
Abstract
Selexipag (Uptravi®) is a highly selective, long-acting, nonprostanoid, prostacyclin receptor agonist that is being developed by Actelion Pharmaceuticals Ltd and Nippon Shinyaku. Oral selexipag is approved in the USA for the treatment of pulmonary arterial hypertension (PAH; WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH. It has subsequently been approved in Canada for the long-term treatment of PAH, and received a positive opinion in the EU for the treatment of PAH in adult patients with WHO functional class II–III. Selexipag received orphan drug designation for the treatment of PAH in Japan in 2014 and is in undergoing regulatory review in several countries for use in this indication. In the large, event-driven, phase III GRIPHON trial, selexipag reduced the risk of the primary composite endpoint of death or a complication related to PAH (whichever occurred first) by 40 % compared with placebo in patients with PAH (80 % were also receiving stable dosages of an endothelin receptor antagonist and/or a phosphodiesterase 5 inhibitor). This article summarizes the milestones in the development of selexipag leading to this first approval for PAH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Pulmonary arterial hypertension (PAH), a rare, chronic, progressive disease, is associated with significant morbidity and mortality, and is characterized by increased pulmonary vascular resistance (PVR) resulting in right ventricular overload, hypertrophy and, ultimately right heart failure and premature death [1, 2]. An improved understanding of the pathogenesis of PAH has led to the development of drugs that target three major signalling pathways involved in the pathogenesis of PAH; namely, the prostacyclin (e.g. prostacyclin analogues), endothelin (e.g. endothelin receptor antagonists) and nitric oxide [e.g. phosphodiesterase 5 (PDE-5) inhibitors] pathways [2].
Selexipag (Uptravi®) is an orally available prostacyclin (PGI2) IP receptor agonist that is under development by Actelion Pharmaceuticals Ltd and Nippon Shinyaku for use in PAH [3]. Selexipag is a long-acting IP receptor agonist, with all other currently licensed drugs that target the prostacyclin pathway having a shorter half-life and, for the most part, being administered as an intravenous or subcutaneous infusion or by inhalation [3].
Selexipag was approved in December 2015 by the US FDA for the treatment of PAH (WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH [4]. The recommended starting dosage is 200 μg twice daily, with the dosage incremented at weekly intervals by 200 μg twice daily to the highest tolerated dosage up to 1600 μg twice daily. The maintenance dosage is determined by tolerability. In patients with moderate hepatic impairment, the starting dosage is 200 μg once daily, with the dosage incremented at weekly intervals by 200 μg once daily to the highest tolerated dosage up to 1600 μg [4].

Clinical development of selexipag for the treatment of patients with pulmonary arterial hypertension. Flags indicate key milestones in approval of selexipag. EMA European Medicines Agency, Est estimated completion, MAA Marketing Authorization Authority, NDA New Drug Application, PAH pulmonary arterial hypertension
In January 2016, selexipag was granted approval in Canada for long-term treatment of PAH [5], and received a positive opinion in the EU for the treatment of PAH in adult patients with WHO functional class II–III [6]. Selexipag was granted orphan drug designation in Japan in September 2014 for use in PAH [7] and is in undergoing regulatory review in several countries including, Australia, New Zealand, South Korea, Switzerland, Taiwan and Turkey for use in PAH [8].
1.1 Company Agreements
In April 2008, Actelion Pharmaceuticals Ltd and Nippon Shinyaku entered into a collaboration agreement with respect to selexipag [9]. Under the terms of the agreement, Actelion will be responsible for the global development and commercialization of selexipag outside of Japan. The two companies will jointly develop and commercialize the therapeutic in Japan. Nippon Shinyaku will receive an upfront payment and additional milestone payments from Actelion and will also be entitled to royalties on sales [9].
2 Scientific Summary
2.1 Pharmacodynamics
PGI2 plays an important role in the maintenance of vascular homeostasis and inhibition of vascular smooth muscle cell differentiation, proliferation and migration, with downregulation of this pathway implicated in the pathogenesis of several vascular diseases, including PAH [3].
Selexipag has a similar mode of action to that of endogenous PGI2, but is a nonprostanoid IP receptor agonist rather than a PGI2 analogue (e.g. iloprost, beraprost and treprostinil) [3, 8, 10]. Selexipag is hydrolysed to its active metabolite ACT-333679, which is ≈37-fold as potent as selexipag [4]. In preclinical studies, selexipag exhibited 130-fold higher selectivity for the human IP receptor over other prostanoid receptors [10]. Unlike iloprost and beraprost-induced vasodilation, ACT-333679-induced vasodilation was not attenuated by antagonism of prostaglandin E3 (EP3) receptors [8]. As some gastric side effects (e.g. nausea and vomiting) associated with PGI2 analogues are mediated via EP3 receptor-dependent mechanisms, selexipag has the potential for improved gastric tolerability [11]. In vitro, ACT-333679 induced intracellular cAMP accumulation (≈100-fold increase [12]) in pulmonary artery smooth muscle cells and inhibited cell proliferation [12, 13]. Unlike iloprost, beraprost and treprostinil, ACT-333679 did not induce IP receptor desensitization and internalization; receptor internalization is a potential mechanism whereby repeated exposure to IP receptor agonists may lead to tachyphylaxis [13]. In a rat model of systemic hypertension, repeated administration of selexipag did not induce tachyphylaxis [14].

Chemical structure of selexipag
Selexipag and its active metabolite induced concentration-dependent inhibition of platelet aggregation in vitro, with 50 % inhibition occurring at concentrations of 5.5 and 0.21 μmol, respectively [4].
In a phase II, proof-of-concept study in patients with PAH receiving stable dosages of endothelin receptor antagonists and/or PDE-5 inhibitor therapy, relative to placebo (n = 10), concomitant selexipag (titrated to the maximum tolerated dosage up to 800 μg twice daily; n = 33) significantly reduced mean PVR by 30.3 % (95 % CI −44.7 to −12.2 %; p = 0.0045) and significantly increased the cardiac index (a measure of the median treatment effect) by 0.41 L/min/m2 (95 % 0.10–0.71) at week 17 [4, 15].
At the maximum tolerated dosage of 1600 μg twice daily, selexipag did not prolong the QT interval to any clinically relevant extent in a thorough QT study in healthy volunteers [4, 16].
In healthy volunteers, selexipag 400 μg twice daily did not alter the pharmacodynamic effect of warfarin on the international normalized ratio [4, 17].
2.2 Pharmacokinetics
The pharmacokinetics of oral selexipag and its active metabolite (ACT-333679) are dose-proportional with single doses of up to 800 μg dose and multiple doses of up to 1800 μg, with no accumulation of selexipag or ACT-333679 in the plasma after multiple doses [4, 18]. Selexipag is rapidly absorbed, with maximum plasma concentrations of selexipag and ACT-333679 attained within 1–3 and 3–4 h, respectively [4, 18, 19]. At steady state, exposure to selexipag and ACT-333679 was similar in patients with PAH to that in healthy volunteers, with exposure to ACT-333679 about 3- to 4-fold higher than that of selexipag [4, 18]. Steady state was attained within 3 days for both selexipag and its active metabolite [18]. Exposure to selexipag and ACT-333679 were not significantly altered in the presence of food [4]. At steady state, bioequivalence of different dose-strength tablets was demonstrated between eight 200 μg tablets and one 1600 μg tablet in healthy volunteers [20].
Enzymatic hydrolysis of selexipag to its active metabolite is mediated by hepatic carboxylesterase 1 [4]. The drug also undergoes oxidative metabolism via CYP3A4 and CYP2C8 to hydroxylated and dealkylated metabolites, with ACT-333679 undergoing glucuronidation via UGT1A3 and UGT2B7. With the exception of ACT-333679, none of the circulating metabolites in human plasma exceeds 3 % of the total drug-related material. Selexipag is predominantly eliminated via metabolism, with a mean terminal half-life of 0.8–2.5 h and an average apparent oral clearance of 35 L/h; the mean terminal half-life of ACT-333679 is 6.2–13.5 h. In healthy volunteers, ≈93 % of radioactive drug material was eliminated in the faeces and 12 % in the urine; no selexipag or ACT-333679 was found in the urine [4].
No clinically relevant effects of sex, age, race or bodyweight on the pharmacokinetics of selexipag and ACT-333679 have been observed in healthy volunteers or in patients with PAH [4]. No dosage adjustments are required in patients with an estimated glomerular filtration rate (eGFR) of >15 mL/min/1.73 m2; there is no clinical experience with selexipag in patients undergoing dialysis or in those with an eGFR of <15 mL/min/1.73 m2. No dosage adjustments are required in patients with mild hepatic impairment (Child-Pugh class A) [4]. A once-daily rather than a twice-daily regimen is recommended in patients with moderate hepatic impairment (Child-Pugh class B) due to increased exposure to selexipag and ACT-333679 [4, 21]. There is no experience with selexipag in patients with severe hepatic impairment; its use should be avoided in this patient population [4].
There are no clinically relevant drug-to-drug interactions when selexipag is coadministered with lopinavir/ritonavir [4, 22], warfarin [4, 17], endothelin receptor antagonists, PDE-5 inhibitors or a combination of an endothelin receptor antagonist and a PDE-5 inhibitor [4]. Concomitant administration with strong inhibitors of CYP2C8 (e.g. gemfibrozil) may significantly increases exposure to selexipag and ACT-333679; hence, concomitant administration of selexipag and strong CYP2C8 inhibitors should be avoided [4].
Features and properties of selexipag
Alternative names | Uptravi®; NS-304; ACT-293987 |
Class | Acetamides; antihypertensives; pyrazines; small molecules; sulfonamides; vasodilators |
Mechanism of action | Prostacyclin (PGI2) receptor agonist |
Route of administration | Oral |
Pharmacodynamics | Selexipag is hydrolyzed in vivo to an active, highly potent diphenylpyrazine derivative (MRE 269; ACT-333679), which exhibits long-acting agonist activity at the PGI2 receptor |
Pharmacokinetics | Rapidly absorbed and hydrolyzed to ACT-333679; at steady state, the respective mean elimination half-life of selexipag and ACT-333679 was 0.8–2.5 and 6.2–13.5 h |
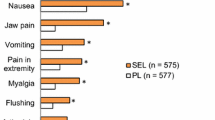
Adverse reactions occurring more frequently (by ≥5 %) than with placebo | Headache, diarrhoea, jaw pain, nausea, myalgia, vomiting, pain in extremity and flushing |
ATC codes | |
WHO ATC code | C02 (antihypertensives); C04 (peripheral vasodilators) |
EphMRA ATC code | C2A (antihypertensives [of non-herbal origin] Plain);C4 (cerebral and peripheral vasotherapeutics) |
Chemical name | 2-{4-[(5,6-Diphenylpyrazin-2-yl)(isopropyl)amino]butox}-N-(methylsulfonyl)acetamide |
2.3 Therapeutic Trials
In the proof-of-concept phase II trial (NCT 00993408), the addition of selexipag improved surrogate haemodynamic markers of PAH compared with the addition of placebo in patients with PAH receiving stable dosages of an endothelin receptor antagonist and/or a PDE-5 inhibitor (see Sect. 2.1) [15].
The event-driven, double-blind, multinational, phase III GRIPHON trial (NCT01106014) was conducted in patients with PAH (n = 1156 randomized), most of whom (≈80 %) were receiving stable dosages of an endothelin receptor antagonist and/or a PDE-5 inhibitor [23]. Patients were randomized to selexipag (≤1600 μg twice daily; initially 200 µg twice daily, with the dosage up-titrated based on tolerability) or placebo, with a respective median duration of treatment at the time of the primary analysis in the selexipag and placebo groups of 70.7 and 63.7 weeks [23].
In the primary analysis, the risk of the primary composite endpoint of death or a complication related to PAH (whichever occurred first) was reduced by 40 % in the selexipag versus the placebo group (hazard ratio 0.60; 99 % CI 0.46–0.78; p < 0.001) [23]. Results of sensitivity analyses of the primary treatment outcome effect were consistent with this primary analysis. Of the 397 primary outcome events, 27.0 % occurred in the selexipag group and 41.6 % in the placebo group, with 81.9 % of primary outcome events involving disease progression and hospitalization. Results for the composite primary endpoint were consistent across prespecified patient subgroups, including in patients who were not receiving treatment at baseline and those who were receiving PAH therapy at baseline [23].
At 26 weeks, selexipag recipients experienced an improvement in 6-min walk distance (increased by 4.0 m), whereas this distance decreased by 9.0 m in the placebo group (treatment effect 12.0 m; 99 % CI 1–24; p = 0.003) [secondary endpoint] [23]. The percentage of patients who experienced no worsening in WHO functional class was similar in the selexipag and placebo groups at 26 weeks (77.8 vs. 74.9 %). Statistical evaluation of secondary endpoints was conducted using a hierarchical testing procedure and thus, all subsequent endpoints were considered exploratory [23].
2.4 Adverse Events
Selexipag was generally well tolerated in patients with PAH participating in the GRIPHON trial, with most common adverse events (e.g. headache, diarrhoea, nausea, jaw pain) consistent with those associated with prostacyclin therapy [23]. These adverse events occurred more frequently during the dose titration phase [4]. Adverse reactions that occurred with at least a 5 % higher incidence in selexipag than placebo recipients were headache, diarrhoea, jaw pain, nausea, myalgia, vomiting, pain in extremity and flushing [4].
In GRIPHON, a similar proportion of selexipag and placebo recipients experienced at least one treatment-emergent adverse event (98.3 vs. 96.9 %) [includes adverse events that occurred during the titration phase], with these events leading to discontinuation of study drug in significantly more selexipag recipients (14.3 vs. 7.1 %; p < 0.001) [23]. The most common adverse events (incidence ≥20 % in either group) occurring in the selexipag and placebo groups were headache (65.2 vs. 32.8 %; p < 0.001), diarrhoea (42.4 vs. 19.1 %; p < 0.001), nausea (33.6 vs. 18.5 %; p < 0.001), pain in jaw (25.7 vs. 6.2 %; p < 0.001), worsening of PAH (21.9 vs. 35.7 %; p < 0.001) and dyspnea (16.0 vs. 21.0 %; p < 0.001). Other adverse events that occurred significantly more frequently in one group than the other were vomiting (18.1 % in the selexipag group vs. 8.5 % in the placebo group; p < 0.001), pain in extremity (16.9 vs. 8.0 %; p < 0.001), myalgia (16.0 vs. 5.9 %; p < 0.001), flushing (12.2 vs. 5.0 %; p < 0.001) and hyperthyroidism (1.4 vs. 0 %; p = 0.004). One selexipag recipient discontinued study drug because of hyperthyroidism [23]. A small reduction (up to a median −0.3 μ/L) in median thyroid-stimulating hormone was observed at most visits in the selexipag group in a phase 3 placebo-controlled study [4]. There were no mean changes in triiodothyronine or thyroxine levels in either treatment group [4].
There was no between-group difference in the percentage of patients experiencing at least one serious adverse event (43.8 % in the selexipag group vs. 47.1 % in the placebo group) [23]. The incidences of individual serious adverse events did not differ between the selexipag and placebo groups (i.e. at a rate >1 % higher) [23].
2.5 Ongoing Clinical Trials
The extension phase (NCT01112306) of the GRIPHON trial (NCT01106014) is currently ongoing, with an estimated completion date of February 2018. The phase IIIb TRANSIT-1 trial (NCT02471183) will evaluate the tolerability and safety of the transition from inhaled trepostinil to oral selexipag in patients with PAH, with this study currently recruiting participants. The phase IIIb TRITON trial (NCT02558231) will evaluate the efficacy and safety of initial triple versus initial dual oral combination therapy in patients with newly diagnosed PAH; this study is not yet open for participant recruitment.
3 Current Status
Oral selexipag received its first global approval on 21 December 2015 for the treatment of PAH (WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH in the USA.
References
Delcroix M, Howard L. Pulmonary arterial hypertension: the burden of disease and impact on quality of life. Eur Respir Rev. 2015;24:621–9.
Mandras SA, Gilkin RJ, Pruett JA, et al. Pulmonary arterial hypertension: progress and challenges in the modern treatment era. Am J Manag Care. 2014;20(9):S191–9.
Asaki T, Kuwano K, Morrison K, et al. Selexipag: an oral and selective IP prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. J Med Chem. 2015;58(18):7128–37.
Actelion Pharmaceuticals US Inc. UPTRAVI® (selexipag) tablets, for oral use: US prescribing information. 2015. http://www.fda.gov/. Accessed 5 Jan 2016.
FirstWord Pharma. Actelion receives Health Canada approval for Uptravi (selexipag) for the long-term treatment of pulmonary arterial hypertension. 2016. http://www.firstwordpharma.com/node/1353734#axzz3yTk90Lnp. Accessed 28 Jan 2016.
European Medicines Agency. CHMP summary of positive opinion for Uptravi. 2016. http://www.ema.europa.eu/. Accessed 2 Feb 2016.
Nippon Shinyaku. Orphan drug designation of pulmonary arterial hypertension drug selexipag [media release]. 2014. http://www.nippon-shinyaku.co.jp. Accessed 23 Dec 2015.
Kuwano K, Hashino A, Noda K, et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetamide (NS-304), ameliorates rat pulmonary hypertension with unique relaxant responses of its active form, {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}acetic acid (MRE-269), on rat pulmonary artery. J Pharmacol Exp Ther. 2008;326(3):691–9.
Actelion Ltd, Nippon Shinyaku. Actelion and Nippon Shinyaku enter into a license agreement on novel PAH compound [media release]. 2008. http://www.actelion.com. Accessed 1 Jan 2016.
Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-Diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetamide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007;322(3):1181–8.
Morrison K, Ernst R, Hess P, et al. Selexipag: a selective prostacyclin receptor agonist that does not affect rat gastric function. J Pharmacol Exp Ther. 2010;335(1):249–55.
Gatfield J, Menyhart K, Nayler O. Anti-contractile and anti-proliferative responses in human pulmonary arterial smooth muscle cells induced by ACT-333679, the active metabolite of selexipag, a non-prostanoid selective prostacyclin receptor agonist [abstract no. P2353]. Eur Respir J. 2014;44(Suppl 58):P2353.
Gatfield J, Menyhart K, Morrison K, et al. The non-prostanoid prostacyclin receptor agonist ACT-333679, the active metabolite of selexipag, is characterized by low beta-arrestin recruitment and receptor internalization activity [abstract no. 1110–174]. J Am Coll Cardiol. 2015;65(10 Suppl):A1542.
Morrison K, Wanner D, Gatfield J, et al. Repeated oral administration of the selective prostacyclin receptor agonist selexipag does not cause tachyphylaxis in spontaneously hypertensive rats [abstract]. J Am Coll Cardiol. 2015;65(10 Suppl):A1558.
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–80.
Hoch M, Darpo B, Remenova T, et al. A thorough QT study in the context of an uptitration regimen with selexipag, a selective oral prostacyclin receptor agonist. Drug Des Devel Ther. 2015;9:175–85.
Niglis S, Bruderer S, Okubo K, et al. Niglis S, Bruderer S, Okubo K, et al. Investigation of potential pharmacodynamic and pharmacokinetic interactions between selexipag and warfarin in healthy male subjects [abstract no. 1690802]. In: American College of Clinical Pharmacology Annual Meeting; 2013.
Bruderer S, Hurst N, Kaufmann P, et al. Multiple-dose up-titration study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of selexipag, an orally available selective prostacyclin receptor agonist, in healthy subjects. Pharmacology. 2014;94(3–4):148–56.
Kaufmann P, Okubo K, Bruderer S, et al. Pharmacokinetics and tolerability of the novel oral prostacyclin IP receptor agonist selexipag. Am J Cardiovasc Drugs. 2015;15(3):195–203.
Baldoni D, Bruderer S, Muhsen N. Bioequivalence of different dose-strength tablets of selexipag, a selective prostacyclin receptor agonist, in a multiple-dose up-titration study. Int J Clin Pharmacol Ther. 2015;53(9):788–98.
Fischer N, Kaufmann P, Bruderer S, et al. Pharmacokinetics of selexipag in subjects with mild, moderate, or severe hepatic impairment compared with healthy subjects [abstract no. PP194]. Clin Ther. 2013;35(8):e77–8.
Kaufmann P, Niglis S, Bruderer S, et al. Effect of lopinavir/ritonavir on the pharmacokinetics of selexipag an oral prostacyclin receptor agonist and its active metabolite in healthy subjects. Br J Clin Pharmacol. 2015;80(4):670–7.
Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–33.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process the manufacturer of selexipag was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Lesley Scott is a salaried employee of Adis, Springer SBM.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Scott, L.J. Selexipag: First Global Approval. Drugs 76, 413–418 (2016). https://doi.org/10.1007/s40265-016-0549-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0549-4