resulting mobilization to the peripheral blood. It was hypothesized that there could be greater antitumor activity if rituximab was able to target these circulating tumor cells. Fifty patients with R/R MCL, all of whom had previously received rituximab-containing regimens, were started on ibrutinib and given rituximab weekly for 4 weeks, followed by monthly for cycles 3–8, and then every other month for up to 2 years. Only 6% of patients experienced lymphocytosis with the combination therapy. Eighty-eight percent of patients had an overall response, with 44% achieving a CR. Of note, Ki67 expression was strongly predictive of response; of the 37 patients with Ki67 below 50%, all achieved an OR; but of the 12 patients with Ki67 of ≥ 50%, only 50% achieved an OR. Side-effect profiles were very similar to ibrutinib monotherapy [63]. In a 4-year follow-up analysis, the CR rate improved to 58%, the median PFS was 43 months, and median OS has not been reached. Lower Ki67 expression remained predictive of durable remissions. Ibrutinib in combination with rituximab (IR) showed a higher CR rate and median OS compared with ibrutinib (58% vs 23%, OS > 4 years vs 22.5 months), although the ability to conclude that the combination improves survival compared to monotherapy is limited by the non-randomized and relatively small sample size of this study [64]. Ibrutinib remains more typically used as a monotherapy in R/R patients.
4.2 Ibrutinib Plus Rituximab in the Upfront Setting
On the basis of these data, several trials were designed to assess IR in the upfront setting. One hundred and thirty-one patients under the age of 65 years received IR as induction therapy for a maximum of 12 cycles until a CR was obtained, then received four cycles of R-Hyper-CVAD/R-MTX for consolidation instead of the traditional eight cycles. Forty-nine percent had a Ki-67 of ≥ 30%, and 15 patients had blastoid/pleomorphic MCL. A median of seven cycles of IR was needed to obtain a CR, and during the IR phase, the CR rate was 88%. Following consolidation, the CR rate was 94%. With a median follow-up of 22 months, median PFS and OS were not reached. These results suggest good responses with chemotherapy-free induction in young patients, with the hope that receiving only four cycles of hyper-CVAD will lead to lower rates of long-term secondary malignancies [65]. The TRIANGLE phase III study is assessing R-CHOP/R-DHAP induction followed by ASCT vs ibrutinib added to R-CHOP and to maintenance regimens following ASCT, vs IR-CHOP/R-DHAP without autologous transplantation. The study is ongoing (NCT02858258), but the completed safety run-in of the first 50 patients confirmed feasibility of the two experimental arms containing ibrutinib [66]. The efficacy of ibrutinib in the front-line setting for patients with TP53 mutations who do not respond well to traditional upfront chemoimmunotherapy will be important to assess in future trials.
Seeking to address the optimal front-line therapy in transplant-ineligible elderly patients who are thought not to be able to tolerate intensive chemoimmunotherapy, IR was trialed in 50 untreated patients over the age of 65 years. Ibrutinib in combination with rituximab was given in monthly cycles (dosing of rituximab as above in the initial phase II trial) until progression or other reason for discontinuation. The ORR was 98%, including a 60% CR rate; the median number of IR cycles to reach CR was 8. At 28 months median follow-up, median PFS and OS were not reached, although notably this trial excluded patients with blastoid histology and elevated Ki67. The most common reason for discontinuation was atrial fibrillation, although only three cases were of new onset [67]. A phase III trial of ibrutinib in combination with BR in elderly patients with newly diagnosed MCL is ongoing (SHINE, NCT 01776840); a phase I/Ib study of this regimen in either the upfront setting or R/R setting has previously shown a 94% RR, including a 76% CR in MCL [68].
4.3 Ibrutinib Plus Venetoclax
B-cell leukemia/lymphoma-2 (BCL-2) is an antiapoptotic protein that promotes cell survival. Its overactivity has been implicated in the pathophysiology of several B-cell lymphomas. Some patients with MCL exhibit high BCL-2 expression via chromosome 18q21 amplification and other mechanisms making the use of BCL-2 inhibitors such as venetoclax a possible therapeutic option [69]. While not FDA approved for monotherapy, venetoclax is now being studied in combination with other agents.
Pre-clinical studies have suggested synergy between ibrutinib and venetoclax [70]. They act through different mechanisms and have relatively separate adverse reaction profiles making them a logical dual therapy. The combination was trialed in 23 patients with R/R MCL and one with untreated MCL in the phase II AIM trial [71]. Patients started with ibrutinib monotherapy for 1 month before adding dose-escalating venetoclax in an attempt to mitigate the risk of tumor lysis syndrome. They then continued the combination until progression or severe adverse events. Notably, these were high-risk patients as 50% of them had a TP53 mutation and or deletion, and 43% had a Ki67 of ≥ 30%. After 4 months, there was a positron emission tomography (PET)-assessed ORR of 71% and a CR of 62%, compared with historical controls of 9% at similar timeframes with ibrutinib monotherapy. The most common adverse effects were diarrhea in 83% of patients (grade 3 occurred in 12%), fatigue (75%), nausea or vomiting in 71%, and bleeding or bruising in 54%, although only one patient had grade 3 bleeding. Patients with Ki67 of ≥ 30% were less likely to have a response, but half of the patients with TP53 aberrations had durable CRs [71]. There is an ongoing phase III study of this combination compared to ibrutinib monotherapy in R/R MCL (NCT03112174, SYMPATICO study).
4.4 Ibrutinib Plus Other Targeted Therapies
A phase II trial assessed the combination of IR plus lenalidomide in R/R MCL on the basis of the above success with ibrutinib, lenalidomide, and rituximab. Fifty patients received induction therapy with all three medications for 12-monthly cycles, followed by a maintenance phase of IR only until progression or unacceptable toxicity. Rituximab was given weekly for the first month, then every other month. Seventy-six percent had an OR, but the 95% confidence interval (63–86) crossed below the 68% historical standard ORR with ibrutinib alone, and it did not improve on the 88% historical ORR of IR, although it is of course challenging to cross-compare study populations. The triplet therapy did have a CR rate of 56%, which was higher than 44% with IR and 19% with ibrutinib alone. The triplet regimen also had greater hematologic toxicity, especially neutropenia, with a 22% rate of grade 3 or 4 infections. Ten percent of patients had TP53 mutations, and their ORR and CR rates were similar to the patients without mutations. Overall, this suggests the triplet regimen is not superior to IR or ibrutinib alone, but there could be a role for it in patients with TP53 mutations [72].
It has been noted that inhibition of CDK4 by the inhibitor palbociclib led to cell-cycle arrest in MCL tumor cells, and induction of this prolonged cell cycle can lead to MCL sensitization to ibrutinib destruction [73, 74]. On this basis, a phase I trial of ibrutinib and palbociclib given for 3 weeks of a monthly cycle to 27 patients with R/R MCL was performed. The ORR was 67%, and the CRR was 37%, with the dose-limiting toxicity being a grade 3 rash. The most common grade 3/4 toxicities were hematologic (neutropenia 41%, thrombocytopenia 30%) [75]. A phase II trial is ongoing (NCT03478514).
4.5 Acalabrutinib Plus Other Targeted Therapies
As the more recently developed BTK inhibitor, there are less published data regarding acalabrutinib in combination with other targeted therapies. A phase Ib trial assessing acalabrutinib with BR in patients with previously treated or untreated MCL assessed 38 patients. Of the 18 treatment-naïve patients, 17 had an overall response, with 13 achieving a CR. In the 20 R/R patients, there were 16 overall responses, with 13 achieving a CR [76]. Given its success as a single agent, multiple clinical trials assessing acalabrutinib in combination with chemotherapy and other targeted therapies in the upfront and R/R setting are ongoing; for example, NCT04115631 is investigating a BR backbone in combination with acalabrutinib, cytarabine, or both as first line-therapies.
5 Emerging Small Molecular Therapies
5.1 PI3K Inhibitors
Phosphoinositide 3-kinase (PI3K) proteins are involved in BCR signaling and deregulation of their pathway has been implicated in lymphoma as well as many other malignancies. This pathway also appears to be upregulated in MCL [77]. Idelalsib was the first PI3K inhibitor studied in MCL; results of a phase I trial showed an ORR of 40%, but the PFS was only 3.7 months. Idelalsib is also known to have serious adverse events, including opportunistic infections and hepatic and gastrointestinal toxicities, raising concerns for safety; this in combination with a short duration of response has made it unlikely to be used in MCL moving forward [78].
Copanlisib was studied in 11 patients with R/R MCL as part of a larger NHL trial, with a reported ORR of 63.6%. Hyperglycemia and hypertension were the most commonly reported adverse events, but the rates of serious toxicities were less than in the idelalsib trial [79]. Copanlisib displayed more effective targeting of MCL cells in vitro, particularly in an ibrutinib/venetoclax dual resistant model; however, further studies are needed to assess responses in resistant tumors in vivo [80].
Upregulation of the PI3K signaling pathway has been implicated in one method of ibrutinib resistance, and subsequent inactivation of the pathway with idelalisib in combination with ibrutinib in mouse models limited the growth of ibrutinib-resistant tumors [81]. Therefore, combinations of PI3K inhibitors with BTK inhibitors represents a possible pathway to overcome resistance. Umbralisib, a next-generation PI3K inhibitor, is being studied in a triplet combination with ublituximab and the novel selective BTK inhibitor TG-1701 in patients with R/R B-cell lymphomas (NCT03671590) [82]. Umbralisib, ublituximab, and venetoclax were shown to have good tolerability and rapid minimal residual disease (MRD)-negative responses in CLL [83] and the trial has since been expanded to include MCL (NCT03379051). Given the improved tolerability of umbralisib in other malignancies, we anticipate this may be a beneficial PI3K option for patients with MCL in the future.
5.2 Syk Inhibitors
Spleen tyrosine kinase (Syk) is also part of the BCR pathway, acting upstream of BTK and PI3K to initiate the BCR survival signal and amplify it through a non-antigen-binding dependent pathway [84]. Fostamatanib, an oral Syk inhibitor, was first trialed in patients with R/R B-cell NHL and CLL, including nine patients with MCL in a phase I/II study. The ORR was 22%; only one patient with MCL had a partial response and four had stable disease, but this represented a novel way to interrupt the BCR pathway [85].
Entospletinib was developed as a more selective kinase inhibitor with the hope that the selectivity would limit the off-target adverse effects [86]. In a phase II trial of entospletinib in R/R NHL, 39 patients with MCL had a modest 18% response rate to the monotherapy with a 4-month PFS of 64%. The toxicities included fatigue, nausea, and diarrhea, with a grade 3 or 4 neutropenia rate of 9%. The authors suggested that Syk inhibition may be more effective when used in combination with other therapies based on in vitro and in vivo studies suggesting synergism [87]. Entospletinib and idelalisib, the PI3K inhibitor, were trialed together in CLL and NHL; however, the study was ended early because of pneumonitis in 18% of patients, including two fatalities, thought to be due to significant cytokine production as a result of PI3K and downstream mTOR inhibition [88]. There are limited ongoing trials of this medication in combination with other currently available agents and thus we do not anticipate that Syk inhibitors will play a significant role in MCL moving forward.
6 Bispecific T-Cell Engagers (BiTEs) and Antibody-Drug Conjugates (ADCs)
Antibody therapy has been crucial in the treatment of B-cell lymphomas. The creation of antibody combinations that can simultaneously bind to two individual antigens, typically an antigen on a malignant cell and a T cell, is an advancement in antibody technology known as a bispecific T-cell engager (BiTe). Once bound to both CD3 on T cells and the antigen on the cancer cell, the T cell is activated, resulting in cytokine release and target cell death [89]. Blinotumomab, a CD3/CD19 BiTE, is FDA approved for B-cell acute lymphoblastic leukemia (B-ALL), and has been studied in NHL given the expression of CD19 in these malignancies. In a phase I study, blinotumomab was trialed in 76 patients with relapsed/refractory NHL, including 24 with MCL. The patients with MCL had an ORR of 71%, including three patients with a CR. The most frequent cause for treatment discontinuation was neurologic events, from headache to encephalopathy. Cytokine release syndrome (CRS) also occurred [90]. In a subsequent paper including long-term follow-up of 38 of these patients, including 13 with MCL, there was no evidence of long-term toxicities, specifically no neurocognitive impairments. Two of the patients with MCL were in ongoing remission after a median follow-up of 4.6 years [91].
Mosunetuzumab, a BiTE construct combining CD3 with CD20, has also been investigated in NHL with favorable results, including in patients who had progressed through CAR-T therapy. A phase Ib study of this agent in indolent and aggressive NHL showed an ORR in the aggressive lymphomas of 37% in these heavily pre-treated patients. In addition, the investigators were able to show an increase in the number of CAR-T present in those patients who had relapsed through CAR-T therapy and then received mosunetuzumab. The authors hypothesized that the BiTE could be increasing the effect of those prior cells. Mantle cell data were not reported as a subgroup [92]. Given the “off-the-shelf” advantage to BiTEs vs CAR-T products, additional studies of blinatumomab, mosunetuzumab, and other BiTE constructions are ongoing in patients with NHL and patients with MCL, including in combination with other therapies.
Another advance in antibody technology has been the advent of antibody-drug conjugates. A toxin is attached to an antibody that binds to a tumor antigen. The complex is then internalized, and the cytotoxic agent is delivered directly to the tumor cell, bypassing non-malignant cells. This precision limits off-target side effects [93]. While there are no approved ADCs for MCL at the present, they are being studied. Loncastuximab tesirine is an ADC that combines an anti-CD19 antibody with the toxin pyrrolobenzodiazepine. A phase I study in 88 patients with relapsed or refractory NHL was performed, including nine patients with MCL, and showed efficacy and tolerability. The patients with MCL had an ORR at all doses of 44.4% with 33.3% achieving a CR [94]. Studies of loncastuximab tesirine in combination with ibrutinib [95, 96] and durvalumab [97] are ongoing in patients with relapsed or refractory NHL including MCL. VLS-101, an ADC that combines an antibody to the receptor tyrosine kinase-like orphan receptor with monomethyl auristain E (MMAE) is being investigated in a phase I trial in previously treated hematologic malignancies including MCL (NCT03833180) [98].
7 CAR-T Therapy
Arguably, the most exciting recent advancement in the treatment of hematologic malignancies is the development of CAR-T. Autologous T cells undergo lentiviral transduction to produce CAR proteins on their surface [99]. These proteins are designed to include an antigen-binding domain that can recognize a tumor antigen irrespective of the major histocompatibility complex presentation and a costimulatory domain that enhances survival and proliferation of CAR-T. The antigen-binding domain can be modified to recognize a variety of different antigens of clinical importance in B-cell malignancies, such as CD19, CD20, and CD22 [100].
Mantle cell lymphoma has the additional challenge of occasional circulating lymphoma cells in the peripheral blood, which limits the ability to manufacture the cells using unselected peripheral blood mononuclear cells [101, 102]. There have also been reports of introducing the CAR gene into leukemic cells that were unintentionally present during the T-cell manufacturing, with subsequent relapse [103]. Brexucabtagene autoleucel (brexu-cel), which is an anti-CD19 CAR-T, incorporates a T-cell enrichment phase in the manufacturing process. Brexu-cel was investigated in the ZUMA-2 phase II trial. Adult patients were eligible if they had received up to five prior therapies for relapsed or refractory MCL. They were not required to have not responded to BTK inhibitor therapy and were allowed to receive bridging therapy prior to cell infusion [104].
Seventy-four patients were enrolled in ZUMA-2 at 20 sites in the USA and Europe. Following leukapheresis, they were conditioned with fludarabine and cyclophosphamide. Brexu-cel was able to be manufactured for 71 patients and 68 patients ultimately received the cells. There was a 93% ORR, including a CR rate of 67% in the primary efficacy analysis of 60 patients. The response rate was similar in sub-group analyses of those with high-risk features such as those refractory to BTK inhibitor therapy, harboring TP53 mutations, those with blastoid morphology, or with a Ki67 of ≥ 50% [104]. At a median follow-up of 17.5 months from the recently reported American Society of Hematology (ASH) abstract, 48% of patients had ongoing responses [105].
Safety has been a concern with CAR-T therapy, owing to the potential for life-threatening CRS and immune effector cell-associated neurotoxicity syndrome (ICANS), in addition to the common side effects following stem cell transplant conditioning therapy of cytopenias and infection. Cytokine release syndrome is a systemic inflammatory response arising from activation of the CAR-T and associated elevation in cytokines such as interleukin-6. This leads to fever, hypoxia, and hypotension of varying severity and is treated with immunosuppression including steroids and the interleukin-6 inhibitor tocilizumab depending on the CRS grade. In ZUMA-2, patients were graded and treated based on the Lee et al. criteria [106]. Immune effector cell-associated neurotoxicity syndrome describes a wide array of neurotoxic adverse events from tremor to encephalopathy to cerebral edema. Patients in ZUMA-2 were graded based on National Cancer Institute Common Terminology Criteria for Adverse Events. These symptoms classically occur during or after CRS symptoms, and the pathophysiology is unclear although cytokines likely play a role. Subsequently, consensus grading has been developed by the American Society for Transplantation and Cellular Therapy for both CRS and ICANS [107].
In ZUMA-2, CRS occurred in 91% of the patients, pyrexia (91%) and hypotension (51%) were the most common manifestations of CRS. Fifty-nine percent received tocilizumab, 22% received glucocorticoids, and only 16% required vasopressors. Neurotoxicity occurred in 63% of patients, of which 32% was grade 1 or 2. Tremor (35%) and encephalopathy (31%) of any grade were the most common symptoms of ICANS. Notably, most symptoms of both CRS and ICANS occurred early and were generally reversible; the incidence of severe symptoms was similar to prior studies. Ninety-four percent of patients had grade 3 or higher cytopenias and 32% developed infections. There were two fatalities due to grade 5 infectious adverse events [104]. This therapy represents an effective and relatively safe option for patients with heavily pretreated MCL, particularly as it does not require patients to enter a remission prior to therapy, and the response rates in high-risk patients were encouraging. On the basis of these data, the FDA approved brexu-cel for patients with relapsed or refractory MCL who have progressed following chemoimmunotherapy and a BTK inhibitor in July of 2020 [108].
Ongoing clinical trials are evaluating other CAR constructs in relapsed MCL. A phase I/II study investigating lisocabtagene maraleucel is currently recruiting participants (NCT02631044). Lisocabtagene maraleucel is designed to have equal numbers of CD4+ and CD8+ T cells with the goal to reduce the severity and frequency of CRS. Initial data have been presented for the MCL cohort containing 32 patients who had received a median of three prior therapies. The ORR was 84%, including a notable 75% response rate in patients with blastoid morphology. Fifty percent of patients experienced CRS and 28% had neurologic events. Further data regarding this evolving construct will be presented; it has not yet been granted FDA approval [109].
8 Sequencing of Targeted Therapies in the Relapsed Setting
There is no defined sequencing approach for these targeted agents in the relapsed setting, and no direct comparison of regimens. In the absence of data, we offer our opinion on sequencing of agents in this setting (Fig. 1). Given the rarity and incurable nature of MCL, clinical trials should be considered and prioritized for this population [110].
We utilize BTK inhibitors as our first option following progression after first-line chemoimmunotherapy given their high efficacy and tolerability. The three FDA-approved BTK inhibitors have never been compared directly in MCL, and thus we do not know if one is superior to the others in terms of efficacy. There is a suggestion in a cross-trial comparison that acalabrutinib may have a longer duration of response, but this study was performed in a less heavily pre-treated population than in the ibrutinib studies. We therefore generally choose based on patient preference for daily vs twice-daily dosing, consideration of adverse effects, and potential drug interactions. Occasionally, insurance coverage will be a factor in the selection of a BTK inhibitor. We have a preference towards acalabrutinib or zanubrutinib given their higher specificity and seemingly lower rates of adverse effects.
We consider CAR-T therapy in patients who have progressed following chemoimmunotherapy and a BTK inhibitor, particularly in those with poor prognostic risk factors such as TP53 mutations and blastoid histology, or who were refractory to or with short response to BTK inhibitors. The durable responses following progression on a BTK inhibitor make this an attractive option for those who are eligible.
In patients who are ineligible for or do not desire CAR-T therapy, we next consider lenalidomide and rituximab as this combination is generally well tolerated and with greater clinical efficacy than lenalidomide monotherapy. Other considerations include combining rituximab with bortezomib or bendamustine if they did not receive these agents in the front-line setting. While not yet FDA approved, venetoclax could also be used in the relapsed setting and we consider this in patients who desire oral therapy only.
We await ongoing trial data regarding the combination of ibrutinib and other BTK inhibitors with rituximab and venetoclax, as we anticipate this will be a consideration for upfront therapy in patients who are unable to tolerate aggressive chemoimmunotherapy, or in those with TP53 mutations and blastoid histology. These combinations will also likely be utilized in the relapsed setting, and future phase III trials will be useful in determining the optimal sequencing of these regimens.
Amongst the novel agents, we feel the data from the more selective PI3K inhibitor umbralisib and the BiTe mosunetuzumab seem the most favorable although there are limited data. In addition, BiTes also have the advantage of being relatively well tolerated and without the production lag time of CARs.
9 Conclusions
Options for more precise treatment of MCL have increased in recent years, with emerging hope for better outcomes accompanied by less toxicity for patients diagnosed with this at times aggressive disease. The advantage of clinical trials and consideration of novel agents for these patients is clear. Ongoing research and trials assure that we can move forward in the quest to turn MCL into a curable disease.
References
Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2017.
Smedby KE, Hjalgrim H. Epidemiology and etiology of mantle cell lymphoma and other non-Hodgkin lymphoma subtypes. Semin Cancer Biol. 2011;21(5):293–8. https://doi.org/10.1016/j.semcancer.2011.09.010.
Romaguera JE, Medeiros LJ, Hagemeister FB, Fayad LE, Rodriguez MA, Pro B, et al. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma. Cancer. 2003;97(3):586–91. https://doi.org/10.1002/cncr.11096.
Dreyling M, Campo E, Hermine O, Jerkeman M, Le Gouill S, Rule S, et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28:iv62–71. https://doi.org/10.1093/annonc/mdx223.
Jain AG, Chang CC, Ahmad S, Mori S. Leukemic non-nodal mantle cell lymphoma: diagnosis and treatment. Curr Treat Opt Oncol. 2019;20(12):85. https://doi.org/10.1007/s11864-019-0684-8.
Mozos A, Royo C, Hartmann E, De Jong D, Baró C, Valera A, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica. 2009;94(11):1555–62. https://doi.org/10.3324/haematol.2009.010264.
Vegliante MC, Palomero J, Pérez-Galán P, Roué G, Castellano G, Navarro A, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175–85. https://doi.org/10.1182/blood-2012-06-438937.
Bernard M, Gressin R, Lefrère F, Drénou B, Branger B, Caulet-Maugendre S, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia. 2001;15(11):1785–91. https://doi.org/10.1038/sj.leu.2402272.
Dreyling M, Klapper W, Rule S. Blastoid and pleomorphic mantle cell lymphoma: still a diagnostic and therapeutic challenge! Blood. 2018;132(26):2722–9. https://doi.org/10.1182/blood-2017-08-737502.
Hoster E, Rosenwald A, Berger F, Bernd HW, Hartmann S, Loddenkemper C, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016;34(12):1386–94. https://doi.org/10.1200/jco.2015.63.8387.
Katzenberger T, Petzoldt C, Höller S, Mäder U, Kalla J, Adam P, et al. The Ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood. 2006;107(8):3407. https://doi.org/10.1182/blood-2005-10-4079.
Hoster E, Dreyling M, Klapper W, Gisselbrecht C, van Hoof A, Kluin-Nelemans HC, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558–65. https://doi.org/10.1182/blood-2007-06-095331.
Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903–10. https://doi.org/10.1182/blood-2017-04-779736.
Kumar A, Ying Z, Alperovich A, Dogan A, Hamlin P, Moskowitz C, et al. Clinical presentation determines selection of patients for initial observation in mantle cell lymphoma. Haematologica. 2019;104(4):e163–6. https://doi.org/10.3324/haematol.2018.201350.
Cohen JB, Han X, Jemal A, Ward EM, Flowers CR. Deferred therapy is associated with improved overall survival in patients with newly diagnosed mantle cell lymphoma. Cancer. 2016;122(15):2356–63. https://doi.org/10.1002/cncr.30068.
Abrisqueta P, Scott DW, Slack GW, Steidl C, Mottok A, Gascoyne RD, et al. Observation as the initial management strategy in patients with mantle cell lymphoma. Ann Oncol. 2017;28(10):2489–95. https://doi.org/10.1093/annonc/mdx333.
Dreyling M, Lenz G, Hoster E, Van Hoof A, Gisselbrecht C, Schmits R, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood. 2005;105(7):2677–84. https://doi.org/10.1182/blood-2004-10-3883.
Gerson JN, Handorf E, Villa D, Gerrie AS, Chapani P, Li S, et al. Survival outcomes of younger patients with mantle cell lymphoma treated in the rituximab era. J Clin Oncol. 2019;37(6):471–80. https://doi.org/10.1200/JCO.18.00690.
Geisler CH, Kolstad A, Laurell A, Jerkeman M, Räty R, Andersen NS, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol. 2012;158(3):355–62. https://doi.org/10.1111/j.1365-2141.2012.09174.x.
Hermine O, Hoster E, Walewski J, Bosly A, Stilgenbauer S, Thieblemont C, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet. 2016;388(10044):565–75. https://doi.org/10.1016/S0140-6736(16)00739-X.
Romaguera JE, Fayad LE, Feng L, Hartig K, Weaver P, Rodriguez MA, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol. 2010;150(2):200–8. https://doi.org/10.1111/j.1365-2141.2010.08228.x.
Merli F, Luminari S, Ilariucci F, Petrini M, Visco C, Ambrosetti A, et al. Rituximab plus HyperCVAD alternating with high dose cytarabine and methotrexate for the initial treatment of patients with mantle cell lymphoma, a multicentre trial from Gruppo Italiano Studio Linfomi. Br J Haematol. 2012;156(3):346–53. https://doi.org/10.1111/j.1365-2141.2011.08958.x.
Bernstein SH, Epner E, Unger JM, Leblanc M, Cebula E, Burack R, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol. 2013;24(6):1587–93. https://doi.org/10.1093/annonc/mdt070.
Lenz G, Dreyling M, Hoster E, Wörmann B, Dührsen U, Metzner B, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23(9):1984–92. https://doi.org/10.1200/jco.2005.08.133.
Rummel MJ, Niederle N, Maschmeyer G, Banat GA, von Grünhagen U, Losem C, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–10. https://doi.org/10.1016/s0140-6736(12)61763-2.
Robak T, Huang H, Jin J, Zhu J, Liu T, Samoilova O, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944–53. https://doi.org/10.1056/NEJMoa1412096.
Le Gouill S, Thieblemont C, Oberic L, Moreau A, Bouabdallah K, Dartigeas C, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017;377(13):1250–60. https://doi.org/10.1056/NEJMoa1701769.
Kluin-Nelemans HC, Hoster E, Hermine O, Walewski J, Trneny M, Geisler CH, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med. 2012;367(6):520–31. https://doi.org/10.1056/NEJMoa1200920.
Hill BT, Switchenko JM, Martin P, Churnetski MC, Sawalha Y, Goyal S, et al. Maintenance rituximab improves outcomes in mantle eell lymphoma patients who respond to induction therapy with bendamustine + rituximab without autologous transplant. Blood. 2019;134(Suppl_1):1525. https://doi.org/10.1182/blood-2019-129404.
Rummel MJ, Knauf W, Goerner M, Soeling U, Lange E, Hertenstein B, et al. Two years rituximab maintenance vs. observation after first-line treatment with bendamustine plus rituximab (B-R) in patients with mantle cell lymphoma: first results of a prospective, randomized, multicenter phase II study (a subgroup study of the StiL NHL7-2008 MAINTAIN trial). J Clin Oncol. 2016;34(15_Suppl.):7503. https://doi.org/10.1200/JCO.2016.34.15_suppl.7503.
Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–74. https://doi.org/10.1200/jco.2006.07.9665.
Goy A, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009;20(3):520–5. https://doi.org/10.1093/annonc/mdn656.
O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2005;23(4):676–84. https://doi.org/10.1200/jco.2005.02.050.
Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, et al. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13(18 Pt 1):5291–4. https://doi.org/10.1158/1078-0432.Ccr-07-0871.
O’Connor OA, Moskowitz C, Portlock C, Hamlin P, Straus D, Dumitrescu O, et al. Patients with chemotherapy-refractory mantle cell lymphoma experience high response rates and identical progression-free survivals compared with patients with relapsed disease following treatment with single agent bortezomib: results of a multicentre phase 2 clinical trial. Br J Haematol. 2009;145(1):34–9. https://doi.org/10.1111/j.1365-2141.2008.07466.x.
Zilioli VR, Gabutti C, Meli E, Minga P, Zancanella M, Cairoli R, et al. Rituximab plus bortezomib still represents an effective treatment option for patients with relapsed or refractory mantle cell lymphoma. Blood. 2017;130(Suppl_1):5162. https://doi.org/10.1182/blood.V130.Suppl_1.5162.5162.
Friedberg JW, Vose JM, Kelly JL, Young F, Bernstein SH, Peterson D, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood. 2011;117(10):2807–12. https://doi.org/10.1182/blood-2010-11-314708.
Habermann TM, Lossos IS, Justice G, Vose JM, Wiernik PH, McBride K, et al. Lenalidomide oral monotherapy produces a high response rate in patients with relapsed or refractory mantle cell lymphoma. Br J Haematol. 2009;145(3):344–9. https://doi.org/10.1111/j.1365-2141.2009.07626.x.
Goy A, Sinha R, Williams ME, Kalayoglu Besisik S, Drach J, Ramchandren R, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol. 2013;31(29):3688–95. https://doi.org/10.1200/jco.2013.49.2835.
Trněný M, Lamy T, Walewski J, Belada D, Mayer J, Radford J, et al. Lenalidomide versus investigator’s choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): a phase 2, randomised, multicentre trial. Lancet Oncol. 2016;17(3):319–31. https://doi.org/10.1016/s1470-2045(15)00559-8.
Wang M, Fayad L, Wagner-Bartak N, Zhang L, Hagemeister F, Neelapu SS, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol. 2012;13(7):716–23. https://doi.org/10.1016/S1470-2045(12)70200-0.
Ruan J, Martin P, Shah B, Schuster SJ, Smith SM, Furman RR, et al. Lenalidomide plus rituximab as initial treatment for mantle-cell lymphoma. N Engl J Med. 2015;373(19):1835–44. https://doi.org/10.1056/NEJMoa1505237.
Ruan J, Martin P, Christos P, Cerchietti L, Tam W, Shah B, et al. Five-year follow-up of lenalidomide plus rituximab as initial treatment of mantle cell lymphoma. Blood. 2018;132(19):2016–25. https://doi.org/10.1182/blood-2018-07-859769.
Merolle MI, Ahmed M, Nomie K, Wang ML. The B cell receptor signaling pathway in mantle cell lymphoma. Oncotarget. 2018;9(38):25332–41. https://doi.org/10.18632/oncotarget.25011.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16. https://doi.org/10.1056/NEJMoa1306220.
Wang ML, Blum KA, Martin P, Goy A, Auer R, Kahl BS, et al. Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood. 2015;126(6):739–45. https://doi.org/10.1182/blood-2015-03-635326.
Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016;387(10020):770–8. https://doi.org/10.1016/S0140-6736(15)00667-4.
Rule S, Jurczak W, Jerkeman M, Rusconi C, Trneny M, Offner F, et al. Ibrutinib versus temsirolimus: 3-year follow-up of patients with previously treated mantle cell lymphoma from the phase 3, international, randomized, open-label RAY study. Leukemia. 2018;32(8):1799–803. https://doi.org/10.1038/s41375-018-0023-2.
Rule S, Dreyling MH, Goy A, Hess G, Auer R, Kahl BS, et al. Long-term outcomes with ibrutinib versus the prior regimen: a pooled analysis in relapsed/refractory (R/R) mantle cell lymphoma (MCL) with up to 7.5 years of extended follow-up. Blood. 2019;134(Suppl_1):1538. https://doi.org/10.1182/blood-2019-124691.
Hess G, Rule S, Jurczak W, Jerkeman M, Santucci Silva R, Rusconi C, et al. Health-related quality of life data from a phase 3, international, randomized, open-label, multicenter study in patients with previously treated mantle cell lymphoma treated with ibrutinib versus temsirolimus. Leuk Lymphoma. 2017;58(12):2824–32. https://doi.org/10.1080/10428194.2017.1326034.
Hershkovitz-Rokah O, Pulver D, Lenz G, Shpilberg O. Ibrutinib resistance in mantle cell lymphoma: clinical, molecular and treatment aspects. Br J Haematol. 2018;181(3):306–19. https://doi.org/10.1111/bjh.15108.
Wiczer TE, Levine LB, Brumbaugh J, Coggins J, Zhao Q, Ruppert AS, et al. Cumulative incidence, risk factors, and management of atrial fibrillation in patients receiving ibrutinib. Blood Adv. 2017;1(20):1739–48. https://doi.org/10.1182/bloodadvances.2017009720.
Caron F, Leong DP, Hillis C, Fraser G, Siegal D. Current understanding of bleeding with ibrutinib use: a systematic review and meta-analysis. Blood Adv. 2017;1(12):772–8. https://doi.org/10.1182/bloodadvances.2016001883.
Rogers KA, Mousa L, Zhao Q, Bhat SA, Byrd JC, El Boghdadly Z, et al. Incidence of opportunistic infections during ibrutinib treatment for B-cell malignancies. Leukemia. 2019;33(10):2527–30. https://doi.org/10.1038/s41375-019-0481-1.
Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2015;374(4):323–32. https://doi.org/10.1056/NEJMoa1509981.
Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659–67. https://doi.org/10.1016/s0140-6736(17)33108-2.
Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Long-term follow-up of acalabrutinib monotherapy in patients with relapsed/refractory mantle cell lymphoma. Blood. 2018;132(Suppl_1):2876. https://doi.org/10.1182/blood-2018-99-110327.
Song Y, Zhou K, Zou D, Zhou J, Hu J, Yang H, et al. Safety and activity of the investigational Bruton tyrosine kinase inhibitor zanubrutinib (BGB-3111) in patients with mantle cell lymphoma from a phase 2 trial. Blood. 2018;132(Suppl_1):148. https://doi.org/10.1182/blood-2018-99-117956.
Song Y, Zhou K, Zou D, Zhou J, Hu J, Yang H, et al. Treatment of patients with relapsed or refractory mantle-cell lymphoma with zanubrutinib, a selective inhibitor of Bruton’s tyrosine kinase. Clin Cancer Res. 2020;26(16):4216–24. https://doi.org/10.1158/1078-0432.Ccr-19-3703.
Tam CS, Opat S, D’Sa S, Jurczak W, Lee H-P, Cull G, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–50. https://doi.org/10.1182/blood.2020006844.
Song Y, Song Y, Liu L, Zhang M, Li Z, Ji C, et al. Safety and efficacy of orelabrutinib monotherapy in Chinese patients with relapsed or refractory mantle cell lymphoma: a multicenter, open-label, phase II study. Blood. 2019;134(Suppl_1):755. https://doi.org/10.1182/blood-2019-126305.
Wang M, Shah NN, Alencar AJ, Gerson JN, Patel MR, Fakhri B, et al. LOXO-305, a next generation, highly selective, non-covalent BTK inhibitor in previously treated mantle cell lymphoma, Waldenström’s macroglobulinemia, and other non-Hodgkin lymphomas: results from the phase 1/2 BRUIN study. Blood. 2020;136(Suppl_1):8–10. https://doi.org/10.1182/blood-2020-134314.
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J, et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol. 2016;17(1):48–56. https://doi.org/10.1016/s1470-2045(15)00438-6.
Jain P, Romaguera J, Srour SA, Lee HJ, Hagemeister F, Westin J, et al. Four-year follow-up of a single arm, phase II clinical trial of ibrutinib with rituximab (IR) in patients with relapsed/refractory mantle cell lymphoma (MCL). Br J Haematol. 2018;182(3):404–11. https://doi.org/10.1111/bjh.15411.
Wang M, Jain P, Zhang S, Nomie K, Wang L, Oriabure O, et al. Ibrutinib with rituximab (IR) and short course R-hypercvad/MTX is very efficacious in previously untreated young pts with mantle cell lymphoma (MCL). Hematol Oncol. 2019;37(S2):42–3. https://doi.org/10.1002/hon.12_2629.
Dreyling M, Ladetto M, Doorduijn JK, Gine E, Jerkeman M, Mey U, et al. Triangle: autologous transplantation after a rituximab/ibrutinib/ara-c containing induction in generalized mantle cell lymphoma: a randomized European MCL Network Trial. Blood. 2019;134(Suppl_1):2816. https://doi.org/10.1182/blood-2019-127863.
Jain P, Lee HJ, Steiner RE, Hagemeister FB, Samaniego F, Westin JR, et al. Frontline treatment with ibrutinib with rituximab (IR) combination is highly effective in elderly (≥ 65 years) patients with mantle cell lymphoma (MCL): results from a phase II trial. Blood. 2019;134(Suppl_1):3988. https://doi.org/10.1182/blood-2019-125800.
Maddocks K, Christian B, Jaglowski S, Flynn J, Jones JA, Porcu P, et al. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood. 2015;125(2):242–8. https://doi.org/10.1182/blood-2014-08-597914.
Davids MS. Targeting BCL-2 in B-cell lymphomas. Blood. 2017;130(9):1081–8. https://doi.org/10.1182/blood-2017-04-737338.
Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, et al. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL 2 pathways. Br J Haematol. 2015;168(5):765–8.
Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–23. https://doi.org/10.1056/NEJMoa1715519.
Jerkeman M, Eskelund CW, Hutchings M, Räty R, Wader KF, Laurell A, et al. Ibrutinib, lenalidomide, and rituximab in relapsed or refractory mantle cell lymphoma (PHILEMON): a multicentre, open-label, single-arm, phase 2 trial. Lancet Haematol. 2018;5(3):e109–16. https://doi.org/10.1016/s2352-3026(18)30018-8.
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38.
Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–35. https://doi.org/10.1158/2159-8290.Cd-14-0098.
Martin P, Bartlett NL, Blum KA, Park S, Maddocks K, Ruan J, et al. A phase 1 trial of ibrutinib plus palbociclib in previously treated mantle cell lymphoma. Blood. 2019;133(11):1201–4. https://doi.org/10.1182/blood-2018-11-886457.
Phillips TJ, Smith SD, Jurczak W, Robak T, Stevens D, Farber CM, et al. Safety and efficacy of acalabrutinib plus bendamustine and rituximab (BR) in patients with treatment-naive (TN) or relapsed/refractory (R/R) mantle cell lymphoma (MCL). Blood. 2018;132(Suppl_1):4144. https://doi.org/10.1182/blood-2018-99-110617.
Curran E, Smith SM. Phosphoinositide 3-kinase inhibitors in lymphoma. Curr Opin Oncol. 2014;26(5):469–75. https://doi.org/10.1097/cco.0000000000000113.
Kahl BS, Spurgeon SE, Furman RR, Flinn IW, Coutre SE, Brown JR, et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398–405. https://doi.org/10.1182/blood-2013-11-537555.
Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28(9):2169–78. https://doi.org/10.1093/annonc/mdx289.
Jiang C, Zhang R, Zhang S, Lee W, Huang S, Liu Y, et al. Targeting PI3K and PLK1 to overcome ibrutinib-venetoclax resistance in mantle cell lymphoma. Blood. 2019;134(Suppl_1):4062. https://doi.org/10.1182/blood-2019-128299.
Zhang L, Nomie K, Zhang H, Bell T, Pham L, Kadri S, et al. B-cell lymphoma patient-derived xenograft models enable drug discovery and are a platform for personalized therapy. Clin Cancer Res. 2017;23(15):4212–23. https://doi.org/10.1158/1078-0432.Ccr-16-2703.
Cheah CY, Wickham N, Yannakou CK, Lewis KL, Hui C-H, Tang PS, et al. Phase 1 study of TG-1701, a selective irreversible inhibitor of Bruton’s tyrosine kinase (BTK), in patients with relapsed/refractory B-cell malignancies. Blood. 2019;134(Suppl_1):4001. https://doi.org/10.1182/blood-2019-126951.
Barr PM, Hill BT, Ma S, Baran AM, Bui A, Meacham PJ, et al. A phase 1/2 study of umbralisib ublituximab and venetoclax in patients with relapsed or refractory chronic lymphocytic leukemia (CLL). Blood. 2019;134(Suppl_1):360. https://doi.org/10.1182/blood-2019-123404.
Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol. 2017;10(1):145. https://doi.org/10.1186/s13045-017-0512-1.
Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–85. https://doi.org/10.1182/blood-2009-08-236471.
Currie KS, Kropf JE, Lee T, Blomgren P, Xu J, Zhao Z, et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J Med Chem. 2014;57(9):3856–73. https://doi.org/10.1021/jm500228a.
Andorsky DJ, Kolibaba KS, Assouline S, Forero-Torres A, Jones V, Klein LM, et al. An open-label phase 2 trial of entospletinib in indolent non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2019;184(2):215–22. https://doi.org/10.1111/bjh.15552.
Barr PM, Saylors GB, Spurgeon SE, Cheson BD, Greenwald DR, O’Brien SM, et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. 2016;127(20):2411–5. https://doi.org/10.1182/blood-2015-12-683516.
Smits NC, Sentman CL. Bispecific T-cell engagers (BiTEs) as treatment of B-cell lymphoma. J Clin Oncol. 2016;34(10):1131–3. https://doi.org/10.1200/jco.2015.64.9970.
Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol. 2016;34(10):1104–11. https://doi.org/10.1200/jco.2014.59.1586.
Dufner V, Sayehli CM, Chatterjee M, Hummel HD, Gelbrich G, Bargou RC, et al. Long-term outcome of patients with relapsed/refractory B-cell non-Hodgkin lymphoma treated with blinatumomab. Blood Adv. 2019;3(16):2491–8. https://doi.org/10.1182/bloodadvances.2019000025.
Schuster SJ, Bartlett NL, Assouline S, Yoon S-S, Bosch F, Sehn LH, et al. Mosunetuzumab induces complete remissions in poor prognosis non-Hodgkin lymphoma patients, including those who are resistant to or relapsing after chimeric antigen receptor T-cell (CAR-T) therapies, and is active in treatment through multiple lines. Blood. 2019;134(Suppl_1):6. https://doi.org/10.1182/blood-2019-123742.
Jagadeesh DMDMPH, Smith MRMDP. Antibody drug conjugates (ADCs): changing the treatment landscape of lymphoma. Curr Treat Opt Oncol. 2016;17(10):1–13. https://doi.org/10.1007/s11864-016-0428-y.
Kahl BS, Hamadani M, Radford J, Carlo-Stella C, Caimi P, Reid E, et al. A phase I study of ADCT-402 (loncastuximab tesirine), a novel pyrrolobenzodiazepine-based antibody–drug conjugate, in relapsed/refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2019;25(23):6986–94. https://doi.org/10.1158/1078-0432.Ccr-19-0711.
Depaus J, Bryan LJ, Ungar D, Dautaj I, Wagner-Johnston ND. Safety and anti-tumor activity study of loncastuximab tesirine and ibrutinib in diffuse large B-cell or mantle cell lymphoma. Blood. 2019;134(Suppl 1):5309. https://doi.org/10.1182/blood-2019-126913.
Depaus JAE-H, Locke B, Magagnoli M, Gritti G, Chao G, Boni J, et al. Interim results of a phase 1/2 study of loncastuximab tesirine (Lonca) combined with ibrutinib in advanced diffuse large B-cell lymphoma (DLBCL) or mantle cell lymphoma (MCL). European Hematology Association; Rochester, NY, 2020.
Moskowitz CH, Bastos-Oreiro M, Ungar D, Dautaj I, Kalac M. Safety and anti-tumor activity study of loncastuximab tesirine and durvalumab in diffuse large B-cell, mantle cell, or follicular lymphoma. Blood. 2019;134(Suppl_1):2807. https://doi.org/10.1182/blood-2019-128036.
Wang M, Barrientos JC, Furman RR, Mei M, Barr PM, Choi MY, et al. VLS-101, a ROR1-targeting antibody-drug conjugate, demonstrates a predictable safety profile and clinical efficacy in patients with heavily pretreated mantle cell lymphoma and diffuse large B-cell lymphoma. Blood. 2020;136(Suppl 1):13–4. https://doi.org/10.1182/blood-2020-136373.
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–98. https://doi.org/10.1158/2159-8290.Cd-12-0548.
Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–26. https://doi.org/10.1111/imr.12131.
Cohen K, Donovan H. Bone marrow and peripheral blood involvement in mantle cell lymphoma. Br J Haematol. 1998;101(2):302–10. https://doi.org/10.1046/j.1365-2141.1998.00684.x.
Sabatino M, Choi K, Chiruvolu V, Better M. Production of anti-CD19 CAR T cells for ZUMA-3 and -4: phase 1/2 multicenter studies evaluating KTE-C19 in patients with relapsed/refractory B-precursor acute lymphoblastic leukemia (R/R ALL). Blood. 2016;128(22):1227. https://doi.org/10.1182/blood.V128.22.1227.1227.
Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499–503. https://doi.org/10.1038/s41591-018-0201-9.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42. https://doi.org/10.1056/NEJMoa1914347.
Wang M, Munoz J, Goy AH, Locke FL, Jacobson CA, Hill BT, et al. One-year follow-up of ZUMA-2, the multicenter, registrational study of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma. Blood. 2020;136(Suppl_1):20–2. https://doi.org/10.1182/blood-2020-136382.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. https://doi.org/10.1182/blood-2014-05-552729.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–38. https://doi.org/10.1016/j.bbmt.2018.12.758.
US FDA. FDA approves brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma. Available from: https://www.fda.gov/drugs/fda-approves-brexucabtagene-autoleucel-relapsed-or-refractory-mantle-cell-lymphoma. Accessed 8 Dec 2020.
Palomba ML, Gordon LI, Siddiqi T, Abramson JS, Kamdar M, Lunning MA, et al. Safety and preliminary efficacy in patients with relapsed/refractory mantle cell lymphoma receiving lisocabtagene maraleucel in Transcend NHL 001. Blood. 2020;136(Suppl_1):10–1. https://doi.org/10.1182/blood-2020-136158.
Cohen JB, Switchenko JM, Shanmugasundaram K, Goyal S, Calzada O, Churnetski MC, et al. Clinical trial participation is associated with improved overall survival in newly diagnosed patients with mantle cell lymphoma. Blood. 2019;134(Suppl_1):3483. https://doi.org/10.1182/blood-2019-128864.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this article.
Conflict of interest
Danielle Wallace has no conflicts of interest that are directly relevant to the content of this article. Patrick M. Reagan declares consultancy work for Kite Pharma, and research funding from Seattle Genetics and Genentech.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data sharing is not applicable to this article as no datasets were generated or analyzed for this review.
Code availability
Not applicable.
Authors’ contributions
D.W. and P.R. conceptualized and wrote the work.
Rights and permissions
About this article

Cite this article
Wallace, D., Reagan, P.M. Novel Treatments for Mantle Cell Lymphoma: From Targeted Therapies to CAR T Cells. Drugs 81, 669–684 (2021). https://doi.org/10.1007/s40265-021-01497-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01497-y