
Abstract
Fentanyl is a synthetic, highly selective opioid with many desirable physicochemical properties, including a high lipophilicity and predictable pharmacokinetics. These properties have an established record in the management of pain in a variety of settings, particularly acute pain and breakthrough cancer pain. Fentanyl was initially developed for parenteral use; however, this is invasive and impractical in the outpatient setting. Unfortunately, the high first-pass metabolism of fentanyl makes oral formulations unfeasible. However, its high lipophilicity allows fentanyl to be absorbed via a number of other routes. Thus new formulations were designed to allow non-invasive methods of administration. Transmucosal and transdermal fentanyl formulations are well established, and have proven useful in the settings of breakthrough cancer pain, emergencies and in the paediatric population. The iontophoretic transdermal system was developed to provide a needle-free system of delivering bolus doses of fentanyl on demand, a novel way of delivering patient-controlled opioid analgesia. Transpulmonary administration of fentanyl remains experimental. The aim of this review is to provide an update on current non-parenteral fentanyl formulations, with attention to their particular pharmacokinetics and features relevant to clinical use in pain management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Fentanyl, originally developed for parenteral intraoperative use in anaesthesia, has increasingly become an opioid used by other routes of administration. |
The high lipidsolubility of fentanyl makes it as well suited for transdermal applications as for fast-acting transmucosal and transpulmonary use. |
Therefore, fentanyl is now used in a wide range of settings from chronic cancer and non-cancer pain to acute postoperative and breakthrough pain. |
1 Introduction
1.1 Initial Development
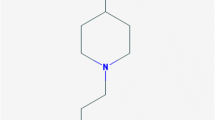
Fentanyl (N-(1-phenethyl-4-piperidyl)propionanilide) was first structurally designed by Dr. Paul Janssen in December 1960 and was introduced into clinical practice in Europe as an analgesic agent in 1963 [1]. The development was explicitly driven by the desire to create a more lipid-soluble opioid than those previously available, as the group realised that lipophilicity was the key to faster onset and higher potency [2]. The use of fentanyl was initially restricted to intraoperative parenteral administration as a component of anaesthesia.
1.2 Pharmacology
Fentanyl is a synthetic phenylpiperidine derivative with potent μ-opioid receptor activity, and some δ and κ-opioid receptor activity [3]. It is at least 100 times more potent than morphine [4].
Fentanyl is a small and highly lipophilic molecule, with a high octanol:water partition coefficient of 816. The low ionization and high lipophilicity favours absorption across biological membranes [5].
It is therefore particularly well suited for use via transdermal and transmucosal routes of delivery, with bioavailability in the range of 50–90%, significantly better than other clinically used opioids [5, 6]. Fentanyl also shows rapid penetration into the CNS, with a transfer half-life (t 1/2ke0) of 4.7–6.6 min [6]. In addition to passive diffusion, active transport systems have been described for fentanyl uptake via the brain endothelium [7].
Fentanyl has a large volume of distribution (6 l/kg) [4]. Fentanyl is rapidly distributed from plasma into highly vascularised compartments, before redistribution to muscle and fat tissue occurs. After equilibration at these sites, fentanyl is released back into the plasma, leading to its long terminal elimination half-life of 3–8 h [8]. Therefore, it has a short duration of action after single bolus administration, but the duration of effect is increased due to accumulation after multiple boluses or with a continuous infusion.
Approximately 80% of fentanyl is bound to plasma proteins, including acute phase proteins, with only free fentanyl able to cross the blood brain barrier [9]. The variable binding of serum fentanyl to plasma proteins may contribute to observed variability of pharmacokinetic and pharmacodynamic parameters between patients.
Fentanyl is primarily metabolised in the liver by the cytochrome P450 (CYP) 3A4 isoenzyme system [10], undergoing rapid and extensive first-pass metabolism via oxidative N-dealkylation to norfentanyl and other inactive metabolites [9, 11]. This accounts for its poor oral bioavailability of less than 30%, and the lack of oral fentanyl formulations [12].
Concomitant administration of fentanyl with drugs that inhibit CYP3A4 activity (e.g., certain protease inhibitors, macrolide antibiotics, azole antifungals, diltiazem and verapamil) may result in an increase in fentanyl exposure due to a reduction in its systemic clearances; this has led to potentially fatal respiratory depression [2]. Approximately 75% of fentanyl is excreted via the kidney, with less than 10% as unchanged fentanyl, while about 9% of metabolites are recovered in the faeces [8, 9].
In opioid-naïve postoperative patients, fentanyl produces rapid and effective analgesia at minimum plasma concentrations between 0.6 and 1.5 ng/ml [13]. Fentanyl displays all opioid-related adverse effects, including nausea and vomiting, pruritus, difficulty concentrating and somnolence at clinically relevant doses [9]. As with all other opioids, dose-dependent respiratory depression is the most serious and potentially life-threatening adverse effect with no ceiling effect [14].
1.3 New Formulations
The need for parenteral administration limited the usefulness of fentanyl in non-perioperative indications. It is unnecessarily invasive, inconvenient and may lead to complications such as haematoma, infection and nerve damage. These limitations created a growing clinical and commercial interest in the development of non-parenteral fentanyl formulations, which began in the mid-1980s [6].
The first of these formulations entered the market in the late 1980s in the form of a transdermal patch, initially developed by the Alza Corporation and then marketed as Duragesic® by Janssen Pharmaceuticals [2]. The main advantage was the achievement of a steady-state fentanyl plasma concentration ideal for the treatment of background cancer pain. It became the commercially most successful analgesic preparation of its time.
Transmucosal preparations were first introduced in the USA in 1993, as Oralet®, a lollipop containing fentanyl for transmucosal absorption in the setting of pediatric anaesthetic premedication [15]. It was not particularly commercially successful [2]. However, the rapid pharmacokinetics of transmucosal fentanyl were found to be ideal for management of breakthrough pain in cancer patients.
Breakthrough pain is defined as a transitory exacerbation of pain that occurs in cancer patients with otherwise persistent pain, which is controlled with long-acting opioid therapy [16]. Up to 50% of cancer patients suffer from multiple, severe episodes of breakthrough pain, with exacerbations having rapid onset within min, but of overall short duration (median 30 min) [17]. These are often undertreated due to concerns about the adverse effects of opioids [18].
Traditionally, immediate-release oral opioids (e.g. morphine, oxycodone) were used for treating breakthrough pain. However, their generally slow onset and prolonged time to maximum analgesic effect (of up to 60 min) made them less than ideal for effective treatments for breakthrough pain [6, 19].
Transmucosal fentanyl displays suitable pharmacokinetics with rapid onset of analgesic effect, which is ideal for the treatment of breakthrough pain [20]. Subsequently, a modified version of the transmucosal fentanyl ‘lollipop’ was launched as Actiq® in the USA in 1998 for the treatment of breakthrough pain in opioid-tolerant cancer patients. It became extremely successful and revolutionised the treatment of breakthrough pain [19].
Based on this success, a whole series of transmucosal fentanyl preparations utilising sublingual, buccal and intranasal routes were developed for the same indication. There is strong evidence that all such fentanyl preparations are superior to immediate-release morphine preparations in the treatment of breakthrough pain [21].
A transdermal system permitting patient-controlled analgesia in the postoperative setting has been developed in the form of iontophoretic transdermal fentanyl and, after initial technical problems and a withdrawal from the market, is now again marketed [22].
More recently, pilot studies have examined the use of inhaled transpulmonary fentanyl, though their role in clinical practice has yet to be determined.
Overall, these newer fentanyl formulations have permitted the use of fentanyl in non-perioperative indications. They have changed the management of pain in a variety of clinical scenarios, including chronic and breakthrough cancer pain, pre-hospital and critical-care settings, paediatric patients and post-surgical patients. A summary of the characteristics of these new formulations is presented in Table 1.
2 Transmucosal Fentanyl
The pharmacokinetics of fentanyl delivered via oral and nasal mucosa have been characterised throughout the development of all the main fentanyl drug products that are approved by the US Food and Drug Administration (FDA) for the treatment of breakthrough cancer pain [23–27]. Knowledge of the pharmacokinetics of different formulations may aid physicians in individualising therapy, depending on the onset and duration of a patient’s pain.
2.1 Oral Transmucosal Fentanyl Citrate (OTFC)
2.1.1 Formulation
Oral transmucosal fentanyl citrate (OTFC) was the first transmucosal fentanyl formulation, introduced as Actiq® in the USA in 1998 and in Europe in 2002. Actiq® is a sweetened lozenge containing fentanyl citrate that is attached to a stick to help the patient sweep the medication across the lining of the cheek [20]. OTFC is currently available in six strengths: 200, 400, 600, 800, 1200, and 1600 μg. Dose-concentration linearity has been demonstrated within this range of doses [28]. OTFC is approved in general for breakthrough pain in adults with cancer who are opioid tolerant [23].
2.1.2 Pharmacokinetics
Transmucosal administration allows intake via the oral route whilst still largely avoiding first-pass metabolism. This provides rapid access into the systemic circulation with a higher bioavailability. Administration of the lozenge takes approximately 15 min [23]. As the lozenge dissolves in the oral cavity, approximately one-quarter of the total dose is absorbed through the buccal mucosa, which is responsible for early peak plasma fentanyl concentrations, rapid onset of effect and high bioavailability. The median time to maximum plasma concentration was 23 min after the start of administration [6].
The overall bioavailability of OTFC also relies on subsequent absorption after ingestion into the gastrointestinal tract. Around three quarters of the total dose is swallowed and slowly absorbed from the gastrointestinal tract. This portion achieves a lower bioavailability of around 40% [29] with total overall bioavailability around 50% [6]. The gradual intestinal absorption maintains fentanyl concentrations in the analgesic range (1–3 ng/ml) for around 2 h [30].
2.1.3 Clinical Use
The rapid onset combined with short duration of effect makes OTFC one attractive option for treatment of breakthrough cancer pain. The recommended starting dose of OTFC is 200 μg up to four times daily, with no more than two lozenges used to treat an individual episode of breakthrough pain [9]. The lozenge design also allows the unit to be removed from the mouth if signs of excessive opioid effects occur during administration [9]. Doses can then be increased as required; suggested dosing guidelines have been published [31]. A detailed analysis of pain intensity, pain relief and duration of analgesia with IV morphine and OTFC suggests a relative potency ratio of 8–14:1 [32].
The efficacy of OTFC has been compared with placebo in a multicentre double-blind, randomised study of breakthrough pain in opioid-tolerant patients with cancer [33]. Compared to placebo, better pain relief was reported with OTFC at all time points from 15 min to 1 h after administration (p < 0.0001).
In a long-term safety study, OTFC (200–1600 μg) was used to treat 38,595 episodes of breakthrough pain in 155 opioid-tolerant patients with cancer [34]. The patients consistently gave global satisfaction ratings indicating the pain relief provided by OTFC was very good or excellent. OTFC was not more effective than placebo in ameliorating dyspnea on exertion in cancer patients [35].
There is significant ‘off-label’ use described in patients other than opioid-tolerant cancer pain patients with breakthrough pain. Such use includes post-operative pain, migraine, sickle cell disease, wound care, burns dressing and procedural pain including in children as well as chronic pain of non-malignant origin [9, 13]; it is of note that OTFC is specifically contraindicated in opioid-naïve patients, including use to manage acute and postoperative pain in view of the high risk of respiratory depression.
There is an ongoing discussion on the potential use of OTFC as an analgesic in the battlefield [36, 37]. This notion is supported by a case series of 286 US soldiers who received OTFC for analgesia and reported overall rapid and safe analgesia [38].
Reported adverse effects of OTFC are typical of opioids as described. Hallucinations and confusion have also been reported in clinical studies of this formulation [39]. The sugar content of the OTFC lozenge improves its palatability for patients, but some concerns have been raised regarding dental problems with prolonged and repeated use [40].
There are several limitations to the OTFC as differences in application technique may result in variable absorption of the fentanyl dose. Absorption is reduced if the patient has reduced saliva, applies the OTFC to the tongue or gums rather than the buccal mucosa, chews the OTFC, has ingested liquids that change the oral pH before OTFC application or applies the product for less than or longer than 15 min [29, 41]. In addition, OTFC may be difficult to administer in patients who are severely disabled, lack dexterity, have poor cognitive function or who are severely fatigued [42–44]. In order to overcome some of these limitations, other transmucosal fentanyl formulations have been developed.
2.2 Fentanyl Buccal Tablet
2.2.1 Formulation
The fentanyl buccal tablet (FBT), marketed as Effectora® in Europe and Fentora® in the USA, is based on an active OraVescent® drug delivery technology to enhance fentanyl dissolution and absorption in the mouth [45]. The five strengths of FBT available are 100, 200, 400, 600, and 800 μg, proportional to increasing disk size. Dose-concentration proportionality has been shown from 100 to 1300 μg [46]. Use of FBT was approved in the USA in 2006 for treatment of breakthrough cancer pain in opioid-tolerant adults. It was subsequently approved for the same indication in Europe in 2008 [47].
2.2.2 Pharmacokinetics
OraVescent® drug technology activates both the dissolution and absorption of fentanyl from the tablet by producing dynamic changes in the local pH [45]. As the tablet dissolves, the local environment becomes more acidic because of the dissolution of citric acid into carbonic acid [28]. In this acidic environment, the available fentanyl becomes almost completely ionized and highly soluble. As the carbonic acid then dissociates into water and carbon dioxide, the environment becomes less acidic. The dissolved fentanyl becomes more lipophilic and available for rapid absorption across the mucosa [48].
The FBT has been designed to be placed within the buccal cavity above a rear molar, between the upper cheek and gum, and retained in place until it disintegrates after 14–25 min [49]. In contrast to OTFC, it does not require active participation from the patient.
Because 50% of the fentanyl in FBT is absorbed transmucosally, first-pass metabolism is bypassed to a greater extent than with OTFC, hence a greater proportion of fentanyl enters the systemic circulation [18]. A pooled analysis of studies relevant to FBT pharmacokinetics, suggests absolute bioavailability of 65%, linear proportional absorption over the 100–800 μg dose range, an average T max of 35–45 min, with 80% of the maximum concentration maintained for 2 h [46].
2.2.3 Clinical Use
The effervescent delivery system of the FBT results in faster and more complete mucosal uptake of fentanyl compared with OTFC. The passive delivery system may also improve drug delivery compared to self-administration of a lozenge [50]. Clinically, this results in FBT providing a slightly faster onset and a higher bioavailability than OTFC. Additionally, FBT is sugar free which reduces the risk of dental caries compared with OTFC.
Key clinical trials investigating the efficacy of FBT have mainly focused on opioid-tolerant cancer patients and compared it against placebo. Double-blinded, randomized placebo-controlled trials in more than 250 patients have demonstrated that FBT provides effective treatment of breakthrough cancer pain in adult cancer patients taking maintenance opioid therapy for their persistent pain, with a significant reduction in pain intensity as early as 10 min after administration [51, 52]. Clinical efficacy has also been demonstrated in patients with chronic, non-cancer related pain including those with chronic back and neuropathic pain, although these are not approved indications [13, 51, 53].
An indirect mixed treatment comparison meta-analysis, which compared the efficacy of buccal fentanyl with OTFC and sublingual fentanyl citrate, indicates that FBT appears to produce a better outcome for patients [21]. Specifically, the results indicated a 2:1 likelihood that FBT would produce better pain relief than sublingual or oral transmucosal fentanyl during the first 60 min after dosing. The apparent advantage has been attributed to the active effervescent action of buccal fentanyl, enhancing the rate and efficiency of absorption.
Adverse effects were similar to those described for opioids in general; however, off-label use and improper selection of patients or dose have led to safety concerns and reports of respiratory depression and deaths [50].
2.3 Fentanyl Buccal Soluble Film
2.3.1 Formulation
Fentanyl buccal soluble film (FBSF) is an alternate formulation of buccal fentanyl, which is approved for treatment of breakthrough pain. It is marketed as Breakyl® in Europe and Onsolis® in the USA [28]. In this formulation, fentanyl is included in a film that adheres to the inside of the cheek and delivers fentanyl across the buccal mucosa. FBSF is currently available as 200, 400, 600, 800, and 1200 μg formulations. There is a dose-concentration proportionality from 200 to 1200 μg [54, 55].
2.3.2 Physicochemical Properties and Pharmacokinetics
FBSF dissolves within 15–30 min, thereby releasing fentanyl, which passively diffuses into the bloodstream through the buccal mucosa. The bio-erodible muco-adhesive delivery technology consists of two different layers made of water-soluble polymeric films, one with a bio-adhesive layer containing the active substance and one inactive. The inactive layer isolates the bio-adhesive layer from the buccal cavity, minimizing the amount of fentanyl that is swallowed [55]. The absolute bioavailability is 71 with 51% absorbed through the buccal mucosa [54].
A low intra-individual variability (7–10%) and a much higher inter-individual variability (23–39%) in pharmacokinetic parameters were found, suggesting a relative reliable dose-to-dose predictability [56]. The inter-individual variability emphasizes the need for dose titration in an individual patient.
2.3.3 Clinical Use
The trials published so far compare FBSF exclusively with placebo and show, not surprisingly, superiority in efficacy here [57]. Comparisons with other transmucosal fentanyl preparations are lacking so far.
2.4 Sublingual Fentanyl
2.4.1 Formulation
The sublingual formulation of fentanyl is a small tablet (FST) composed of a combination of active drug particles and water-soluble carrier particles coated with a muco-adhesive agent [39]. A sublingual spray (FSS) has also been developed. These formulations have been designed to be placed or applied to the deepest part of the sublingual cavity and left to completely dissolve without chewing or sucking.
Two sublingual fentanyl formulations are currently approved for use in the USA. The sublingual tablet formulation, marketed as Abstral®, is associated with rapid dissolution and passive absorption through the mucosa [58]. The tablet rapidly disintegrates when in contact with the sublingual mucosa into an ordered mixture of fentanyl combined with a soluble carrier [59]; the disintegration times are in the range of 70–100 s [60]. Available strengths are 100, 200, 300, 400, 600 and 800 μg. There is a dose-concentration linearity from 100 to 800 μg [61, 62].
Another sublingual formulation in form of a FSS and marketed as Subsys®, was approved by the FDA in 2012. Available strengths are 100, 200, 400, 600 and 800 μg [58].
2.4.2 Pharmacokinetics
These formulations are designed to enable fentanyl to dissolve quickly and utilise the known high vascularity of the sublingual mucosa so the dose of fentanyl is absorbed over about 30 min [18]. The onset of action is similar to that of the fentanyl buccal tablet, with time to first detectable plasma concentration at around 10 min [58]. Any fentanyl dissolved in saliva and swallowed is absorbed orally and subject to first-pass metabolism that decreases the effective dose of fentanyl [28]. The time to T max ranges between 40 and 57 min and mean elimination half-life is reported to be between 3 and 12.5 h [58]. Low salivation affects the pharmacokinetics of sublingual tablets negatively with later onset of effect and lower C max [63].
In a direct comparison of the pharmacokinetics of FSS and OTFC, mean Cmax values (0.81 vs. 0.61 ng/ml) and bioavailability (approximately 76 vs. 51%) were higher with FSS [64]. Effective plasma concentrations were also reached much earlier with FSS and those at 10 min with FSS were equivalent to those with OTFC at 60 min.
2.4.3 Clinical Use
Sublingual fentanyl preparations are generally well tolerated and are recommended for use in management of breakthrough pain in opioid-tolerant adult patients with cancer [65–67]. In a long-term study over 90 days, FSS was effective, safe and well tolerated [68]. However, again direct comparisons to other transmucosal preparations have not been published. In comparison to oral morphine solution, FSS provided faster pain relief with higher patient satisfaction [69]; mean doses required were 38 mg morphine and 235 µg FSS.
2.5 Intranasal Transmucosal Fentanyl
2.5.1 Formulations
The first intranasal fentanyl (INF) formulation available was Instanyl® (Narcomed Danmark), which was approved for use in opioid-tolerant patients with chronic cancer pain in 2009 [70]. Instanyl® is currently available in three concentrations: as 50, 100, and 200 μg diluted in 100 μl per spray. Dose linearity has been demonstrated from 100 to 800 μg [71, 72].
Subsys® (Insys Therapeutics) delivers a nasal spray of fentanyl in units of 100 or 200 μg via a delivery device and was approved in the USA in January 2012. This is a single-use system for the same group of patients that allows for subsequent dose titration to a maximum of 1600 μg [70].
To enhance nasal penetration and lessen local irritation, additives such as pectin, which form a thin gel over the mucosa, have been added into newer spray formulations such as NasalFent®, PecFent® and PecSys® [71].
A wide range of other fentanyl formulations has been used intranasally; even the solution of fentanyl citrate for intravenous administration can be delivered as a nasal spray into one or both nostrils in droplet form, with a therapeutic dose delivered in 1 or 2 sprays [73, 74].
2.5.2 Pharmacokinetics
The nasal cavity is well suited for mucosal administration of drugs because of its vascularity, large surface area and thin barrier creating high tissue permeability [75, 76]. Nasal routes additionally deliver drugs directly to the CNS site of action via the olfactory and trigeminal nerves, vasculature, cerebrospinal fluid and the lymphatic system, resulting in rapid central nervous analgesic effects [77, 78]. The intranasal route may also have advantages in patients with mucosal damage or salivary dysfunction [79]. Plasma concentrations measured after INF administration compare well to concentrations after intravenous injection [80] with bioavailability in the range of 90% [81]. The onset of action is rapid (7 min) with a T max of 12–15 min and duration of around 2 h. This compares favourably for all pharmacokinetic parameters compared to OTFC [82].
The extent of drug absorption from this formulation can vary due to runoff via the pharynx, which is subsequently swallowed and undergoes first-pass metabolism. When used in combination with pectin, there is less potential for runoff into the pharynx due to the gel nature of pectin formulations [28]. However, in a comparative trial this did not translate into clinical advantages [83].
2.5.3 Clinical Use
Intranasal application of drugs is a valuable option in situations where successful intravenous cannulation is difficult to achieve, where nausea and vomiting makes oral dosing problematic, and where training levels or circumstances are not permissive for intravenous pain management [84]. Its non-invasive nature and rapid onset of effect makes INF an increasingly popular choice not only in management of breakthrough cancer pain, but also of acute pain, particularly in out-of-hospital care and within the paediatric population.
Several studies have attempted to compare INF with parenteral morphine in the paediatric population, and results indicate that it is an effective analgesic that causes minimal distress and no serious adverse effects were reported [61]. Other studies have evaluated its use in children with orthopaedic or other acute injuries presenting to an emergency department. A dose of 1.5–2 μg/kg results in maximum pain score reduction after 20–30 min, with good analgesia achieved within 10 min [74, 85, 86]. Efficacy and safety was even shown in 1- to 3-year-old children [87].
INF used for the management of acute pain in adults has also been shown to be safe, effective and generally well tolerated. Among inpatients, several randomized trials among adult orthopaedic, gynaecological and general surgical patients found that INF had similar efficacy and side effects to intravenous fentanyl [88–92]. A randomized, crossover trial comparing INF and intravenous morphine for procedural wound care in adult burns patients showed similar efficacy [86]. However, the study does not provide information about intensity of pain during wound care and it is difficult to draw conclusions from this [84, 86]. A meta-analysis of 16 randomised, controlled trials (RCTs) found no significant analgesic differences between INF and IV fentanyl in treatment of acute and post-operative pain [84].
INF was even used for treatment of labour pain with good efficacy, although some neonatal respiratory depression occurred [93].
INF in the out-of-hospital setting (ambulance service) was safe and effective in 903 patients; a mean cumulative fentanyl dose of 114 µg was used [94].
Superior analgesic efficacy of INF has been demonstrated in the treatment of breakthrough cancer pain compared to OTFC. In an open-label study, the intravenous fentanyl solution delivered via the nasal route (dose 50–200 μg) proved effective, with two-thirds of cancer patients achieving meaningful analgesia more rapidly compared to OTFC 200–1600 μg [95]. More patients preferred the intranasal spray to OTFC and found it easier to use. In a meta-analysis, INF was the most effective treatment of breakthrough cancer pain compared to oral morphine and fentanyl by all other transmucosal routes [96].
Specific adverse effects of intranasal fentanyl include potential nasal congestion, epistaxis and a change of nasociliary function following long-term use [70]. However, a study involving over 40,000 intranasal self-administrations of intravenous fentanyl citrate solution by 356 patients found no evidence of nasal toxicity [97]. This was confirmed by a 6-month observational study, which found no serious adverse effects over this period [98].
2.6 General Considerations on Transmucosal Fentanyl
2.6.1 Comparative Meta-Analyses
In two meta-analyses of all transmucosal fentanyl preparations, they resulted in lower pain intensity and higher pain relief scores at all time points compared to placebo or oral morphine [99, 100]. Transmucosal fentanyl preparations also achieved better global assessment scores, but the lack of head-to-head comparisons in the literature was mentioned.
These findings were in principle confirmed in a network meta-analysis, which was performed to overcome the lack of direct comparisons [19]. Here INF, FBT and OTFC were more effective (greater pain intensity difference PID) than placebo 15 min after intake, with only INF rated as resulting in a clinically meaningful effect (PID ≥2/10) at this point of time. All transmucosal preparations showed this effect at 30 min, but not immediate-release oral morphine, which became superior to placebo only at 45 min.
The same superior efficacy of INF was also found in another meta-analysis, which showed a PID of 1.7/10 at 15 min versus 0.4/10 for OTFC and 0.5/10 for FBT; again immediate release oral morphine was the worst comparator, deemed not an efficacious treatment of breakthrough pain by the investigators [96].
2.6.2 Importance of Dose Titration
It is important to note that the effective dose of both OTFC and buccal fentanyl cannot necessarily be predicted from the dose of maintenance opioid therapy as there is no clinical correlation [101]. For FSS, there is only a moderate correlation even to the background transdermal fentanyl dose [102]. Data from two double-blind, phase III clinical trials with FBT indicate that the final effective dose for individual patients can have a wide range from 100 to 800 μg for each episode of breakthrough cancer pain [51, 52]. Similarly, titration of FSS dose found a range of effective doses with 800 µg (24.5%) and 1200 µg (20.4%) the most commonly identified effective doses; only 2.3% of patients could not be titrated to an effective dose [103]. A large European study on titration of FBT identified, that a 200 µg dose to start titration was as safe as a 100 µg starting dose [104]. Furthermore, the wide inter-individual response to opioids has been well documented in the past. Therefore, the dose of rapid onset fentanyl should be carefully titrated to avoid serious adverse consequences, even in opioid-tolerant patients.
This claim is, however, contradicted by two studies from the same group, which suggest that an effective and safe dose of FBT can be identified without titration by using doses proportional to the background opioid regimen [105, 106]. A more recent publication suggests that an effective and safe breakthrough dose of FST can be calculated from the morphine rescue dose used effectively by applying a conversion ratio of 1:50 [107].
Cautious dose titration is also important when a patient is transitioning from one oral fentanyl product to another. For instance, cases of respiratory depression have been reported when dose-for-dose substitution from OTFC to FBT was implemented [50]. This is not appropriate as the FBT delivers significantly higher concentrations of systemic fentanyl. Sublingual fentanyl has an even higher bioavailability of up to 75%, which may pose the same risks without careful dose titration.
2.6.3 Pharmacoeconomics
Concerns have been raised regarding the cost effectiveness of transmucosal fentanyl products in comparison with oral morphine for treatment of breakthrough cancer pain. In some comparative studies with oral opioids, the number of doses needed to obtain relief at 15 min was greater than ten [99]. For this reason and possibly others, the National Institute for Health and Care Excellence guidelines from 2012 explicitly recommend a trial of morphine before utilising rapid onset fentanyl in the treatment of breakthrough cancer pain [108]. However, the European Association of Palliative Care released guidelines in the same year, recommending rapid onset fentanyl (either buccal or nasal) as the treatment of choice for patients with breakthrough cancer pain [109]. Given the difference in time to onset, the authors suggest that fentanyl should be offered for patients suffering with unpredictable fast onset and/or short duration breakthrough pain.
A number of cost-effectiveness analyses on this topic have been published. One found an incremental cost-effectiveness ratio of INF of 10,140 euros/quality-adjusted life-year (QALY), thereby undercutting a threshold of 30,000 euros/QALY [110]. In Sweden, a study found with high certainty that INF is the most cost-effective intervention for breakthrough cancer pain [111]. Overall, there is a lack of useful pharmacoeconomic studies on the treatment of breakthrough cancer pain [112].
2.6.4 Safety
As outlined above, the high potency of fentanyl and the transmucosal delivery systems with rapid absorption increase the risk of adverse effects, in particular when used in opioid-naïve patients. The American College of Medical Toxicology has therefore published a position statement on fentanyl preparations highlighting some of these concerns, which are also contained in the transmucosal immediate release fentanyl (TIRF) risk evaluation and mitigation strategy (REMS) for use with transmucosal fentanyl formulations released by the FDA [113].
2.6.5 Abuse Potential
Similar to all full μ-opioid agonists, fentanyl is subject to misuse and abuse with high addictive potential. It continues to be one of the most widely available and therefore one of most highly abused prescribed opioids in the community.
According to recent reports released by the US Center for Health Statistics [114], fentanyl is amongst the top ten drugs most frequently involved in overdose deaths from the period 2010–2014. What is perhaps most alarming is that the rate of drug overdose deaths involving fentanyl is rising significantly, and more than doubled in a single year from 1905 deaths in 2013 to 4200 deaths 2014.
Within the range of different fentanyl formulations, the transdermal fentanyl devices, particularly those with a drug reservoir containing high doses of fentanyl, are especially prone to abuse. Previously worn transdermal devices may contain 28–84% of the initial drug [115], thus safe disposal of used devices or patches is also paramount in preventing abuse. Given that there is limited evidence for the use of fentanyl outside the setting of acute pain or cancer pain, the importance of appropriate patient selection must be emphasized; and use outside these boundaries should discouraged.
3 Transdermal Fentanyl
3.1 Transdermal Therapeutic Systems
3.1.1 Formulation
Transdermal fentanyl patches containing fentanyl in a reservoir have been in clinical use since the 1990s [116]. There are two types of transdermal delivery systems currently available. The original transdermal therapeutic system (TTS) was produced by Janssen, named Duragesic®; it was followed by many generic formulations. Duragesic® patches are available in 12, 25, 50, 75 or 100 μg/h concentrations.
The initial TTS formulations consisted of a liquid fentanyl drug reservoir separated from the adhesive layer by a rate-limiting membrane, and ethanol as co-absorbent to provide controlled drug release [117]. However, these reservoir systems carried a risk of accidental leakage resulting in release of the entire dose within a short period of time [118]. They also carried a higher risk of abuse, due to the high concentration of fentanyl present in the liquid reservoir, which could be easily syphoned off [119].
Therefore, newer passive transdermal fentanyl formulations exclusively use the matrix technique, with the drug dissolved in an inert polymer matrix that controls drug release [117]. This method diminishes the risk of incidental drug leakage and complicates the extraction of the drug for abuse [120]. However, abuse, overdose and even death can still occur by applying the patches sublingually [121], ingesting them [122] or by injection of fentanyl extracted by boiling [123]. The high risk of ingestion is confirmed by an in vitro study, which revealed that an average of 26 and 41% of 7.65 mg of fentanyl contained within a 75 µg/h patch was released into gastric and intestinal fluid in 3 h, respectively [124]. The newer matrix patch consists of only two functional layers and a protective peel strip. The pharmacokinetics and clinical effects of the matrix transdermal device are similar to that of the original transdermal device [125].
3.1.2 Pharmacokinetics
Transdermal fentanyl patches constantly deliver a certain dose per hour over 72 h, so that theoretically a zero-order delivery with a rate constant k0 should be provided [6]. However, the initial movement of fentanyl into the systemic circulation is not zero-order, as the drug needs to cross skin and underlying tissue.
Fentanyl becomes detectable in the serum within 1–2 h of application and therapeutic serum fentanyl concentrations are achieved approximately 12–16 h after transdermal device applications [126–129]; steady-state concentrations are usually only achieved with the second patch administration at 24 h [125]. Peak analgesic effect may vary with local conditions such as skin condition and body temperature; there is a risk of unintentional opioid overdose in febrile patients [130, 131] or in patients with impaired skin integrity, e.g. by eczema [132]. Increased absorption leading to toxicity has also been described with exposure to high temperatures [133]. Absorption of TTS fentanyl in cancer patients has also been found to be highly variable, with lower absorption in older patients, and variations depending on the type of cancer, with breast or gastrointestinal cancer associated with higher absorption than lung cancer [134, 135].
The C max achieved ranges from 0.3 ng/ml for a 12 μg/h transdermal device to 2.6 ng/ml for a 100 μg/h transdermal device [126, 136]. In cancer pain patients, a dose of 100 µg/h of TTS fentanyl was equivalent to 240 mg morphine/day [137]. Bioavailability of transdermal fentanyl is approximately 92%; towards the end of the delivery interval, the concentration gradient between the transdermal therapeutic system and the skin decreases until the delivery ceases [138] leading to ‘end-of-dose failure’ in some patients; poor adhesion may be another explanation here [139]. The apparent half-life is around 16–22 h, due to continued absorption from the subcutaneous depot during the elimination phase [126, 129, 136].
3.1.3 Clinical Use
TTS fentanyl is well established in the treatment of cancer pain [140] and may offer advantages over other opioids [141]. The maintenance of a relatively steady serum concentration results in reduced side effects and improved efficacy [142]. Factors such as ease of application and convenience in an outpatient setting also lead to improved therapeutic compliance. As the release of fentanyl is continuous over 72 h, TTS is more suitable for chronic pain management. TTS has, therefore, also been used in chronic pain of non-malignant origin, although the usefulness of opioids in general in this indication is limited [143]. In a comparative systematic review of transdermal opioids in this indication, buprenorphine caused fewer adverse effects than fentanyl [144]. It is not suited to situations where pain may fluctuate, such as in the acute postoperative phase as its long half-life is impractical for rapid adjustments. Here, potentially life-threatening respiratory depression has been observed [145].
The overall safety and tolerability of transdermal fentanyl is favourable. Most reported adverse effects are typical of opioids. Mild irritations at the application site have been reported in around 25% of cases [146]. This can be ameliorated by rotating the patch to different skin sites. There appears to be a lower incidence of constipation compared with slow-release oral morphine in patients with cancer pain [140]. As with other opioids, concurrent use of other sedatives, opioids, hypnotics or alcohol can cause oversedation or unwanted respiratory depression and the dose should be adjusted [147]. The period of greatest risk is in the initiation phase of therapy prior to the development of steady-state pharmacokinetics. Clinically relevant respiratory depression in patients with chronic pain on opioid analgesics was not observed in three randomised trials [148–150].
Economic evaluation has been evaluated in the treatment of chronic moderate to severe pain, where transdermal fentanyl was compared with two long-acting oral opioids [151]. In a conservative modelled analysis, the fentanyl transdermal system led to increased quality-adjusted life-days (QALDs) at a nominal increased cost.
3.2 Iontophoretic Therapeutic Systems
3.2.1 Formulation
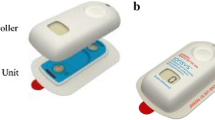
Iontophoresis is a method for transdermal administration of ionisable drugs in which electrically charged components are propelled through the skin by an external electric field [152]. This process uses an imperceptible, low-intensity, electric current to drive ionisable fentanyl molecules from a reservoir across the stratum corneum and into the systemic circulation [153]. Serum levels C max and AUC increase with increasing current [154, 155]. The iontophoretic therapeutic system (ITS) device offers a pre-programmed, credit card-sized, needle-free system that is designed to be activated by pressing the recessed button twice within 3 s, with an audible beep and light-emitting diode flash indicating a successful activation [156].
The original fentanyl ITS comprised of a drug-containing hydrogel sandwiched between two electrodes arranged parallel to the skin surface, with the lower electrode attached closely to the skin via an adhesive layer [157]. It received European marketing authorisation in January 2006 and FDA approval in May 2006 for this indication. However, it was never launched in the USA due to the detection of corrosion in a small number of samples during storage caused by moisture from the hydrogels and battery voltage being applied to the electronics during storage [155]. There was also a high incidence of respiratory depression when used for treatment of acute pain [13]. The device was voluntarily withdrawn from the market worldwide in September 2008 [158, 159]. At the time of withdrawal, over 10,000 integrated fentanyl ITS devices had been used in the EU by patients for postoperative pain management [22].
The recently approved two-component fentanyl ITS IonSys® device consists of a controller unit and a drug unit designed to be assembled just prior to use. The controller unit contains the electronics including an activation button, a red light-emitting diode, a green light-emitting diode and a dose counting display [22]. The drug unit contains two hydrogel reservoirs, with the bottom of the unit covered with a skin adhesive for attachment to the patient. The separated system ensures that the hydrogels in the drug unit and the electronic circuit of the controller are kept apart during manufacture and storage, thereby preventing corrosion of the electronics [159]. The new system is bioequivalent to the previous version [155].
3.2.2 Pharmacokinetics
The standard demand dose of fentanyl ITS is 40 μg delivered over a 10-min period, hence allowing for up to six doses per hour. Each system lasts for 24 h or a maximum of 80 doses, whichever occurs first. Bioequivalence has been established between the original integrated fentanyl ITS and the current two component fentanyl ITS device. Dose-finding studies using an intravenous fentanyl patient-controlled analgesia (PCA) system, deduced an ideal bolus dose of 40 μg to maximise efficacy and minimize respiratory depression [160].
Iontophoretic systems may provide both rapid onset (within 15 min) and prolonged duration of analgesia [161]. After an initial 40 μg bolus administered over 10 min, fentanyl plasma concentrations continue to increase for approximately 5 min, resulting in a quick onset of effect at approximately 15 min [6]. Steady-state absorption can be attained approximately 10–12 h after initiation of treatment; allowing fentanyl to accumulate within tissues, creating a longer elimination half-life and prolonged duration of analgesia [156, 162]. In postsurgical patients, average serum concentrations of fentanyl over a 24-h dosing period ranged from 0.4 to 1.5 ng/ml with a median T max of around 23 h [162].
Absorption of fentanyl after ITS activation is similar when the system is applied to the upper outer arm or chest, but is approximately 20% lower when applied to the lower inner arm [163]. Unlike traditional transdermal absorption, iontophoresis does not form a drug depot within the skin and passive absorption is minimal [164], thus no clinically relevant fentanyl absorption occurs unless an electric current is applied [13].
3.2.3 Clinical Use
Fentanyl ITS is indicated for the management of acute postoperative pain in adults requiring opioid analgesia in the hospital setting. The design of fentanyl HCl ITS adds convenience and ease of use to postoperative pain management practices. Unlike intravenous PCA methods, ITS does not require tubing, poles or a pump apparatus, which hinders patient mobility. It adheres to the patient’s upper outer arm or chest using an adhesive backing, making it less invasive than other patient controlled analgesic options. Dosing parameters are fixed, which reduces the risk of operator errors in programming [13]. As with all PCAs, patients are able to titrate their own analgesia to individual needs [153].
In well-designed, multicentre clinical trials, fentanyl ITS was an effective and generally well tolerated method for managing acute postoperative pain in inpatients who had undergone major abdominal, thoracic or orthopaedic surgery [165]. Overall, fentanyl ITS provided equivalent analgesia efficacy to that with morphine patient-controlled intravenous analgesia (PCIA) [166], but was perceived to be more convenient and easier to use than morphine PCIA by patients, nurses [167] and physical therapists [168].
Fentanyl ITS has been shown to be superior to placebo and therapeutically equivalent to morphine intravenous PCA as a method of pain control for the treatment of acute postoperative pain in adult patients [165, 169]. In randomised, double-blind trials, adult patients who had undergone major abdominal, orthopaedic or thoracic surgery showed superiority to placebo on all efficacy measures [170]. Patients receiving fentanyl ITS also had a greater ability to mobilize after surgery than patients receiving morphine PCIA [171, 172].
The convenience offered by the fentanyl ITS could also be viewed as a double edged sword. Whilst it may be better for patient mobility, it also has the potential to be more easily concealed, stolen or abused compared with other forms of PCA fentanyl [169]. Another limitation is that it may only be used for a maximum of 24 h before it must be discarded and replaced with a new system, which has the potential to result in analgesic gaps [13]. It may also result in unnecessary costs if a system is applied and not used within 24 h [173]. Fixed dosing may be a limitation in patients requiring a lower dose (e.g. hepatic impairment, elderly patients) and those who require a higher dose or background infusion (e.g. opioid-tolerant patients).
Adverse events associated with fentanyl ITS are similar to those associated with general opioid administration. Nausea is the most frequently reported adverse event in Phase III clinical trials, with incidence ranging from 31 to 67% compared to 25 to 60% in those receiving placebo [170, 173]. Incidence of nausea, vomiting, pruritus and headache is similar to that observed with intravenous morphine PCA [174], although a later study from the same group showed a lower incidence of opioid-related adverse events with fentanyl ITS [175]. Fentanyl ITS has been tested under X-ray exposure levels beyond those associated with diagnostic X-rays and CT scans and under exposure to radiofrequency fields and found to function correctly [176].
4 Transpulmonary Fentanyl
4.1 Formulations
Pilot studies have examined the use of metered dose inhalers or nebulised intravenous fentanyl solution as well as liposome encapsulated formulations in human volunteers [86, 89, 90, 177, 178].
4.2 Pharmacokinetics
Inhaled fentanyl can result in rapid analgesia, with a T max of 2 min after administration of 300 μg intravenous fentanyl citrate solution [179]. Inhalation of liposome encapsulated fentanyl achieves a T max of 22 min [180].
The absorption of transpulmonary fentanyl is unreliable, with bioavailability ranging from 12% with liposome-encapsulated fentanyl to close to 100% with free fentanyl, with substantial inter-individual variability [13]. Duration of action and half-life of transpulmonary fentanyl are prolonged compared to intravenous fentanyl [70].
4.3 Clinical Use
Transpulmonary fentanyl for use in management of acute or chronic pain remains experimental, although it is appealing as a non-invasive method of delivering drugs directly to highly vascularised tissue with a very large surface area (~90 m2). It has been used successfully within the context of palliation of patients with chronic lung disease to relieve dyspnoea [181]. Although there have been no reported significant adverse events reported within the small pilot studies; recommendations cannot be made without a larger body of evidence to support its use.
5 Conclusion
Over the past half century, fentanyl has proven to be one of the most successful opioid analgesics. While initially designed for parenteral use as a component of anaesthesia, a large range of fentanyl formulations have shown usefulness in a variety of clinical scenarios, including management of acute and breakthrough pain in patients with cancer, postsurgical patients, paediatric patients, pre-hospital and critical-care settings. Transdermal formulations of fentanyl are in particular suitable for use in management of cancer pain. However, in view of increasing concerns about the inappropriate use of opioids in the management of many chronic pain conditions, in particular in Canada, the USA and Australia, this indication needs to be considered with extreme caution [145].
As with use of any potent opioid agonist, the importance of cautious dose titration cannot be overstated, as it is essential to tailor the treatment to the individual patient and reduce adverse effects. Fentanyl in its available formulations is well tolerated in general with a good safety profile when used responsibly; however, the high potency and ease of administration carries a risk of inappropriate use and the rapid effect a risk of abuse and addiction.
The more recently developed and now again available iontophoretic system utilises modern technology to allow fentanyl to be delivered in a needle-free and patient-controlled setting.
Due to its unique and favourable pharmacokinetics, fentanyl is likely to remain a mainstay of treatment of pain in many clinical scenarios. Newer fentanyl formulations continue to offer promising features that produce effective pain relief with a good safety and tolerability profile.
References
Stanley TH, Egan TD, Van Aken H. A tribute to Dr. Paul A. J. Janssen: entrepreneur extraordinaire, innovative scientist, and significant contributor to anesthesiology. Anesth Analg. 2008;106(2):451–62. doi:10.1213/ane.0b013e3181605add.
Stanley TH. The fentanyl story. J Pain. 2014;15(12):1215–26. doi:10.1016/j.jpain.2014.08.010.
Maguire P, Tsai N, Kamal J, Cometta-Morini C, Upton C, Loew G. Pharmacological profiles of fentanyl analogs at mu, delta and kappa opiate receptors. Eur J Pharmacol. 1992;213(2):219–25.
Peng PW, Sandler AN. A review of the use of fentanyl analgesia in the management of acute pain in adults. Anesthesiology. 1999;90(2):576–99.
Chen C, Gupta A. Clinical and pharmacokinetic considerations of novel formulations of fentanyl for breakthrough cancer pain. Pain Manag. 2014;4(5):339–50. doi:10.2217/pmt.14.32.
Lötsch J, Walter C, Parnham MJ, Oertel BG, Geisslinger G. Pharmacokinetics of non-intravenous formulations of fentanyl. Clin Pharmacokinet. 2013;52(1):23–36. doi:10.1007/s40262-012-0016-7.
Henthorn TK, Liu Y, Mahapatro M, Ng KY. Active transport of fentanyl by the blood-brain barrier. J Pharmacol Exp Ther. 1999;289(2):1084–9.
Mather LE. Clinical pharmacokinetics of fentanyl and its newer derivatives. Clin Pharmacokinet. 1983;8(5):422–46.
Mystakidou K, Katsouda E, Parpa E, Vlahos L, Tsiatas ML. Oral transmucosal fentanyl citrate: overview of pharmacological and clinical characteristics. Drug Deliv. 2006;13(4):269–76. doi:10.1080/10717540500394661.
Labroo RB, Paine MF, Thummel KE, Kharasch ED. Fentanyl metabolism by human hepatic and intestinal cytochrome P450 3A4: implications for interindividual variability in disposition, efficacy, and drug interactions. Drug Metab Dispos. 1997;25(9):1072–80.
Poklis A, Backer R. Urine concentrations of fentanyl and norfentanyl during application of Duragesic transdermal patches. J Anal Toxicol. 2004;28(6):422–5.
Darwish M, Kirby M, Robertson P Jr, Tracewell W, Jiang JG. Absolute and relative bioavailability of fentanyl buccal tablet and oral transmucosal fentanyl citrate. J Clin Pharmacol. 2007;47(3):343–50.
Grape S, Schug SA, Lauer S, Schug BS. Formulations of fentanyl for the management of pain. Drugs. 2010;70(1):57–72. doi:10.2165/11531740-000000000-00000.
Dahan A, Yassen A, Bijl H, Romberg R, Sarton E, Teppema L, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825–34. doi:10.1093/bja/aei145.
Streisand JB, Stanley TH, Hague B, van Vreeswijk H, Ho GH, Pace NL. Oral transmucosal fentanyl citrate premedication in children. Anesth Analg. 1989;69(1):28–34.
Fine PG, Narayana A, Passik SD. Treatment of breakthrough pain with fentanyl buccal tablet in opioid-tolerant patients with chronic pain: appropriate patient selection and management. Pain Med. 2010;11(7):1024–36. doi:10.1111/j.1526-4637.2010.00891.x.
Deandrea S, Corli O, Consonni D, Villani W, Greco MT, Apolone G. Prevalence of breakthrough cancer pain: a systematic review and a pooled analysis of published literature. J Pain Symptom Manage. 2014;47(1):57–76. doi:10.1016/j.jpainsymman.2013.02.015.
Elsner F, Zeppetella G, Porta-Sales J, Tagarro I. Newer generation fentanyl transmucosal products for breakthrough pain in opioid-tolerant cancer patients. Clin Drug Investig. 2011;31(9):605–18. doi:10.2165/11592910-000000000-00000.
Zeppetella G, Davies A, Eijgelshoven I, Jansen JP. A network meta-analysis of the efficacy of opioid analgesics for the management of breakthrough cancer pain episodes. J Pain Symptom Manage. 2014;47(4):772–85. doi:10.1016/j.jpainsymman.2013.05.020.
Smith H. A comprehensive review of rapid-onset opioids for breakthrough pain. CNS Drugs. 2012;26(6):509–35. doi:10.2165/11630580-000000000-00000.
Jandhyala R, Fullarton J. Various formulations of oral transmucosal fentanyl for breakthrough cancer pain: an indirect mixed treatment comparison meta-analysis. BMJ Support Palliat Care. 2012;2(2):156–62. doi:10.1136/bmjspcare-2011-000139.
Scott LJ. Fentanyl iontophoretic transdermal system: a review in acute postoperative pain. Clin Drug Investig. 2016;36(4):321–30. doi:10.1007/s40261-016-0387-x.
Drug Approval Package: Actiq (oral transmucosal fentanyl citrate) NDA# 02747. In: US Food and Drug Administration. http://www.accessdata.fda.gov/drugsatfda_docs/nda/98/20747_Actiq.cfm. Accessed 19 Mar 2017.
Drug Approval Package: Fentota (fentanyl) NDA #021947. In: US Food and Drug Administration. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021947s000FentoraTOC.cfm. Accessed 19 Mar 2017.
Drug Approval Package: Onsolis NDA #022266. In: US Food and Drug Administration. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022266_onsolis_toc.cfm. Accessed 19 Mar 2017.
Drug Approval Package: Abstral (fentanyl) NDA #022510. In: US Food and Drug Administration. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022510_abstral_toc.cfm. Accessed 19 Mar 2017.
Drug Approval Package: Subsys (fentanyl sublingual) NDA #202788. In: US Food and Drug Administration. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202788_subsys_toc.cfm. Accessed 19 Mar 2017.
Moore N, Darwish M, Amores X, Schneid H. A review of the pharmacokinetic profile of transmucosal fentanyl formulations. Curr Med Res Opin. 2012;28(11):1781–90. doi:10.1185/03007995.2012.735227.
Streisand JB, Varvel JR, Stanski DR, Le Maire L, Ashburn MA, Hague BI, et al. Absorption and bioavailability of oral transmucosal fentanyl citrate. Anesthesiology. 1991;75(2):223–9.
Zhang H, Zhang J, Streisand JB. Oral mucosal drug delivery: clinical pharmacokinetics and therapeutic applications. Clin Pharmacokinet. 2002;41(9):661–80. doi:10.2165/00003088-200241090-00003.
Aronoff GM, Brennan MJ, Pritchard DD, Ginsberg B. Evidence-based oral transmucosal fentanyl citrate (OTFC) dosing guidelines. Pain Med. 2005;6(4):305–14.
Lichtor JL, Sevarino FB, Joshi GP, Busch MA, Nordbrock E, Ginsberg B. The relative potency of oral transmucosal fentanyl citrate compared with intravenous morphine in the treatment of moderate to severe postoperative pain. Anesth Analg. 1999;89(3):732–8.
Farrar JT, Cleary J, Rauck R, Busch M, Nordbrock E. Oral transmucosal fentanyl citrate: randomized, double-blinded, placebo-controlled trial for treatment of breakthrough pain in cancer patients. J Natl Cancer Inst. 1998;90(8):611–6.
Payne R, Coluzzi P, Hart L, Simmonds M, Lyss A, Rauck R, et al. Long-term safety of oral transmucosal fentanyl citrate for breakthrough cancer pain. J Pain Symptom Manage. 2001;22(1):575–83.
Pinna MA, Bruera E, Moralo MJ, Correas MA, Vargas RM. A randomized crossover clinical trial to evaluate the efficacy of oral transmucosal fentanyl citrate in the treatment of dyspnea on exertion in patients with advanced cancer. Am J Hosp Palliat Care. 2015;32(3):298–304. doi:10.1177/1049909113513063.
Butler FK, Kotwal RS, Buckenmaier CC 3rd, Edgar EP, O’Connor KC, Montgomery HR, et al. A Triple-Option Analgesia Plan for Tactical Combat Casualty Care: TCCC Guidelines Change 13-04. J Spec Oper Med. 2014;14(1):13–25.
Blankenstein TN, Gibson LM, Claydon MA. Is intramuscular morphine satisfying frontline medical personnels’ requirement for battlefield analgesia in Helmand Province, Afghanistan? A questionnaire study. Br J Pain. 2015;9(2):115–21. doi:10.1177/2049463714535563.
Wedmore IS, Kotwal RS, McManus JG, Pennardt A, Talbot TS, Fowler M, et al. Safety and efficacy of oral transmucosal fentanyl citrate for prehospital pain control on the battlefield. J Trauma Acute Care Surg. 2012;73(6 suppl 5):S490–5. doi:10.1097/TA.0b013e3182754674.
Coluzzi PH, Schwartzberg L, Conroy JD, Charapata S, Gay M, Busch MA, et al. Breakthrough cancer pain: a randomized trial comparing oral transmucosal fentanyl citrate (OTFC) and morphine sulfate immediate release (MSIR). Pain. 2001;91(1–2):123–30.
Mandel L, Carunchio MJ. Rampant caries from oral transmucosal fentanyl citrate lozenge abuse. J Am Dent Assoc. 2011;142(4):406–9 ([pii] 142/4/406).
Gordon D, Schroeder M. Oral transmucosal fentanyl citrate–OTFC (ACTIQ) #103. J Palliat Med. 2008;11(4):633–4. doi:10.1089/jpm.2008.9922.
Davies A. Current thinking in cancer breakthrough pain management. Eur J Palliat Care. 2005;12(suppl):4–6.
Jacobsen R, Møldrup C, Christrup L. Rationales behind the choice of administration form with fentanyl: Delphi survey among Danish general practitioners. J Opioid Manag. 2010;6(4):259–68.
Laverty D. Treating cancer-related breakthrough pain: the oral transmucosal route. Int J Palliat Nurs. 2007;13(7):326–31. doi:10.12968/ijpn.2007.13.7.24344.
Pather I, Siebert JM, Hontz J, Khankari R, Kumbale R, Gupte S. Enhanced buccal delivery of fentanyl using the OraVescent drug delivery system. Drug Deliv Technol. 2001;1(1):54–7.
Darwish M, Xie F. Pharmacokinetics of fentanyl buccal tablet: a pooled analysis and review. Pain Pract. 2012;12(4):307–14. doi:10.1111/j.1533-2500.2011.00491.x.
European Public Assessment Report (EPAR): Effentora. European Medicines Agency; 2008.
Durfee S, Messina J, Khankari R. Fentanyl effervescent buccal tablets. Am J Drug Deliv. 2006;4(1):1–5. doi:10.2165/00137696-200604010-00001.
FENTORA (fentanyl) buccal tablet [package insert]. Cephalon; 2013.
Taylor DR. Fentanyl buccal tablet: rapid relief from breakthrough pain. Expert Opin Pharmacother. 2007;8(17):3043–51. doi:10.1517/14656566.8.17.3043.
Portenoy RK, Taylor D, Messina J, Tremmel L. A randomized, placebo-controlled study of fentanyl buccal tablet for breakthrough pain in opioid-treated patients with cancer. Clin J Pain. 2006;22(9):805–11. doi:10.1097/01.ajp.0000210932.27945.4a.
Slatkin NE, Xie F, Messina J, Segal TJ. Fentanyl buccal tablet for relief of breakthrough pain in opioid-tolerant patients with cancer-related chronic pain. J Support Oncol. 2007;5(7):327–34.
Lister N, Warrington S, Boyce M, Eriksson C, Tamaoka M, Kilborn J. Pharmacokinetics, safety, and tolerability of ascending doses of sublingual fentanyl, with and without naltrexone. Japanese subjects. J Clin Pharmacol. 2011;51(8):1195–204. doi:10.1177/0091270010379410.
Vasisht N, Gever LN, Tagarro I, Finn AL. Single-dose pharmacokinetics of fentanyl buccal soluble film. Pain Med. 2010;11(7):1017–23. doi:10.1111/j.1526-4637.2010.00875.x.
Finn AL, Vasisht N, Stark JG, Gever LN, Tagarro I. Dose proportionality and pharmacokinetics of fentanyl buccal soluble film in healthy subjects: a phase I, open-label, three-period, crossover study. Clin Drug Investig. 2012;32(1):63–71. doi:10.2165/11594670-000000000-00000.
Davies A, Finn A, Tagarro I. Intra- and interindividual variabilities in the pharmacokinetics of fentanyl buccal soluble film in healthy subjects: a cross-study analysis. Clin Drug Investig. 2011;31(5):317–24. doi:10.2165/11533540-000000000-00000.
Garnock-Jones KP. fentanyl buccal soluble film: a review in breakthrough cancer pain. Clin Drug Investig. 2016;36(5):413–9. doi:10.1007/s40261-016-0394-y.
Chwieduk CM, McKeage K. Fentanyl sublingual: in breakthrough pain in opioid-tolerant adults with cancer. Drugs. 2010;70(17):2281–8. doi:10.2165/11200910-000000000-00000.
Bredenberg S, Duberg M, Lennernäs B, Lennernäs H, Pettersson A, Westerberg M, et al. In vitro and in vivo evaluation of a new sublingual tablet system for rapid oromucosal absorption using fentanyl citrate as the active substance. Eur J Pharm Sci. 2003;20(3):327–34.
Nalamachu S. An evaluation of total disintegration time for three different doses of sublingual fentanyl tablets in patients with breakthrough pain. Pain Ther. 2013;2(2):121–8. doi:10.1007/s40122-013-0019-6.
Murphy A, O’Sullivan R, Wakai A, Grant TS, Barrett MJ, Cronin J, et al. Intranasal fentanyl for the management of acute pain in children. Cochrane Database Syst Rev. 2014;2014(10):CD009942. doi:10.1002/14651858.CD009942.pub2.
Abstral Sublingual Tablets—Summary of Product Characteristics. In: Electronic Medicines Compendium. http://www.medicines.org.uk/EMC/medicine/21371/SPC/Abstral%2BSublingual%2BTablets/.
Davies A, Mundin G, Vriens J, Webber K, Buchanan A, Waghorn M. The influence of low salivary flow rates on the absorption of a sublingual fentanyl citrate formulation for breakthrough cancer pain. J Pain Symptom Manage. 2016;51(3):538–45. doi:10.1016/j.jpainsymman.2015.11.018.
Parikh N, Goskonda V, Chavan A, Dillaha L. Single-dose pharmacokinetics of fentanyl sublingual spray and oral transmucosal fentanyl citrate in healthy volunteers: a randomized crossover study. Clin Ther. 2013;35(3):236–43. doi:10.1016/j.clinthera.2013.02.017.
Rauck R, Reynolds L, Geach J, Bull J, Stearns L, Scherlis M, et al. Efficacy and safety of fentanyl sublingual spray for the treatment of breakthrough cancer pain: a randomized, double-blind, placebo-controlled study. Curr Med Res Opin. 2012;28(5):859–70. doi:10.1185/03007995.2012.683111.
Rauck RL, Tark M, Reyes E, Hayes TG, Bartkowiak AJ, Hassman D, et al. Efficacy and long-term tolerability of sublingual fentanyl orally disintegrating tablet in the treatment of breakthrough cancer pain. Curr Med Res Opin. 2009;25(12):2877–85. doi:10.1185/03007990903368310.
Uberall MA, Muller-Schwefe GH. Sublingual fentanyl orally disintegrating tablet in daily practice: efficacy, safety and tolerability in patients with breakthrough cancer pain. Curr Med Res Opin. 2011;27(7):1385–94. doi:10.1185/03007995.2011.583231.
Minkowitz H, Bull J, Brownlow RC, Parikh N, Rauck R. Long-term safety of fentanyl sublingual spray in opioid-tolerant patients with breakthrough cancer pain. Support Care Cancer. 2016;24(6):2669–75. doi:10.1007/s00520-015-3056-3.
Velazquez Rivera I, Munoz Garrido JC, Garcia Velasco P, de Enciso IEX, Clavarana LV. Efficacy of sublingual fentanyl vs. oral morphine for cancer-related breakthrough pain. Adv Ther. 2014;31(1):107–17. doi:10.1007/s12325-013-0086-4.
Paech MJ, Bloor M, Schug SA. New formulations of fentanyl for acute pain management. Drugs Today. 2012;48(2):119–32. doi:10.1358/dot.2012.48.2.1745275.
Fisher A, Watling M, Smith A, Knight A. Pharmacokinetics and relative bioavailability of fentanyl pectin nasal spray 100–800 µg in healthy volunteers. Int J Clin Pharmacol Ther. 2010;48(12):860–7.
European Public Assessment Report (EPAR): PecFent. European Medicines Agency; 2010.
Gizurarson S. The relevance of nasal physiology to the design of drug absorption studies. Adv Drug Deliv Rev. 1993;11(3):329–47. doi:10.1016/0169-409X(93)90015-V.
Crellin D, Ling RX, Babl FE. Does the standard intravenous solution of fentanyl (50 microg/ml) administered intranasally have analgesic efficacy? Emerg Med Australas. 2010;22(1):62–7. doi:10.1111/j.1742-6723.2010.01257.x.
Dale O, Hjortkjaer R, Kharasch ED. Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand. 2002;46(7):759–70.
Illum L. Nanoparticulate systems for nasal delivery of drugs: a real improvement over simple systems? J Pharm Sci. 2007;96(3):473–83. doi:10.1002/jps.20718.
Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–73. doi:10.1002/jps.21924.
Illum L, Davis SS, Pawula M, Fisher AN, Barrett DA, Farraj NF, et al. Nasal administration of morphine-6-glucuronide in sheep–a pharmacokinetic study. Biopharm Drug Dispos. 1996;17(8):717–24. doi:10.1002/(SICI)1099-081X(199611)17:8<717:AID-BDD985>3.0.CO;2-I.
Mercadante S. Pharmacotherapy for breakthrough cancer pain. Drugs. 2012;72(2):181–90. doi:10.2165/11597260-000000000-00000.
Moksnes K, Fredheim OM, Klepstad P, Kaasa S, Angelsen A, Nilsen T, et al. Early pharmacokinetics of nasal fentanyl: is there a significant arterio-venous difference? Eur J Clin Pharmacol. 2008;64(5):497–502. doi:10.1007/s00228-007-0444-8.
Panagiotou I, Mystakidou K. Intranasal fentanyl: from pharmacokinetics and bioavailability to current treatment applications. Expert Rev Anticancer Ther. 2010;10(7):1009–21. doi:10.1586/era.10.77.
Nave R, Schmitt H, Popper L. Faster absorption and higher systemic bioavailability of intranasal fentanyl spray compared to oral transmucosal fentanyl citrate in healthy subjects. Drug Deliv. 2013;20(5):216–23. doi:10.3109/10717544.2012.762435.
Mercadante S, Prestia G, Adile C, Casuccio A. Intranasal fentanyl versus fentanyl pectin nasal spray for the management of breakthrough cancer pain in doses proportional to basal opioid regimen. J Pain. 2014;15(6):602–7. doi:10.1016/j.jpain.2014.02.002.
Hansen MS, Mathiesen O, Trautner S, Dahl JB. Intranasal fentanyl in the treatment of acute pain—a systematic review. Acta Anaesthesiol Scand. 2012;56(4):407–19. doi:10.1111/j.1399-6576.2011.02613.x.
Saunders M, Adelgais K, Nelson D. Use of intranasal fentanyl for the relief of pediatric orthopedic trauma pain. Acad Emerg Med. 2010;17(11):1155–61. doi:10.1111/j.1553-2712.2010.00905.x.
Finn J, Wright J, Fong J, Mackenzie E, Wood F, Leslie G, et al. A randomised crossover trial of patient controlled intranasal fentanyl and oral morphine for procedural wound care in adult patients with burns. Burns. 2004;30(3):262–8. doi:10.1016/j.burns.2003.10.017.
Cole J, Shepherd M, Young P. Intranasal fentanyl in 1-3-year-olds: a prospective study of the effectiveness of intranasal fentanyl as acute analgesia. Emerg Med Australas. 2009;21(5):395–400. doi:10.1111/j.1742-6723.2009.01216.x.
Shelley K, Paech MJ. The clinical applications of intranasal opioids. Curr Drug Deliv. 2008;5(1):55–8.
Striebel HW, Oelmann T, Spies C, Rieger A, Schwagmeier R. Patient-controlled intranasal analgesia: a method for noninvasive postoperative pain management. Anesth Analg. 1996;83(3):548–51.
Toussaint S, Maidl J, Schwagmeier R, Striebel HW. Patient-controlled intranasal analgesia: effective alternative to intravenous PCA for postoperative pain relief. Can J Anaesth. 2000;47(4):299–302. doi:10.1007/BF03020941.
Christrup LL, Foster D, Popper LD, Troen T, Upton R. Pharmacokinetics, efficacy, and tolerability of fentanyl following intranasal versus intravenous administration in adults undergoing third-molar extraction: a randomized, double-blind, double-dummy, two-way, crossover study. Clin Ther. 2008;30(3):469–81. doi:10.1016/j.clinthera.2008.03.001.
Galinkin JL, Fazi LM, Cuy RM, Chiavacci RM, Kurth CD, Shah UK, et al. Use of intranasal fentanyl in children undergoing myringotomy and tube placement during halothane and sevoflurane anesthesia. Anesthesiology. 2000;93(6):1378–83.
Kerr D, Taylor D, Evans B. Patient-controlled intranasal fentanyl analgesia: a pilot study to assess practicality and tolerability during childbirth. Int J Obstet Anesth. 2015;24(2):117–23. doi:10.1016/j.ijoa.2014.11.006.
Karlsen AP, Pedersen DM, Trautner S, Dahl JB, Hansen MS. Safety of intranasal fentanyl in the out-of-hospital setting: a prospective observational study. Ann Emerg Med. 2014;63(6):699–703. doi:10.1016/j.annemergmed.2013.10.025.
Mercadante S, Radbruch L, Davies A, Poulain P, Sitte T, Perkins P, et al. A comparison of intranasal fentanyl spray with oral transmucosal fentanyl citrate for the treatment of breakthrough cancer pain: an open-label, randomised, crossover trial. Curr Med Res Opin. 2009;25(11):2805–15. doi:10.1185/03007990903336135.
Vissers D, Stam W, Nolte T, Lenre M, Jansen J. Efficacy of intranasal fentanyl spray versus other opioids for breakthrough pain in cancer. Curr Med Res Opin. 2010;26(5):1037–45. doi:10.1185/03007991003694340.
Portenoy RK, Burton AW, Gabrail N, Taylor D, Fentanyl Pectin Nasal Spray 043 Study G. A multicenter, placebo-controlled, double-blind, multiple-crossover study of fentanyl pectin nasal spray (FPNS) in the treatment of breakthrough cancer pain. Pain. 2010;151(3):617–24. doi:10.1016/j.pain.2010.07.028.
Mercadante S, Vellucci R, Cuomo A, Adile C, Cortegiani A, Valle A, et al. Long-term efficacy and tolerability of intranasal fentanyl in the treatment of breakthrough cancer pain. Support Care Cancer. 2015;23(5):1349–54. doi:10.1007/s00520-014-2491-x.
Jandhyala R, Fullarton JR, Bennett MI. Efficacy of rapid-onset oral fentanyl formulations vs. oral morphine for cancer-related breakthrough pain: a meta-analysis of comparative trials. J Pain Symptom Manage. 2013;46(4):573–80. doi:10.1016/j.jpainsymman.2012.09.009.
Zeppetella G, Davies AN. Opioids for the management of breakthrough pain in cancer patients. Cochrane Database Syst Rev. 2013;10:4311. doi:10.1002/14651858.CD004311.pub3.
Nalamachu SR, Parikh N, Dillaha L, Rauck R. Lack of correlation between the effective dose of fentanyl sublingual spray for breakthrough cancer pain and the around-the-clock opioid dose. J Opioid Manag. 2014;10(4):247–54. doi:10.5055/jom.2014.0212.
Alberts DS, Smith CC, Parikh N, Rauck RL. Fentanyl sublingual spray for breakthrough cancer pain in patients receiving transdermal fentanyl. Pain Manag. 2016;6(5):427–34. doi:10.2217/pmt-2015-0009.
Rauck R, Bull J, Parikh N, Dillaha L, Stearns L. Effective dose titration of fentanyl sublingual spray in patients with breakthrough cancer pain. Pain Pract. 2015. doi:10.1111/papr.12360.
Kleeberg UR, Davies A, Jarosz J, Mercadante S, Poulain P, O’Brien T, et al. Pan-European, open-label dose titration study of fentanyl buccal tablet in patients with breakthrough cancer pain. Eur J Pain. 2015;19(4):528–37. doi:10.1002/ejp.577.
Mercadante S, Gatti A, Porzio G, Lo Presti C, Aielli F, Adile C, et al. Dosing fentanyl buccal tablet for breakthrough cancer pain: dose titration versus proportional doses. Curr Med Res Opin. 2012;28(6):963–8. doi:10.1185/03007995.2012.683112.
Mercadante S, Porzio G, Aielli F, Averna L, Ficorella C, Casuccio A. The use of fentanyl buccal tablets for breakthrough pain by using doses proportional to opioid basal regimen in a home care setting. Support Care Cancer. 2013;21(8):2335–9. doi:10.1007/s00520-013-1799-2.
Shimoyama N, Gomyo I, Teramoto O, Kojima K, Higuchi H, Yukitoshi N, et al. Efficacy and safety of sublingual fentanyl orally disintegrating tablet at doses determined from oral morphine rescue doses in the treatment of breakthrough cancer pain. Jpn J Clin Oncol. 2015;45(2):189–96. doi:10.1093/jjco/hyu182.
Bennett MI, Graham J, Schmidt-Hansen M, Prettyjohns M, Arnold S, Guideline Development G. Prescribing strong opioids for pain in adult palliative care: summary of NICE guidance. BMJ. 2012;344:e2806.
Gaertner J, Schiessl C. Cancer pain management: what’s new? Curr Pain Headache Rep. 2013;17(4):328. doi:10.1007/s11916-013-0328-9.
Ruggeri M, Turriziani A, Oradei M. Cost-effectiveness analysis of transnasal fentanyl citrate for the treatment of breakthrough cancer pain. Expert Rev Pharmacoecon Outcomes Res. 2014;14(3):459–64. doi:10.1586/14737167.2014.904750.
Vissers DC, Lenre M, Tolley K, Jakobsson J, Sendersky V, Jansen JP. An economic evaluation of short-acting opioids for treatment of breakthrough pain in patients with cancer. Value Health. 2011;14(2):274–81. doi:10.1016/j.jval.2010.09.007.
Kuo KL, Saokaew S, Stenehjem DD. The pharmacoeconomics of breakthrough cancer pain. J Pain Palliat Care Pharmacother. 2013;27(2):167–75. doi:10.3109/15360288.2013.787137.
American College of Medical T. ACMT Position Statement: safety issues regarding prescription fentanyl products. J Med Toxicol. 2016;12(2):211–2. doi:10.1007/s13181-016-0535-y.
Warner M, Trinidad J, Bastian B. Drugs most frequently involved in drug overdose deaths: United States, 2010–2014. Hyattsville, MD: National Center for Health Statistics; 2016.
Marquardt KA, Tharratt RS, Musallam NA. Fentanyl remaining in a transdermal system following three days of continuous use. Ann Pharmacother. 1995;29(10):969–71.
Zernikow B, Michel E, Anderson B. Transdermal fentanyl in childhood and adolescence: a comprehensive literature review. J Pain. 2007;8(3):187–207. doi:10.1016/j.jpain.2006.11.008.
Prodduturi S, Sadrieh N, Wokovich AM, Doub WH, Westenberger BJ, Buhse L. Transdermal delivery of fentanyl from matrix and reservoir systems: effect of heat and compromised skin. J Pharm Sci. 2010;99(5):2357–66. doi:10.1002/jps.22004.
Forsgren J, Jämstorp E, Bredenberg S, Engqvist H, Strømme M. A ceramic drug delivery vehicle for oral administration of highly potent opioids. J Pharm Sci. 2010;99(1):219–26. doi:10.1002/jps.21814.
Lilleng PK, Mehlum LI, Bachs L, Morild I. Deaths after intravenous misuse of transdermal fentanyl. J Forensic Sci. 2004;49(6):1364–6.
Marier J-F, Lor M, Potvin D, Dimarco M, Morelli G, Saedder EA. Pharmacokinetics, tolerability, and performance of a novel matrix transdermal delivery system of fentanyl relative to the commercially available reservoir formulation in healthy subjects. J Clin Pharmacol. 2006;46(6):642–53. doi:10.1177/0091270006286901.
Moore PW, Palmer RB, Donovan JW. Fatal fentanyl patch misuse in a hospitalized patient with a postmortem increase in fentanyl blood concentration. J Forensic Sci. 2015;60(1):243–6. doi:10.1111/1556-4029.12559.
Mrvos R, Feuchter AC, Katz KD, Duback-Morris LF, Brooks DE, Krenzelok EP. Whole fentanyl patch ingestion: a multi-center case series. J Emerg Med. 2012;42(5):549–52. doi:10.1016/j.jemermed.2011.05.017.
Schauer CK, Shand JA, Reynolds TM. The fentanyl patch boil-up—a novel method of opioid abuse. Basic Clin Pharmacol Toxicol. 2015;117(5):358–9. doi:10.1111/bcpt.12412.
Arroyo Plasencia AM, Mowry J, Smith J, Quigley K. In vitro release of fentanyl from transdermal patches in gastric and intestinal fluid. Clin Toxicol (Phila). 2014;52(9):945–7. doi:10.3109/15563650.2014.967399.
Freynhagen R, von Giesen HJ, Busche P, Sabatowski R, Konrad C, Grond S. Switching from reservoir to matrix systems for the transdermal delivery of fentanyl: a prospective, multicenter pilot study in outpatients with chronic pain. J Pain Symptom Manage. 2005;30(3):289–97. doi:10.1016/j.jpainsymman.2005.03.015.
Gourlay GK, Kowalski SR, Plummer JL, Cherry DA, Gaukroger P, Cousins MJ. The transdermal administration of fentanyl in the treatment of postoperative pain: pharmacokinetics and pharmacodynamic effects. Pain. 1989;37(2):193–202.
Varvel JR, Shafer SL, Hwang SS, Coen PA, Stanski DR. Absorption characteristics of transdermally administered fentanyl. Anesthesiology. 1989;70(6):928–34.
Ross JR, Quigley C. Transdermal fentanyl: informed prescribing is essential. Eur J Pain. 2003;7(5):481–3. doi:10.1016/S1090-3801(02)00148-9.
Holley FO, van Steennis C. Postoperative analgesia with fentanyl: pharmacokinetics and pharmacodynamics of constant-rate i.v. and transdermal delivery. Br J Anaesth. 1988;60(6):608–13.
DURAGESIC (fentanyl transdermal system) for transdermal administration [package insert]. Janssen Pharmaceuticals; 2014.
Carter KA. Heat-associated increase in transdermal fentanyl absorption. Am J Health Syst Pharm. 2003;60(2):191–2.
Doris MK, Sandilands EA. Life-threatening opioid toxicity from a fentanyl patch applied to eczematous skin. BMJ Case Rep. 2015. doi:10.1136/bcr-2014-208945.
Sindali K, Sherry K, Sen S, Dheansa B. Life-threatening coma and full-thickness sunburn in a patient treated with transdermal fentanyl patches: a case report. J Med Case Rep. 2012;6:220. doi:10.1186/1752-1947-6-220.
Solassol I, Bressolle F, Caumette L, Garcia F, Poujol S, Culine S, et al. Inter- and intraindividual variabilities in pharmacokinetics of fentanyl after repeated 72-hour transdermal applications in cancer pain patients. Ther Drug Monit. 2005;27(4):491–8.
Solassol I, Caumette L, Bressolle F, Garcia F, Thézenas S, Astre C, et al. Inter- and intra-individual variability in transdermal fentanyl absorption in cancer pain patients. Oncol Rep. 2005;14(4):1029–36.
Portenoy RK, Southam MA, Gupta SK, Lapin J, Layman M, Inturrisi CE, et al. Transdermal fentanyl for cancer pain. Repeated dose pharmacokinetics. Anesthesiology. 1993;78(1):36–43.
Reddy A, Yennurajalingam S, Reddy S, Wu J, Liu D, Dev R, et al. The opioid rotation ratio from transdermal fentanyl to “strong” opioids in patients with cancer pain. J Pain Symptom Manage. 2016;51(6):1040–5. doi:10.1016/j.jpainsymman.2015.12.312.
Bentley JB, Borel JD, Nenad RE, Gillespie TJ. Age and fentanyl pharmacokinetics. Anesth Analg. 1982;61(12):968–71.
Arnet I, Schacher S, Balmer E, Koeberle D, Hersberger KE. Poor adhesion of fentanyl transdermal patches may mimic end-of-dosage failure after 48 h and prompt early patch replacement in hospitalized cancer pain patients. J Pain Res. 2016;9:993–9. doi:10.2147/JPR.S116091.
Hadley G, Derry S, Moore RA. Wiffen PJ (2013) Transdermal fentanyl for cancer pain. Cochrane Database Syst Rev. 2013;10:CD010270. doi:10.1002/14651858.CD010270.pub2.
Koyyalagunta D, Bruera E, Solanki DR, Nouri KH, Burton AW, Toro MP, et al. A systematic review of randomized trials on the effectiveness of opioids for cancer pain. Pain Phys. 2012;15(3 suppl):ES39–58.
Nelson L, Schwaner R. Transdermal fentanyl: pharmacology and toxicology. J Med Toxicol. 2009;5(4):230–41.
Freynhagen R, Geisslinger G, Schug SA. Opioids for chronic non-cancer pain. BMJ. 2013;346:f2937. doi:10.1136/bmj.f2937.
Wolff RF, Aune D, Truyers C, Hernandez AV, Misso K, Riemsma R, et al. Systematic review of efficacy and safety of buprenorphine versus fentanyl or morphine in patients with chronic moderate to severe pain. Curr Med Res Opin. 2012;28(5):833–45. doi:10.1185/03007995.2012.678938.
Centers For Disease C, Prevention Public Health Service USDOH, Human S. Guideline for prescribing opioids for chronic pain. J Pain Palliat Care Pharmacother. 2016;30(2):138–40. doi:10.3109/15360288.2016.1173761.
Hostynek JJ, Maibach HI. Fentanyl transdermal patches: overview of cutaneous adverse effects in humans. Cutan Ocul Toxicol. 2010;29(4):241–6. doi:10.3109/15569527.2010.492487.
Catterall RA. Problems of sweating and transdermal fentanyl. Palliat Med. 1997;11(2):169–70.
Roy SD, Flynn GL. Transdermal delivery of narcotic analgesics: pH, anatomical, and subject influences on cutaneous permeability of fentanyl and sufentanil. Pharm Res. 1990;7(8):842–7.
Collins JJ, Dunkel IJ, Gupta SK, Inturrisi CE, Lapin J, Palmer LN, et al. Transdermal fentanyl in children with cancer pain: feasibility, tolerability, and pharmacokinetic correlates. J Pediatr. 1999;134(3):319–23.
Grond S, Radbruch L, Lehmann KA. Clinical pharmacokinetics of transdermal opioids: focus on transdermal fentanyl. Clin Pharmacokinet. 2000;38(1):59–89. doi:10.2165/00003088-200038010-00004.
Neighbors DM, Bell TJ, Wilson J, Dodd SL. Economic evaluation of the fentanyl transdermal system for the treatment of chronic moderate to severe pain. J Pain Symptom Manage. 2001;21(2):129–43.
Schröder B, Nickel U, Meyer E, Lee G. Transdermal delivery using a novel electrochemical device, part 2: in vivo study in humans. J Pharm Sci. 2012;101(6):2262–8. doi:10.1002/jps.23108.
Chelly JE. An iontophoretic, fentanyl HCl patient-controlled transdermal system for acute postoperative pain management. Expert Opin Pharmacother. 2005;6(7):1205–14. doi:10.1517/14656566.6.7.1205.
Sathyan G, Jaskowiak J, Evashenk M, Gupta S. Characterisation of the pharmacokinetics of the fentanyl HCl patient-controlled transdermal system (PCTS): effect of current magnitude and multiple-day dosing and comparison with IV fentanyl administration. Clin Pharmacokinet. 2005;44(Suppl 1):7–15.
Phipps JB, Joshi N, Regal KA, Li J, Sinatra RS. Pharmacokinetic characteristics of fentanyl iontophoretic trandermal system over a range of applied current. Expert Opin Drug Metab Toxicol. 2015;11(4):481–9. doi:10.1517/17425255.2015.1020296.
IONSYS (fentanyl iontophoretic transdermal system) [package insert]. The Medicines Company; 2015.
Schröder B, Nickel U, Meyer E, Lee G. Transdermal delivery using a novel electrochemical device, part 1: device design and in vitro release/permeation of fentanyl. J Pharm Sci. 2012;101(1):245–55. doi:10.1002/jps.22765.
Minkowitz HS, Danesi H, Ding L, Jones JB. Development of the fentanyl iontophoretic transdermal system (ITS) for patient-controlled analgesia of postoperative pain management. Pain Manag. 2015;5(5):327–37. doi:10.2217/pmt.15.26.
Joshi N, Lemke J, Danesi H. Design and functionality of a smart fentanyl iontophoretic transdermal system for the treatment of moderate-to-severe postoperative pain. Pain Manag. 2016;6(2):137–45. doi:10.2217/pmt.15.70.
Camu F, Van Aken H, Bovill JG. Postoperative analgesic effects of three demand-dose sizes of fentanyl administered by patient-controlled analgesia. Anesth Analg. 1998;87(4):890–5.
Panchal SJ, Damaraju CV, Nelson WW, Hewitt DJ, Schein JR. System-related events and analgesic gaps during postoperative pain management with the fentanyl iontophoretic transdermal system and morphine intravenous patient-controlled analgesia. Anesth Analg. 2007;105(5):1437–41. doi:10.1213/01.ane.0000281442.36582.81 (table of contents).
European Public Assessment Report (EPAR): IonSys. European Medicines Agency; 2015.
Gupta SK, Hwang S, Southam M, Sathyan G. Effects of application site and subject demographics on the pharmacokinetics of fentanyl HCl patient-controlled transdermal system (PCTS). Clin Pharmacokinet. 2005;44(Suppl 1):25–32.
Sathyan G, Phipps B, Gupta SK. Passive absorption of fentanyl from the fentanyl HCl iontophoretic transdermal system. Curr Med Res Opin. 2009;25(2):363–6. doi:10.1185/03007990802594941.
Viscusi ER, Reynolds L, Tait S, Melson T, Atkinson LE. An iontophoretic fentanyl patient-activated analgesic delivery system for postoperative pain: a double-blind, placebo-controlled trial. Anesth Analg. 2006;102(1):188–94. doi:10.1213/01.ane.0000183649.58483.77.
Sinatra RS, Viscusi ER, Ding L, Danesi H, Jones JB, Grond S. Meta-analysis of the efficacy of the fentanyl iontophoretic transdermal system versus intravenous patient-controlled analgesia in postoperative pain management. Expert Opin Pharmacother. 2015;16(11):1607–13. doi:10.1517/14656566.2015.1054279.
Hartrick CT, Pestano CR, Ding L, Danesi H, Jones JB. Patient considerations in the use of transdermal iontophoretic fentanyl for acute postoperative pain. J Pain Res. 2016;9:215–22. doi:10.2147/JPR.S89278.
Hartrick CT, Abraham J, Ding L. Ease-of-care from the physical therapists’ perspective comparing fentanyl iontophoretic transdermal system versus morphine intravenous patient-controlled analgesia in postoperative pain management. J Comp Eff Res. 2016;5(6):529–37. doi:10.2217/cer-2016-0038.
Viscusi ER. Patient-controlled drug delivery for acute postoperative pain management: a review of current and emerging technologies. Reg Anesth Pain Med. 2008;33(2):146–58. doi:10.1016/j.rapm.2007.11.005.
Chelly JE, Grass J, Houseman TW, Minkowitz H, Pue A. The safety and efficacy of a fentanyl patient-controlled transdermal system for acute postoperative analgesia: a multicenter, placebo-controlled trial. Anesth Analg. 2004;98(2):427–33 (table of contents).
Langford RM, Chang KY, Ding L, Abraham J. Comparison of fentanyl iontophoretic transdermal system and routine care with morphine intravenous patient-controlled analgesia in the management of early postoperative mobilisation: results from a randomised study. Br J Pain. 2016;10(4):198–208. doi:10.1177/2049463716668905.
Oliashirazi A, Wilson-Byrne T, Shuler FD, Parvizi J. Patient-controlled fentanyl iontophoretic transdermal system improved postoperative mobility compared to intravenous patient-controlled analgesia morphine: a pooled analysis of randomized, controlled trials. Pain Pract. 2016. doi:10.1111/papr.12432.
Brown CR, Moodie JE, Bisley EJ, editors. Safety and efficacy of Transfenta in the treatment of post-operative pain: a double-blind, single-center, placebo-controlled trial. In: 17th Annual Scientific Meeting of the American Pain Society. San Diego, CA; 1998 (1998/11/05/8).
Viscusi ER, Siccardi M, Damaraju CV, Hewitt DJ, Kershaw P. The safety and efficacy of fentanyl iontophoretic transdermal system compared with morphine intravenous patient-controlled analgesia for postoperative pain management: an analysis of pooled data from three randomized, active-controlled clinical studies. Anesth Analg. 2007;105(5):1428–36. doi:10.1213/01.ane.0000281913.28623.fd.
Viscusi ER, Grond S, Ding L, Danesi H, Jones JB, Sinatra RS. A comparison of opioid-related adverse events with fentanyl iontophoretic transdermal system versus morphine intravenous patient-controlled analgesia in acute postoperative pain. Pain Manag. 2016;6(1):19–24. doi:10.2217/pmt.15.49.
Lemke J, Sardariani E, Phipps JB, Patel N, Itri LM, Caravelli J, et al. Fentanyl iontophoretic transdermal system (IONSYS((R))) can be Safely used in the Hospital Environment with X-Rays, computerized tomography and radiofrequency identification devices. Adv Ther. 2016;33(9):1649–59. doi:10.1007/s12325-016-0381-y.
Wong P, Chadwick FD, Karovits J. Intranasal fentanyl for postoperative analgesia after elective Caesarean section. Anaesthesia. 2003;58(8):818–9.
Rickard C, O’Meara P, McGrail M, Garner D, McLean A, Le Lievre P. A randomized controlled trial of intranasal fentanyl vs intravenous morphine for analgesia in the prehospital setting. Am J Emerg Med. 2007;25(8):911–7. doi:10.1016/j.ajem.2007.02.027.
Worsley MH, MacLeod AD, Brodie MJ, Asbury AJ, Clark C. Inhaled fentanyl as a method of analgesia. Anaesthesia. 1990;45(6):449–51.
Hung OR, Whynot SC, Varvel JR, Shafer SL, Mezei M. Pharmacokinetics of inhaled liposome-encapsulated fentanyl. Anesthesiology. 1995;83(2):277–84.
Zeppetella G. An assessment of the safety, efficacy, and acceptability of intranasal fentanyl citrate in the management of cancer-related breakthrough pain: a pilot study. J Pain Symptom Manage. 2000;20(4):253–8.
Acknowledgements
The editorial assistance of Ms. Peta Gjedsted, Anaesthesiology Unit of UWA, is kindly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Both authors (SAS and ST) have no conflicts of interest that are directly relevant to the content of this review.
Funding
No funding or assistance was received for the preparation of this review.
Rights and permissions
About this article

Cite this article
Schug, S.A., Ting, S. Fentanyl Formulations in the Management of Pain: An Update. Drugs 77, 747–763 (2017). https://doi.org/10.1007/s40265-017-0727-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0727-z