
Abstract
Rheumatoid arthritis (RA) is a systemic, autoimmune disease that affects joints and extra-articular structures. In the last decade, the management of this chronic disease has dramatically changed with the introduction of several targeted mechanisms of action, such as tumor necrosis factor-α inhibition, T-cell costimulation inhibition, B-cell depletion, interleukin-6 blockade, and Janus kinase inhibition. Beyond its well-known hematopoietic role on the proliferation and differentiation of myeloid cells, granulocyte-monocyte colony-stimulating factor (GM-CSF) is a proinflammatory mediator acting as a cytokine, with a proven pathogenetic role in autoimmune disorders such as RA. In vitro studies clearly demonstrated the effect of GM-CSF in the communication between resident tissue cells and activated macrophages at chronic inflammation sites, and confirmed the elevation of GM-CSF levels in inflamed synovial tissue of RA subjects compared with healthy controls. Moreover, a pivotal role of GM-CSF in the perception of pain has been clearly confirmed. Therefore, blockade of the GM-CSF pathway by monoclonal antibodies directed against the cytokine itself or its receptor has been investigated in refractory RA patients. Overall, the safety profile of GM-CSF inhibitors seems to be very favorable, with a particularly low incidence of infectious complications. The efficacy of this new mechanism of action is comparable with main competitors, even though the response rates reported in phase II randomized controlled trials (RCTs) appear to be numerically lower than the response rates observed with other biological disease-modifying antirheumatic drugs already licensed for RA. Mainly because of this reason, nowadays the development program of most GM-CSF blockers for RA has been discontinued, with the exception of otilimab, which is under evaluation in two phase III RCTs with a head-to head non-inferiority design against tofacitinib. These studies will likely be useful for better defining the potential role of GM-CSF inhibition in the therapeutic algorithm of RA. On the other hand, the potential role of GM-CSF blockade in the treatment of other rheumatic diseases is now under investigation. Phase II trials are ongoing with the aim of evaluating mavrilimumab for the treatment of giant cell arteritis, and namilumab for the treatment of spondyloarthritis. Moreover, GM-CSF inhibitors have been tested in osteoarthritis and diffuse subtype of systemic sclerosis. This review aims to describe in detail the available evidence on the GM-CSF blocking pathway in RA management, paving the way to a possible alternative treatment for RA patients. Novel insights regarding the potential use of GM-CSF blockers for alternative indications will be also addressed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The increasing number of available therapeutic options in the field of targeted agents has dramatically improved the management of rheumatoid arthritis (RA), but the complexity and variety of the pathogenetic mechanisms accounting for RA manifestations still limit the proportion of patients achieving the treatment target of clinical remission, suggesting the need for the identification of novel mechanisms of action. |
Besides its well-known hematopoietic role, granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine involved in the modulation of differentiation, polarization, and activation of immune cells as macrophages, dendritic cells, and lymphocytes, resulting in a strong influence on the typical immune/inflammatory cascade of chronic autoimmune diseases, including RA. |
The available data on the use of GM-CSF inhibitors in RA, with respect to the peculiar mechanism of action, preclinical experience, efficacy and safety profile, and rapidity of response, are encouraging and warrant phase III trials in order to better establish the future positioning of these novel drugs in the therapeutic algorithm of RA. |
1 Introduction
Rheumatoid arthritis (RA) is a complex, chronic, systemic inflammatory autoimmune disease that affects approximately 0.5–1% of the population worldwide [1, 2]. Even if the etiology is unknown, environmental factors and several genetic polymorphisms have been associated with increased susceptibility and disease severity [3]. RA primarily affects peripheral joints, with aberrant synovial inflammation and proliferation of the synovial tissue, leading to bone and cartilage erosion [1, 4]. Considering the systemic inflammatory burden, RA can also be complicated by extra-articular manifestations such as interstitial lung disease, chronic anemia, and fatigue, as well as comorbidities such as increased cardiovascular disease, osteoporosis, type 2 diabetes mellitus, psychological impairment, infections, and cancer [5, 6]. As a consequence, RA can lead to progressive disability over time and is associated with an increased risk of mortality compared with the general population [7]. The pathogenesis of RA is the result of the complex interaction of a number of different immune cells (in particular T and B lymphocytes) and proinflammatory mediators (mainly tumor necrosis factor (TNF)-α and interleukin (IL)-6), which represent eligible goals for the development of targeted therapies [8, 9]. According to main international recommendations for the management of RA [10, 11], methotrexate is still considered the anchor drug for the treatment of newly diagnosed RA patients [12]. Moreover, in the last decades, the introduction of biological disease-modifying antirheumatic drugs (bDMARDs) focused on cells and molecules involved in RA pathogenesis has dramatically changed the management of the disease, improving the application of more recent treatment strategies and making low disease activity and remission achievable targets, even in methotrexate-insufficient responder patients [13]. Nowadays, five TNF inhibitors, two IL-6 blockers, one T-cell costimulation modulator, one IL-1 soluble receptor, and one B-cell-depleting monoclonal antibody are licensed for the treatment of RA. In addition, the therapeutic armamentarium for RA has recently been enriched by the development of Janus kinase (JAK) inhibitors, active on the transduction into the cell of the signal produced by the interaction between some proinflammatory mediators and their specific transmembrane receptors on immune cells [14]. Nevertheless, despite the expanding number of available treatment options, in real-life experience about 50–70% of treated patients still fail to achieve clinical remission or to maintain an initially good response over time [15,16,17,18,19,20]. Moreover, real-world registries are still populated by a non-negligible proportion of patients presenting a ‘difficult-to-treat’ RA pattern refractory to the majority of available mechanisms of action [21, 22], as a result of the complexity and variety of the pathogenetic mechanisms accounting for RA clinical manifestation. In this scenario, the right choice of the first-line targeted agent in methotrexate-insufficient responder patients [23], and the strategy for managing bDMARD failures, still remain critical unmet needs in the treatment of RA [24,25,26]. Therefore, research toward the identification of novel potential pathways aimed at discovering further therapeutic options is still crucial for improving the better application of a tailored strategy based on precision medicine [27].
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine mostly acting as a hemopoietic growth factor on the proliferation and differentiation of myeloid cells from bone marrow progenitors [28]. Besides its well-known hematopoietic role, GM-CSF is involved in the modulation of differentiation, polarization, and activation of immune cells as macrophages, dendritic cells, and lymphocytes, resulting in a strong influence on the typical immune/inflammatory cascade of chronic autoimmune diseases [29,30,31,32]. Therefore, GM-CSF blockade could be expected to hamper RA inflammation through decreasing leukocyte activation, as confirmed by the available results from preclinical and clinical studies targeting GM-CSF or its receptor [33].
In this review, we describe in detail the pathogenic rationale for blockade of the GM-CSF pathway in RA, and summarize the results of the main clinical trials conducted to explore the potential role of GM-CSF inhibitors in the treatment of RA. Furthermore, we address the role of GM-CSF in other rheumatological disorders, exploring the potential use of the inhibition of this pathway for alternative indications.
2 Granulocyte-Monocyte Colony-Stimulating Factor (GM-CSF) Pathway and Its Biological Functions
GM-CSF, also known as colony-stimulating factor 2 (CSF2), was first identified as a hematopoietic growth factor, as mentioned above [34]. GM-CSF has shown to be relevant in regulating mature myeloid cell populations in homeostatic and inflammatory conditions [35], and exerts several effects on myeloid cells, including survival, activation, differentiation, and mobilization [36,37,38]. GM-CSF circulating levels are quite low in homeostatic conditions but can quickly rise in a few circumstances, such as infections or inflammation [39]. In fact, several cell types can produce GM-CSF, including endothelial cells, macrophages, fibroblasts, DCs, neutrophils, eosinophils, resident tissue cells, T cells, and cancer cells [40,41,42]. In inflammatory milieu, expression of GM-CSF is induced by several cytokines, such as IL-1α and β, IL-12, and TNFα, whereas it is suppressed by IL-4, IL-10, and interferon (IFN)-γ [40, 41, 43,44,45]. Under inflammatory conditions, GM-CSF can recruit and activate myeloid and resident tissue cells, such as endothelial, epithelial, fibroblast, and T-cell populations. Moreover, in vitro studies demonstrated the capability of GM-CSF, when combined with other proinflammatory agents, to polarize macrophages to the proinflammatory M1-like phenotype [46, 47], which is responsible for the production of several proinflammatory cytokines such as IL-1, IL-2, IL-6, IL-23, IL-1, and TNF [28]. In addition, GM-CSF is able to activate resident macrophage-like microglia and induce central nervous system (CNS) inflammation through the upregulation of CD14 and toll-like receptor 4 [48]. Moreover, GM-CSF effects on myeloid-lineage include DCs, by increasing their uptake capacity and promoting non-lymphoid tissue homeostasis [49], and osteoclasts, by stimulating the differentiation of osteoclast precursors and by stopping their late-stage maturation [50]. GM-CSF also plays a pivotal role in lung physiology maintenance and local resistance to infections. In fact, pulmonary epithelial cells produce GM-CSF, leading to alveolar macrophage maturation and favoring the clearing of surfactant lipids and proteins from lung surface [51, 52]. GM-CSF-deficient mice have altered phagocytosis and other immune defects, with accumulation of surfactant-like proteins, leading to peribronchovascular infiltrate and increasing susceptibility to infections [53]. This function impairment causes alveolar proteinosis and enhanced mortality [54].
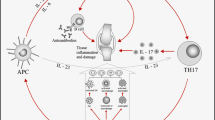
GM-CSF works through the binding to its receptor (GM-CSFR), a heterodimeric molecule composed by one α chain, specific for the ligand, and one β chain, specific for the transduction of the signal into the cell, which is shared with IL-3 and IL-5 [55, 56]. Once GM-CSF binds to its receptor, the signal is transduced not only mainly through the JAK-2/STAT pathway but also through the mitogen-activated protein kinase (MAPK), nuclear factor (NF)-κB, and phosphatidylinositol 3 kinase (PI3K) pathways [30, 56,57,58,59,60] (Fig. 1).

Biological (depicted in normal font) and pathological (depicted in italics) roles of GM-CSF. In the nervous system, GM-CSF contributes to the regulation of satiety and prevents weight gain. Moreover, GM-CSFR is expressed on pain-sensing neurons. In sensory nerves, activation of the GM-CSFR induces neurite sprouting, nerve hypertrophy, and sensitization to nociceptive stimuli. In lungs, a transitional resident population, the innate-like B cells, can produce GM-CSF and express GM-CSFRα. This autocrine signaling loop produces natural IgM and provides an efficient line of defense against pathogens. GM-CSF contributes, to maintain barrier immune homeostasis. When this balance subsides, there is a propensity for infections. In the gastrointestinal tract, GM-CSF promotes the differentiation of monocytes into a specific subtype, CD11b+, capable of phagocytosing and cross-presenting antigens to prime T-cell responses. The role of GM-CSF in resolution of inflammation is highlighted by studies of mice deficient in GM-CSF, IL-3, and IFNγ, which have persistent, low-level inflammation resulting from improper responses to commensal microbes and pathogenes. In an inflamed synovial joint affected by RA, GM-CSF induces activation, differentiation, survival, and proliferation of circulating monocytes, macrophages and resident tissue phagocytes, contributing to articular damage. GM-CSF contributes to the expansion of immunosuppressive myeloid cells at the resolution of inflammation to promote wound healing and tissue repair. GM-CSF promotes trafficking of myeloid cells through activated endothelium of blood vessels, generating the accumulation of monocytes and macrophages in blood vessels. Furthermore, many tumors secrete GM-CSF directly from malignant cells or from cells in the microenvironment, influencing tissue-resident macrophages and increasing systemic myelopoiesis. GM-CSF granulocyte-monocyte colony-stimulating factor, GM-CSFR granulocyte-monocyte colony-stimulating factor receptor, Ig immunoglobulin, IL interleukin, IFN interferon, RA rheumatoid arthritis
3 Targeting GM-CSF in Rheumatoid Arthritis (RA)
CSF family members (macrophage CSF (M-CSF), granulocyte CSF (G-CSF), and GM-CSF), the so-called ‘CSF network’, are able to mediate the communication between resident tissue cells and activated macrophages at chronic inflammation sites. This crosstalk can regulate the proinflammatory network in the setting of autoimmune diseases [28], and in particular RA, whose pathogenesis is deeply driven by the enhanced activity of synovial tissue macrophages responsible for proinflammatory cytokine release [61]. In fact, GM-CSF levels are elevated in inflamed synovial tissue of RA subjects compared with healthy controls [62,63,64,65], and a preclinical study demonstrated by immunohistochemistry that selected clusters of synovial macrophages (especially CD68+ and CD163+) expressed high levels of GM-CSFRα in RA and psoriatic arthritis subjects compared with osteoarthritis (OA) patients and healthy controls [66]. Moreover, several data demonstrated that GM-CSF production could be enhanced by synovial fibroblasts and human chondrocytes in response to TNFα and IL-1 [67, 68]. A recent report highlighted that CD25+ IL-33Ra+ GATA+, a subtype of innate lymphoid cells, is the prevalent cell type in inflamed joints and actively produce GM-CSF [69]. In addition, GM-CSF+ B cells could contribute to autoantibody production in RA patients. In fact, a higher frequency of T and B cells expressing GM-CSF has been described in the peripheral blood of RA subjects [70]. Furthermore, administration of GM-CSF in mouse models of arthritis has been associated with exacerbations of the disease [71], and patients undergoing chemotherapy [72] or affected by Felty’s syndrome [73] treated with recombinant GM-CSF as a growth factor experienced flares of RA. Moreover, GM-CSF has been demonstrated to act on differentiation and activation of T helper (Th) 17 cells that have a central role in RA pathogenesis [41, 74]. The study by Donatien et al. suggests a pivotal role of GM-CSF in pain management [75]. The activation of GM-CSFR in sensory nerves causes neurite sprouting, nerve hypertrophy, and sensitization to nociceptive stimuli that alter the production of proinflammatory chemokines (including TNF) in mouse dorsal root ganglion neurons, through JAK/STAT signaling [76, 77]. GM-CSF has a proinflammatory role in the CNS, where it is synthesized by astrocytes and can modulate the function of microglia and neurons [78]. Furthermore, GM-CSF can cross the blood–brain and blood–spinal cord barrier, mediating pronociceptive and pathogenic effects in CNS inflammation [79].
Considering all this biologic evidence, GM-CSF blockade can interfere with macrophage function and provide clinical benefit in RA. In fact, the administration of a monoclonal antibody against GM-CSFRα (CAM3003) blocked disease progression of collagen-induced arthritis, reducing synovial inflammation and joint destruction [66]. Moreover, GM-CSFRα blockade had a different ability to reduce inflammatory macrophages/monocyte-derived dendritic cells compared with TNF or IL-6 blockade, acting on inflammatory modulation in different ways [80]. Again, CAM3003-treated mouse models reported a reduction in the number of F4/80+ macrophages in antigen-induced arthritis [80].
4 Clinical Trials Using GM-CSF Blockade in RA
All the abovementioned preclinical evidence opened the way for the development of several blockers of the GM-CSF pathway, targeting either GM-CSFRα (mavrilimumab) or the cytokine directly [MORAb-022, MOR103, lenzilumab (KB003), namilumab (MT203)] [33].
4.1 Mavrilimumab
Mavrilimumab (CAM-3001) is a human immunoglobulin (Ig) G4 monoclonal antibody with high affinity to the GM-CSFRα chain and low ability of complement activation because of its IgG4 Fc isotype [81]. The efficacy and safety profiles of mavrilimumab have been investigated in a phase I trial and subsequently extensively assessed in the EARTH clinical development program, embracing three phase IIa–IIb randomized controlled trials (RCTs; EARTH, EARTH EXPLORER 1 and EARTH EXPLORER 2), as reported in Table 1. The pharmaceutical industry manufacturer discontinued the development program on mavrilimumab in RA, although it remains active for giant cell arteritis (https://www.kiniksa.com/our-pipeline).
4.1.1 Phase I Studies
The first randomized, placebo-controlled, double-blind, dose-escalating, phase I study with mavrilimumab (ClinicalTrials.gov identifier: NCT00771420) was conducted in 32 RA patients with active disease despite a stable methotrexate dose (10–25 mg/week) [82]. Patients were randomized 5:1 to receive a single, escalating intravenous dose of mavrilimumab (0.01, 0.03, 0.1, 0.3, 1.0, 3.0 and 10.0 mg/kg) or placebo and were followed up for 24 weeks [82]. The Disease Activity Score 28-joint (DAS28) at week 4 was significantly decreased in patients treated with mavrilimumab compared with controls, along with a significant decrease in C-reactive protein (CRP) levels over 4 weeks in patients receiving mavrilimumab with elevated baseline CRP levels (> 5 mg/L). Overall, adverse events (AEs) were mild or moderate, were reported with similar frequency in all treatment subgroups (74% and 80% for mavrilimumab and placebo, respectively), and were not related to the drug dose. None of the included patients had significant hematological changes or lung disorders [82].
4.1.2 Phase II Studies
The EARTH study (ClinicalTrials.gov identifier: NCT01050998) was a phase II, randomized, double-blind, placebo-controlled trial evaluating the use of mavrilimumab in 233 RA patients randomized to receive subcutaneous mavrilimumab 10, 30, 50, 100 mg, or placebo every other week [83]. At week 12, a greater proportion of patients receiving mavrilimumab achieved the primary endpoint compared with placebo (55.7% vs. 34.7%, p = 0.003), with the 100 mg dose showing a higher effect on American College of Rheumatology (ACR) response versus placebo (ACR20: 40 vs. 69.2%, p =0.005; ACR50: 12 vs. 30.8%, p =0.021; ACR70: 4 vs. 17.9%, p =0.030). Five serious AEs (SAEs) were reported (none were considered treatment related): four (one spontaneous abortion, one intervertebral disc disorder, one fracture of the patella, and one fracture of the humerus) in the mavrilimumab group, and one (worsening of RA) in the placebo group. Low-titer and transient anti-mavrilimumab antibodies were observed in 23 patients (20 of 158 in the active treatment group, and 3 of 75 in the placebo group), but with no apparent impact on the pharmacokinetics. The Japanese cohort of the same study was evaluated separately (ClinicalTrials.gov identifier: NCT01050998) and confirmed similar efficacy and safety results despite the smaller sample size [84].
The phase IIb, multicenter, placebo-controlled EARTH EXPLORER 1 study (ClinicalTrials.gov identifier: NCT01706926) was conducted in 236 RA patients with moderate to severe active RA (mean disease duration 7.8 years, mean baseline DAS28-erythrocyte sedimentation rate (DAS28-ESR) 6.6, > 70% rheumatoid factor (RF)- and/or anticitrullinated protein antibody (ACPA)-positive) despite stable treatment with methotrexate (7.5–25.0 mg/week), randomized 1:1:1:1 to subcutaneous mavrilimumab 150, 100, 30 mg, or placebo, on top of methotrexate, for a 24-week, double-blind phase followed by a long-term, open-label extension [85]. ACR20 response rates were significantly higher in all active treatment arms (73.4, 61.2, and 50.6% for mavrilimumab 150, 100, and 30 mg, respectively) compared with placebo (24.7%, p <0.001 for all subgroups). Similarly, the DAS28-CRP score significantly decreased in all mavrilimumab subgroups compared with placebo (p =0.001), with the best response reported in the mavrilimumab 150 mg arm. Both CRP and ESR levels decreased in a dose-dependent, rapid (since week 1) and sustained (till week 24) manner. The overall incidence of AEs was similar in the mavrilimumab and placebo cohorts, and only two patients discontinued treatment because of treatment-related AEs (one angioedema and one pneumonia). No evident increase in pulmonary toxicity (dose–response changes in oxygen saturation, dyspnea score, or pulmonary function) was reported in mavrilimumab-treated patients. Moreover, no suspected cases of pulmonary alveolar proteinosis (PAP) were observed in patients receiving mavrilimumab [85].
The EARTH EXPLORER II study (ClinicalTrials.gov identifier: NCT01715896) was a phase II, double-blind, randomized trial evaluating the use of mavrilimumab (100 mg every other week, n =70) in combination with methotrexate, in long-standing, active RA patients (mean disease duration 6.7 years, mean baseline DAS28-ESR 6.5, > 70% RF- and/or ACPA-positive) who had not previously responded to a conventional synthetic DMARD (csDMARD) or TNF inhibitor [86]. The study was designed by including a parallel treatment arm treated with the anti-TNF monoclonal antibody golimumab (50 mg every 4 weeks, n =68), even if the study was not powered to perform a statistical head-to-head comparison versus mavrilimumab. At week 24, ACR20, ACR50, and ACR70 responses rates (primary endpoints) were similar in the mavrilimumab and golimumab treatment arms in the overall population (62.0%, 34.8%, and 16.1% vs. 65.6%, 43.4%, and 25.9%, respectively) and in the TNF inhibitor inadequate response (IR) subgroup (72.3%, 33.5%, and 23.5% vs. 61.2%, 42.2%, and 24.2%, respectively), but apparently lower in the mavrilimumab-treatment group compared with golimumab (53.8%, 35.9%, and 10.3% vs. 69.4%, 44.4%, and 27.8%, respectively) in the csDMARD-IR subgroup [86]. The main reason for these controversial results could lie in the use of a suboptimal mavrilimumab dose (100 mg every other week) in this trial compared with the most effective dose (150 mg every other week), confirmed by the EARTH EXPLORER 1 study [85]. The percentage of patients experiencing any treatment-emergent AE (TEAE) was numerically greater in the mavrilimumab arm compared with the golimumab arm (51.4% vs. 42.6%), but consistent with what has been reported in previous studies [82, 85]. The majority of AEs were classified as mild and were not considered as drug-related. The only two SAEs were observed in the golimumab group (pneumocystis pneumonia and another lung disorder). Once again, no significant pulmonary concerns were observed in patients exposed to GM-CSF blockade [86].
4.1.3 Long-Term Efficacy and Safety Profile of Mavrilimumab (Study 1109)
All patients who completed the double-blind phase of the EARTH EXPLORER 1 and 2 trials had the opportunity to enter the open-label extension analysis (study 1109; ClinicalTrials.gov identifier: NCT01712399) and to receive mavrilimumab 100 mg every other week plus methotrexate for a 3-year follow-up period [87]. The study included 442 subjects with a cumulative exposure of 899 patient-years and a median treatment duration of 2.5 years (range 0.1–3.3 years). At week 122, 65.0% and 40.6% patients achieved a DAS28-CRP score of < 3.2 and < 2.6, respectively. Moreover, 68% of mavrilimumab patients had no radiographic progression (≤ 0.5-point change in modified total Sharp score (mTSS) compared with baseline values) at week 74 [87]. Similarly, the safety profile was consistent with the experience observed in the double-blind phase. Only 10% of the overall as-treated patients reported a TEAE of grade 3 or higher severity. No cases of monocytopenia were reported, and mavrilimumab was not associated with any important long-term negative effects on pulmonary safety (no cases of PAP or pulmonary-related deaths), whereas neutropenia was observed in four patients only [87].
The infection risk observed with mavrilimumab [only 14 patients showed serious infections (1.56 per 100 patient-years)] seems to be lower compared with other conventional and targeted DMARDs, even if this evidence is the result of an indirect comparison only. In fact, in a recent review, the estimated incidence rates of serious infections for the pooled population of RCTs conducted with abatacept, rituximab, tocilizumab, TNF inhibitors, and tofacitinib were 3.04, 3.72, 5.45, 4.90, and 3.02 per 100 patient-years, respectively [88]. Similarly, a longitudinal study of a population-based cohort including 27,710 RA patients reported an incidence rate of serious infection of 4.52 per 100 patient-years in the subgroup receiving methotrexate alone [89].
In conclusion, the overall safety profile of mavrilimumab seems to be very reassuring, in particular with regard to infections, even though a longer follow-up period is needed for a more comprehensive analysis of long-term toxicity.
4.1.4 Potential Biomarkers Predicting Clinical Response to Mavrilimumab
Considering the increasing number of therapeutic options for the treatment of RA, as well as the limited number of head-to-head trials directly comparing two different targeted drugs [90], the identification of specific biomarkers useful for predicting clinical response to the available mechanism of action is still a major unmet need in the management of RA [91,92,93].
Two exploratory post hoc analyses of both EARTH EXPLORER studies have been conducted to better investigate the role of the GM-CSF pathway in RA. In the first analysis, whole blood gene expression profiles and serum biomarkers were analyzed by whole genome microarray and protein immunoassay [94]. The administration of mavrilimumab was associated with significant downregulation of type IV collagen formation markers (P4NP7S), macrophage-derived chemokines [C–C motif chemokine 22 (CCL22)], IL-2 receptor-α (IL-2Rα), and IL-6 compared with placebo, with a decreased expression of transcripts enriched in macrophage and IL-22/IL-17 signaling pathways. The suppression of IL-2Rα- and IL-17/IL-22-associated transcripts seems to indicate an indirect suppressive effect of mavrilimumab on T-cell activation, while IL-6 and CCL22 downregulation could reflect a direct role of GM-CSFR inhibition on the release of proinflammatory cytokines by myeloid cells [94].
In the EARTH EXPLORER 2 study, patients in the mavrilimumab arm showed suppressed serum levels of CCL22 and C–C motif chemokine 17 (CCL17), whereas levels of intercellular adhesion molecule-1 (ICAM-1) and C–X–C motif ligand 13 (CXCL13) were decreased in the golimumab-treated group [95]. Moreover, both drugs produced early and sustained suppression of serum markers of disease activity (such as CRP, serum amyloid A, IL-6, CD163, vascular endothelial growth factor (VEGF), IL-2RA, and matrix metalloproteinase (MMP) 1 and 3) in DMARD‐IR patients only. In the TNF-IR population, this effect was transient for golimumab and was maintained over time by mavrilimumab only, despite a similar clinical response in the two treatment arms, suggesting a theoretical, more widespread and upstream effect of GM-CSF inhibition compared with TNF blockade [95].
In the pooled population of the EARTH and EARTH EXPLORER 1 trials, the presence of antibodies against peptidyl-arginine deiminase 4 (PAD-4) in the serum of patients treated with mavrilimumab (150 mg) was associated with a worse clinical response compared with negative subjects [96].
Another post hoc analysis of the EARTH EXPLORER 1 study investigated the changes of a biomarker of activated macrophage activity [citrullinated and MMP degraded vimentin fragment (VICM)] and the blood expression of MMP-9 transcripts and PAD-2 after treatment with mavrilimumab [71]. VICM was significantly (p <0.01) and dose-dependently inhibited by mavrilimumab, and this suppression was supported by a decreased expression of PAD-2 and MMP9 transcripts in patients receiving mavrilimumab. Thus, the authors suggested the potential use of VICM as a novel biomarker of anti-GM-CSF response [97].
4.2 Gimsilumab (MORAb-022)
Gimsilumab (MORAb-022) is a human IgG1 monoclonal antibody against GM-CSF, developed by Morphotek, Inc. To date, the drug has only been evaluated through a phase I, randomized, double-blind, placebo-controlled, single-dose, dose-escalation trial conducted in patients with active RA who were randomized into four arms (including five subjects each) receiving an intravenous infusion of MORAb-022 at increasing doses of 0.36, 0.7, 1, 3, or 10 mg/kg (ClinicalTrials.gov identifier: NCT01357759). The primary outcome was assessing the safety and tolerability of the compound, however the only available results were reported in an abstract [98]. Gimsilumab was well tolerated, both in healthy subjects and RA patients. Mean DAS28-CRP score decreased according to dose regimen, reaching a median decrease of 1.2 units by day 2 for the highest-dose regimen only. A gimsilumab 10 mg/kg dose reported an ACR response, maintained over 1 month, however the drug has not been progressed into phase II, despite completion of the phase I study in 2014.
4.3 Otilimab (GSK3196165)
Otilimab (GSK3196165), previously known as MOR103, is a human, high-affinity, recombinant IgG1 monoclonal antibody against GM-CSF, produced by GlaxoSmithKline and investigated in completed RCTs, with a few published data.
4.3.1 Phase I Study
NCT01023256 was a double-blind, placebo-controlled, multidose, dose-escalation phase Ib/IIa trial evaluating 96 RA patients randomized to receive placebo or three MOR103 doses (0.3, 1.0, and 1.5 mg/kg) for 16 weeks [99]. In the exploratory efficacy analyses, the highest doses of MOR103 (1.5 and 1.0 mg/kg) significantly improved all efficacy outcomes (ACR and European League Against Rheumatism (EULAR) response) compared with placebo. The overall rate of TEAEs (in the majority of cases, mild or moderate) was higher in the MOR103 groups (60.0%) than in the placebo group (44.4%). No pulmonary function test abnormalities were detected [99].
4.3.2 Phase II Studies
Otilimab was subsequently evaluated in two other phase II trials. The first was a double-blind, placebo-controlled, parallel group, phase IIa study (ClinicalTrials.gov identifier: NCT02799472) evaluating the efficacy of subcutaneous otilimab 180 mg once weekly for 5 weeks compared with placebo, and then every other week until week 10 [100]. Patients receiving the active treatment showed a significant decrease in synovial inflammation, and no progression of structural joint damage was detected by magnetic resonance imaging (MRI). The overall incidence of AEs was similar between the otilimab-treated arm and the placebo arm (39.3% vs. 36.4%), and no SAEs, serious infections, or pulmonary events were observed. Moreover, otilimab use did not seem to be complicated by the development of antidrug antibodies [100].
Another trial (ClinicalTrials.gov identifier: NCT02504671) was a phase IIb, double-blind, placebo-controlled, dose-adaptive study aiming to evaluate five different doses of otilimab (22.5, 45, 90, 135, or 180 mg subcutaneously weekly) versus placebo in active RA despite methotrexate [101]. A dose-related treatment effect was observed in DAS-CRP change from baseline to week 12. Compared with placebo, the highest otilimab dose (180 mg) showed a significantly higher (p <0.001) ACR20 response (11 vs. 51%) and a significantly greater (p <0.001) mean change from baseline of DAS28 (− 0.6 vs. − 1.87), visual analog scale (VAS) pain (− 7.07 vs. − 25.01), and patient global assessment (− 6.72 vs. − 23.2). Pharmacodynamic analysis suggested a theoretical benefit from increased exposure to a weekly dose regimen, but further studies are needed to confirm this. Otilimab was well tolerated, without reported TEAEs, and no differences across the treatment groups and no infections and/or pulmonary events were observed [101]. Buckley and coauthors reported the effect of different dose regimens of otilimab on patient-reported outcomes (PROs), such as VAS, Patient’s Global Assessment of Arthritis (PtGA), Health Assessment Questionnaire–Disability Index (HAQ-DI), Brief Fatigue Inventory (BFI) Question 3, the Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F), and the 36-item Short Form Health Survey (SF-36), in moderate-to-severely active RA [101]. Even though these data have only been published as an abstract, otilimab substantially improved the scores of a range of PRO measures among RA patients, particularly pain, as expected according to the previously described pathogenetic role of GM-GSF in pain perception [102].
4.3.3 Phase III Studies
Two phase III RCTs are ongoing (ClinicalTrials.gov) with the aim of comparing otilimab with placebo and tofacitinib in a population of moderate to severe RA patients who have an IR to methotrexate (NCT03980483) or cs/bDMARDs (NCT03970837). Both studies have been similarly designed, with a double-blinded randomization to one of six intervention arms in a ratio of 6:6:3:1:1:1 (two active arms receiving otilimab 90 or 150 mg weekly, one active comparator arm receiving tofacitinib 5 mg twice daily, and three placebo arms), the same primary endpoint (ACR20 response superiority vs. placebo at week 12), the same secondary endpoints (including ACR20 non-inferiority vs. tofacitinib at week 12), and the same duration (52 weeks). The head-to-head design against an innovative drug such as tofacitinib might be useful to clarify the efficacy and safety profile of otilimab, driving the positioning of the drug in the therapeutic landscape of RA.
4.4 Namilumab
Namilumab (AMG203) is a human IgG1 monoclonal antibody that binds to the GM-CSF ligand with high affinity. The drug has been developed by Takeda and, to date, the available data have come from two clinical trials only.
4.4.1 Phase I Study
The PRIORA study (ClinicalTrials.gov identifier: NCT01317797) was a phase Ib double-blind, placebo-controlled, randomized, dose-escalating trial investigating the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of two different subcutaneous dosages of namilumab (150 or 300 mg) on days 1, 15, and 29 in patients with mild to moderate RA receiving treatment with stable methotrexate [103]. Although the study was designed to evaluate the safety profile, the observed clinical response was favorable (especially in the 300 mg arm) according to DAS44 and DAS28-CRP score changes from baseline and ACR20 response rates. The incidence of TEAEs was similar across the three groups: namilumab 150 mg: 63%; namilumab 300 mg: 57%; placebo: 56%. Nasopharyngitis (17%) and exacerbation/worsening of RA (13%) were the most frequent TEAEs (≥ 10% of patients). No anti-namilumab antibodies were detected [103].
4.4.2 Phase II Study
The NEXUS study was a phase II trial (ClinicalTrials.gov identifier: NCT02379091) conducted to evaluate three different doses of namilumab (20, 80, 150 mg by subcutaneous injection) compared with placebo in combination with methotrexate in patients who previously did not respond to methotrexate or TNF inhibitors. The mean change in DAS28-CRP score from baseline at week 12 (primary endpoint) was significantly higher in the namilumab 150 mg group compared with placebo (− 1.69 vs. − 0.77, p = 0.010), with a dose–response effect observed from week 2. The incidence of SAEs was similar among different namilumab doses. The most frequent AEs were upper respiratory tract infections (in the 150 mg arm: nasopharyngitis 17.9% and bronchitis 3.6%), but no serious infections were observed [104].
A second phase II study was started but was subsequently discontinued (ClinicalTrials.gov identifier: NCT02393378). That study evaluated the efficacy of namilumab 150 mg subcutaneously in combination with methotrexate over 24 weeks in patients with moderate to severe RA. The study had adalimumab 40 mg as the active comparator. The primary endpoint was change from baseline in synovitis, erosion, and bone marrow edema, resulting in an underlying osteitis score at MRI at week 24. Even though some trial results have been reported, recruitment status was terminated as a result of a strategic decision. As a further explanation, there is a need to understand data from the psoriasis study (ClinicalTrials.gov identifier: NCT02129777) and to wait for the results of a formal proof-of-concept study (Clinical Trials.gov identifier: NCT02379091. However, the drug does not appear on Takeda’s pipeline any more, generating reasonable doubts on future drug development.
4.5 Lenzilumab (KB003)
KB003 is a high-affinity, recombinant, anti-GM-CSF IgG1κ monoclonal antibody produced by Kalobios Pharmaceuticals. A phase II trial (ClinicalTrials.gov identifier: NCT00995449) aiming to evaluate the safety and efficacy profile of three different intravenous regimens of KB003 (70, 200, and 600 mg × five doses) in active RA patients who have previously not responded to biologic therapy, was terminated due to a refocus of the program development.
5 GM-CSF Blocking in Rheumatology Beyond RA
The GM-CSF pathway has also been investigated in other rheumatological conditions, beyond RA (Table 2).
Moving from the rapid effect on pain reduction by GM-CSF in an experimental OA model [105], GSK3196165 is now under investigation as a potential therapeutic option for human hand OA. In a multicenter, double-blind, placebo-controlled, phase IIa trial (ClinicalTrials.gov identifier: NCT02683785), 44 patients with inflammatory hand OA were randomized to receive five weekly subcutaneous administrations of GSK3196165 (180 mg) or placebo, followed by three further administrations every other week [106]. Patients who received GSK3196165 reported a numerically larger, although not statistically significant, decrease in hand pain at all time points compared with placebo. In particular, a 30% and 50% reduction in pain was observed in 23% and 27% of patients, respectively [106].
In July 2018, Kiniksa Pharmaceuticals initiated a trial to evaluate the use of mavrilimumab (KPL 301) in patients with giant cell arteritis (GCA). The trial is a phase II, randomized, double-blind, placebo-controlled proof-of-concept study evaluating the efficacy and safety of mavrilimumab coadministered with a 26-week corticosteroid taper in subjects with GCA. The screening period consists of up to 6 weeks, followed by a 26-week, double-blind, placebo-controlled period during which participants will receive blinded mavrilimumab or placebo coadministered with a 26-week corticosteroid taper, followed by a 12-week washout/safety follow-up. The trial is currently recruiting (Clinicaltrials.gov identifier: NCT03827018) and no results have yet been published.
Another active area of research is focused on the potential role of GM-CSF in the pathogenesis of systemic sclerosis (SSc). An in vitro study demonstrated higher GM-CSF production by B cells (both naive and memory subsets) from patients with SSc (especially the diffuse subtype) compared with healthy donors. Moreover, under Th2 conditions and with transforming growth factor-β, B cells facilitated the differentiation from CD14+ monocytes to DC-SIGN+CD1a+CD14−CD86+ cells, a subset previously reported in skin and mouse models of SSc [107]. Further studies are needed to clarify this relationship and the potential role of GM-CSF blockade in the treatment of the disease.
Lastly, a phase IIa, proof-of-concept, randomized, double-blind, placebo-controlled study (ClinicalTrials.gov identifier: NCT03622658) is evaluating the efficacy, safety, and tolerability profile of four subcutaneous injections of namilumab (150 mg) in moderate to severely active axial spondyloarthritis (ax-SpA). The primary efficacy outcome is 20% improvement in ankylosing spondylitis assessment (ASAS20) at week 12. No published data from this clinical trial are yet available.
6 Conclusions
Considering the persistence of patients unresponsive to one or more targeted drugs despite the increasing abundance of therapeutic options, the introduction of a new mechanism of action could be useful for improving some unmet needs in the management of RA. Besides its well-known hematopoietic effect, both in vitro and in vivo studies indicated GM-CSF as one of the proinflammatory mediators involved in the activation process of autoimmune diseases such as RA, especially in the early phase of the disease. Moreover, the well-established effect of GM-CSF on pain perception can strengthen the rationale for the use of GM-CSF blockade in the treatment of RA, and can partially account for the good clinical response observed in RCTs conducted with monoclonal antibodies targeted on this pathway.
Overall, data on the use GM-CSF inhibitors from clinical trials seem to be favorable, even if the rates of ACR responses observed in phase II RCTs conducted with mavrilimumab and otilimab are numerically lower than those reported with other bDMARDs already licensed for RA. In this regard, it is important to note that a robust indirect comparison is not completely feasible because of the lack of completed phase III trials of both mavrilimumab and otilimab. Indeed, these controversial findings may have played a part in the decision by manufacturers to discontinue the development program of many GM-CSF inhibitors for RA. Hopefully, the ongoing phase III trials comparing otilimab with tofacitinib in a non-inferiority head-to-head design will provide crucial information for the potential positioning of this new compound in the therapeutic algorithm of RA. Of note, some intriguing insights regarding potential biomarkers able to predict the clinical response in patients treated with GM-CSF blockers have been suggested by some post hoc analyses of mavrilimumab RCTs. These preliminary results, if also confirmed for otilimab, might be crucial for the identification of candidates to receive GM-CSF blockade instead of other mechanisms of action. In this context, the low immunogenicity and favorable safety profile observed in the clinical trials are encouraging in regard to continuation of the development program of GM-CSF inhibitors. In particular, the low incidence of serious infections encountered in preliminary experience with anti-GM-CSF can be a strong point in favor of this new compound compared with other biologic and synthetic DMARDs targeted on different mechanisms of action. In addition, considering the role of GM-CSF in the homeostasis of alveolar surfactant, the lack of reported cases of PAP in clinical experience is very reassuring with regard to the potential lung toxicity of this product.
In conclusion, the available data on this new drug class, with respect to the peculiar mechanism of action, preclinical experience, efficacy and safety profile, rapidity of response, and relevant analgesic effect, are encouraging in view of the phase III trials already ongoing, which will be important to better establish the future role of these novel drugs in the treatment landscape of RA.
References
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358(9285):903–11.
Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S265–72.
Lefevre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15(12):1414–20.
Avina-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheumatol. 2008;59(12):1690–7.
Firestein GS. The disease formerly known as rheumatoid arthritis. Arthritis Res Ther. 2014;16(3):114.
Holmqvist M, Ljung L, Askling J. Mortality following new-onset rheumatoid arthritis: has modern rheumatology had an impact? Ann Rheum Dis. 2018;77(1):85–91.
Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-alpha on bone homeostasis. Front Immunol. 2014;5:48.
Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–108.
Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–77.
Singh JA, Saag KG, Bridges SL, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(1):1–26.
Favalli EG, Biggioggero M, Meroni PL. Methotrexate for the treatment of rheumatoid arthritis in the biologic era: still an “anchor” drug? Autoimmun Rev. 2014;13(11):1102–8.
Chighizola CB, Favalli EG, Meroni PL. Novel mechanisms of action of the biologicals in rheumatic diseases. Clin Rev Allergy Immunol. 2014;47(1):6–16.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17(1):78.
Favalli EG, Pregnolato F, Biggioggero M, Becciolini A, Penatti AE, Marchesoni A, et al. Twelve-year retention rate of first-line tumor necrosis factor inhibitors in rheumatoid arthritis: real-life data from a local registry. Arthritis Care Res (Hoboken). 2016;68(4):432–9.
Iannone F, Ferraccioli G, Sinigaglia L, Favalli EG, Sarzi-Puttini P, Atzeni F, et al. Real-world experience of tocilizumab in rheumatoid arthritis: sub-analysis of data from the Italian biologics’ register GISEA. Clin Rheumatol. 2018;37(2):315–21.
Favalli EG, Biggioggero M, Marchesoni A, Meroni PL. Survival on treatment with second-line biologic therapy: a cohort study comparing cycling and swap strategies. Rheumatology (Oxford). 2014;53(9):1664–8.
Iannone F, Sinigaglia L, Favalli EG, Sarzi-Puttini P, Atzeni F, Caporali R, et al. Drug survival of adalimumab in patients with rheumatoid arthritis over 10 years in the real-world settings: high rate remission together with normal function ability. Clin Rheumatol. 2016;35(11):2649–56.
Biggioggero M, Favalli EG. Ten-year drug survival of anti-TNF agents in the treatment of inflammatory arthritides. Drug Dev Res. 2014;75(Suppl 1):S38–41.
Sarzi-Puttini P, Antivalle M, Marchesoni A, Favalli EG, Gorla R, Filippini M, et al. Efficacy and safety of anti-TNF agents in the Lombardy rheumatoid arthritis network (LORHEN). Reumatismo. 2008;60(4):290–5.
de Hair MJH, Jacobs JWG, Schoneveld JLM, van Laar JM. Difficult-to-treat rheumatoid arthritis: an area of unmet clinical need. Rheumatology (Oxford). 2017. https://doi.org/10.1093/rheumatology/kex349.
Conigliaro P, Triggianese P, De Martino E, Fonti GL, Chimenti MS, Sunzini F, et al. Challenges in the treatment of rheumatoid arthritis. Autoimmun Rev. 2019;18(7):706–13.
Cantini F, Niccoli L, Nannini C, Cassarà E, Kaloudi O, Giulio Favalli E, et al. Tailored first-line biologic therapy in patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis. Semin Arthritis Rheum. 2016;45(5):519–32.
Favalli EG, Raimondo MG, Becciolini A, Crotti C, Biggioggero M, Caporali R. The management of first-line biologic therapy failures in rheumatoid arthritis: Current practice and future perspectives. Autoimmun Rev. 2017;16(12):1185–95.
Todoerti M, Favalli EG, Iannone F, Olivieri I, Benucci M, Cauli A, et al. Switch or swap strategy in rheumatoid arthritis patients failing TNF inhibitors? Results of a modified Italian Expert Consensus. Rheumatology (Oxford). 2018;57(57 Suppl 7):vii42–53.
Cantini F, Niccoli L, Nannini C, Cassarà E, Kaloudi O, Giulio Favalli E, et al. Second-line biologic therapy optimization in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Semin Arthritis Rheum. 2017;47(2):183–92.
Selmi C, Kon E, De Santis M, Favalli EG, Cimaz R, Generali E, et al. How advances in personalized medicine will change rheumatology. Per Med. 2018;15(2):75–8.
Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–44.
Cornish AL, Campbell IK, McKenzie BS, Chatfield S, Wicks IP. G-CSF and GM-CSF as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):554–9.
Dougan M, Dranoff G, Dougan SK. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity. 2019;50(4):796–811.
Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediat Inflamm. 2015;2015:568543.
Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. 2016;12(1):37–48.
Crotti C, Raimondo MG, Becciolini A, Biggioggero M, Favalli EG. Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: evidence to date. Drug Des Dev Ther. 2017;11:211–23.
Metcalf D. The colony-stimulating factors and cancer. Cancer Immunol Res. 2013;1(6):351–6.
Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34(2):81–9.
Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15(4):557–67.
Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100(12):4193–200.
Bezbradica JS, Gordy LE, Stanic AK, Dragovic S, Hill T, Hawiger J, et al. Granulocyte-macrophage colony-stimulating factor regulates effector differentiation of invariant natural killer T cells during thymic ontogeny. Immunity. 2006;25(3):487–97.
Williamson DJ, Begley CG, Vadas MA, Metcalf D. The detection and initial characterization of colony-stimulating factors in synovial fluid. Clin Exp Immunol. 1988;72(1):67–73.
Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol. 2012;188(7):3107–15.
El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–75.
Gasson JC. Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood. 1991;77(6):1131–45.
Jansen JH, Wientjens GJ, Fibbe WE, Willemze R, Kluin-Nelemans HC. Inhibition of human macrophage colony formation by interleukin 4. J Exp Med. 1989;170(2):577–82.
Ozawa H, Aiba S, Nakagawa S, Tagami H. Interferon-gamma and interleukin-10 inhibit antigen presentation by Langerhans cells for T helper type 1 cells by suppressing their CD80 (B7-1) expression. Eur J Immunol. 1996;26(3):648–52.
Sagawa K, Mochizuki M, Sugita S, Nagai K, Sudo T, Itoh K. Suppression by IL-10 and IL-4 of cytokine production induced by two-way autologous mixed lymphocyte reaction. Cytokine. 1996;8(6):501–6.
Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178(8):5245–52.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55.
Parajuli B, Sonobe Y, Kawanokuchi J, Doi Y, Noda M, Takeuchi H, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation. 2012;9:268.
Zhan Y, Vega-Ramos J, Carrington EM, Villadangos JA, Lew AM, Xu Y. The inflammatory cytokine, GM-CSF, alters the developmental outcome of murine dendritic cells. Eur J Immunol. 2012;42(11):2889–900.
Lari R, Fleetwood AJ, Kitchener PD, Cook AD, Pavasovic D, Hertzog PJ, et al. Macrophage lineage phenotypes and osteoclastogenesis—complexity in the control by GM-CSF and TGF-beta. Bone. 2007;40(2):323–36.
Avci AB, Feist E, Burmester GR. Targeting GM-CSF in rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(4 Suppl 98):39–44.
Ryan PC, Sleeman MA, Rebelatto M, Wang B, Lu H, Chen X, et al. Nonclinical safety of mavrilimumab, an anti-GMCSF receptor alpha monoclonal antibody, in cynomolgus monkeys: relevance for human safety. Toxicol Appl Pharmacol. 2014;279(2):230–9.
Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol. 2009;21(5):514–21.
Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA. 1994;91(12):5592–6.
Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte–macrophage colony-stimulating factor. Crit Rev Immunol. 2005;25(5):405–28.
Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134(3):496–507.
Jenkins BJ, Blake TJ, Gonda TJ. Saturation mutagenesis of the beta subunit of the human granulocyte-macrophage colony-stimulating factor receptor shows clustering of constitutive mutations, activation of ERK MAP kinase and STAT pathways, and differential beta subunit tyrosine phosphorylation. Blood. 1998;92(6):1989–2002.
Sato N, Sakamaki K, Terada N, Arai K, Miyajima A. Signal transduction by the high-affinity GM-CSF receptor: two distinct cytoplasmic regions of the common beta subunit responsible for different signaling. EMBO J. 1993;12(11):4181–9.
Broughton SE, Nero TL, Dhagat U, Kan WL, Hercus TR, Tvorogov D, et al. The betac receptor family—structural insights and their functional implications. Cytokine. 2015;74(2):247–58.
Hercus TR, Dhagat U, Kan WL, Broughton SE, Nero TL, Perugini M, et al. Signalling by the betac family of cytokines. Cytokine Growth Factor Rev. 2013;24(3):189–201.
Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheumatol. 1996;39(1):115–24.
Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1993;52(12):870–5.
Bell AL, Magill MK, McKane WR, Kirk F, Irvine AE. Measurement of colony-stimulating factors in synovial fluid: potential clinical value. Rheumatol Int. 1995;14(5):177–82.
Berenbaum F, Rajzbaum G, Amor B, Toubert A. Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur Cytokine Netw. 1994;5(1):43–6.
Wright HL, Bucknall RC, Moots RJ, Edwards SW. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford). 2012;51(3):451–9.
Greven DE, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, et al. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis. 2015;74(10):1924–30.
Leizer T, Cebon J, Layton JE, Hamilton JA. Cytokine regulation of colony-stimulating factor production in cultured human synovial fibroblasts: I. Induction of GM-CSF and G-CSF production by interleukin-1 and tumor necrosis factor. Blood. 1990;76(10):1989–96.
Campbell IK, Novak U, Cebon J, Layton JE, Hamilton JA. Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J Immunol. 1991;147(4):1238–46.
Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity. 2018;48(6):1220–32.e5.
Makris A, Adamidi S, Koutsianas C, Tsalapaki C, Hadziyannis E, Vassilopoulos D. Increased frequency of peripheral B and T cells expressing granulocyte monocyte colony-stimulating factor in rheumatoid arthritis patients. Front Immunol. 2017;8:1967.
Campbell IK, Bendele A, Smith DA, Hamilton JA. Granulocyte-macrophage colony stimulating factor exacerbates collagen induced arthritis in mice. Ann Rheum Dis. 1997;56(6):364–8.
de Vries EG, Willemse PH, Biesma B, Stern AC, Limburg PC, Vellenga E. Flare-up of rheumatoid arthritis during GM-CSF treatment after chemotherapy. Lancet. 1991;338(8765):517–8.
Hazenberg BP, Van Leeuwen MA, Van Rijswijk MH, Stern AC, Vellenga E. Correction of granulocytopenia in Felty’s syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood. 1989;74(8):2769–70.
Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–7.
Donatien P, Anand U, Yiangou Y, Sinisi M, Fox M, MacQuillan A, et al. Granulocyte-macrophage colony-stimulating factor receptor expression in clinical pain disorder tissues and role in neuronal sensitization. Pain Rep. 2018;3(5):e676.
Bali KK, Venkataramani V, Satagopam VP, Gupta P, Schneider R, Kuner R. Transcriptional mechanisms underlying sensitization of peripheral sensory neurons by granulocyte-/granulocyte-macrophage colony stimulating factors. Mol Pain. 2013;9:48.
Stösser S, Schweizerhof M, Kuner R. Hematopoietic colony-stimulating factors: new players in tumor–nerve interactions. J Mol Med (Berl). 2011;89(4):321–9.
Malipiero UV, Frei K, Fontana A. Production of hemopoietic colony-stimulating factors by astrocytes. J Immunol. 1990;144(10):3816–21.
McLay RN, Kimura M, Banks WA, Kastin AJ. Granulocyte-macrophage colony-stimulating factor crosses the blood–brain and blood–spinal cord barriers. Brain. 1997;120(Pt 11):2083–91.
Cook AD, Louis C, Robinson MJ, Saleh R, Sleeman MA, Hamilton JA. Granulocyte macrophage colony-stimulating factor receptor alpha expression and its targeting in antigen-induced arthritis and inflammation. Arthritis Res Ther. 2016;18(1):287.
Nair JR, Edwards SW, Moots RJ. Mavrilimumab, a human monoclonal GM-CSF receptor-α antibody for the management of rheumatoid arthritis: a novel approach to therapy. Expert Opin Biol Ther. 2012;12(12):1661–8.
Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-α, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70(9):1542–9.
Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis. 2013;72(9):1445–52.
Takeuchi T, Tanaka Y, Close D, Godwood A, Wu CY, Saurigny D. Efficacy and safety of mavrilimumab in Japanese subjects with rheumatoid arthritis: findings from a Phase IIa study. Mod Rheumatol. 2015;25(1):21–30.
Burmester GR, McInnes IB, Kremer J, Miranda P, Korkosz M, Vencovsky J, et al. A randomised phase IIb study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1020–30.
Weinblatt ME, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Guo X, et al. A randomized phase IIb study of mavrilimumab and golimumab in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(1):49–59.
Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovský J, Godwood A, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor α monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70(5):679–89.
Strand V, Ahadieh S, French J, Geier J, Krishnaswami S, Menon S, et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res Ther. 2015;17:362.
Lacaille D, Guh DP, Abrahamowicz M, Anis AH, Esdaile JM. Use of nonbiologic disease-modifying antirheumatic drugs and risk of infection in patients with rheumatoid arthritis. Arthritis Rheumatol. 2008;59(8):1074–81.
Favalli EG, Bugatti S, Biggioggero M, Caporali R. Treatment comparison in rheumatoid arthritis: head-to-head trials and innovative study designs. Biomed Res Int. 2014;2014:831603.
Ingegnoli F, Favalli EG, Meroni PL. Does polymorphysm of genes coding for pro-inflammatory mediators predict the clinical response to tnf alpha blocking agents? A review analysis of the literature. Autoimmun Rev. 2011;10(8):460–3.
Monti S, Klersy C, Gorla R, Sarzi-Puttini P, Atzeni F, Pellerito R, et al. Factors influencing the choice of first- and second-line biologic therapy for the treatment of rheumatoid arthritis: real-life data from the Italian LORHEN Registry. Clin Rheumatol. 2017;36(4):753–61.
Atzeni F, Bongiovanni S, Marchesoni A, Filippini M, Caporali R, Gorla R, et al. Predictors of response to anti-TNF therapy in RA patients with moderate or high DAS28 scores. Joint Bone Spine. 2014;81(1):37–40.
Guo X, Higgs BW, Bay-Jensen AC, Wu Y, Karsdal MA, Kuziora M, et al. Blockade of GM-CSF pathway induced sustained suppression of myeloid and T cell activities in rheumatoid arthritis. Rheumatology (Oxford). 2018;57(1):175–84.
Guo X, Wang S, Godwood A, Close D, Ryan PC, Roskos LK, et al. Pharmacodynamic biomarkers and differential effects of TNF- and GM-CSF-targeting biologics in rheumatoid arthritis. Int J Rheum Dis. 2019;22(4):646–53.
Grant E, Schwickart M, Godwood A, Moate R, Song E, Chavez C, et al. Lack of autoantibodies to peptidyl arginine deiminase 4 predict increased efficacy of mavrilimumab in rheumatoid arthritis. Arthritis Rheumatol. 2016;68(Suppl 10):1–2.
Mortensen JH, Guo X, De Los Reyes M, Dziegiel MH, Karsdal MA, Bay-Jensen AC, et al. The VICM biomarker is released from activated macrophages and inhibited by anti-GM-CSFRα-mAb treatment in rheumatoid arthritis patients. Clin Exp Rheumatol. 2019;37(1):73–80.
Kivitz A, Hazan L, Hoffman K, Wallin BA. MORAb-022, an anti-granulocyte macrophage-colony stimulating factor (GM-CSF) monoclonal antibody (MAB): results of the first study in patients with mild-to-moderate rheumatoid arthritis (RA). Ann Rheum Dis. 2016;75(Suppl 2):507.
Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74(6):1058–64.
Genovese MC, Berkowitz M, Conaghan PG, Davy K, Inman D, Fisheleva E, et al. A Phase IIa mechanistic study of anti-GM-CSF (GSK3196165) with methotrexate treatment in patients with rheumatoid arthritis (RA) and an inadequate response to methotrexate [abstract]. Arthritis Rheumatol. 2018;70(Suppl):10.
Buckley C, Simon Campos JA, Yakushin S, Zhdan V, Davy K, Inman D, et al. A phase IIb dose-ranging study of anti-GM-CSF with methotrexate treatment in patients with rheumatoid arthritis (RA) and an inadequate response to methotrexate [abstract]. Arthritis Rheumatol. 2018;70(Suppl):10.
Chris B, Simon CJ, Vyacheslav Z, Brandon B, Deven C, Katherine D, et al. GSK3196165 an investigational anti-GM-CSF monoclonal antibody, improves patient reported outcomes in a phase IIb study of patients with rheumatoid arthritis (RA). Ann Rheum Dis. 2019;78(Suppl 2):A191.
Huizinga TW, Batalov A, Stoilov R, Lloyd E, Wagner T, Saurigny D, et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):53.
Taylor PC, Saurigny D, Vencovsky J, Takeuchi T, Nakamura T, Matsievskaia G, et al. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: a randomized, controlled trial. Arthritis Res Ther. 2019;21(1):101.
Cook AD, Pobjoy J, Steidl S, Durr M, Braine EL, Turner AL, et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther. 2012;14(5):R199.
Schett G, Bainbridge C, Berkowitz M, Davy K, Fernandes S, Griep E, et al. A phase IIa study of anti-GM-CSF antibody GSK3196165 in subjects with inflammatory hand osteoarthritis [abstract]. Arthritis Rheumatol. 2018;70(Suppl):10.
Higashioka K, Ota Y, Nakayama T, Mishima K, Ayano M, Kimoto Y, et al. GM-CSF-producing B cells: a novel B cell subset involved in the pathogenesis of systemic sclerosis [abstract]. Arthritis Rheumatol. 2017;69(Suppl):10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Chiara Crotti, Elena Agape, Andrea Becciolini, Martina Biggioggero and Ennio Giulio Favalli declare no conflicts of interest.
Funding
None.
Rights and permissions
About this article

Cite this article
Crotti, C., Agape, E., Becciolini, A. et al. Targeting Granulocyte-Monocyte Colony-Stimulating Factor Signaling in Rheumatoid Arthritis: Future Prospects. Drugs 79, 1741–1755 (2019). https://doi.org/10.1007/s40265-019-01192-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01192-z