
Abstract
Vorapaxar (Zontivity®) is a first-in-class, potent and orally-active protease-activated receptor 1 (PAR-1) antagonist that blocks thrombin-mediated platelet activation without interfering with thrombin-mediated fibrin deposition. The long-term efficacy of once-daily vorapaxar added to standard antiplatelet therapy (aspirin with or without clopidogrel) in the secondary prevention of atherothrombotic events in patients with a history of myocardial infarction (MI), ischaemic stroke or peripheral arterial disease was investigated in the large, multinational TRA 2°P-TIMI 50 trial. Compared with placebo, vorapaxar significantly reduced the risk of the composite endpoints of cardiovascular (CV) death, MI or stroke, and CV death, MI, stroke or urgent coronary revascularization in the overall trial population. Vorapaxar also significantly reduced the risk of these composite endpoints in the subgroup of patients with prior MI (the largest qualifying disease cohort) and the subset of post-MI patients with no history of stroke or transient ischaemic attack (TIA). Vorapaxar significantly increased the risk of GUSTO moderate and/or severe bleeding in the overall trial population and all key subgroups (including post-MI patients with no history of stroke or TIA). Vorapaxar also significantly increased the risk of intracranial haemorrhage (ICH) in the overall trial population and the subgroup of patients with prior stroke, but not the subgroup of post-MI patients or the subset of post-MI patients with no history of stroke or TIA. Based on these results, vorapaxar has been approved in the EU as an adjunctive treatment for the secondary prevention of atherothrombotic events in patients with prior MI who do not have a history of stroke, TIA or ICH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
First-in-class, potent, orally-active, once-daily PAR-1 antagonist |
Reduces the risk of major CV events in patients with a history of MI when used as an adjunct to standard antiplatelet therapy |
Adding vorapaxar to standard antiplatelet therapy increases risk of major bleeding complications |
Contraindicated in patients with a history of stroke, TIA or ICH |
1 Introduction
Chronic atherosclerotic disease is often asymptomatic (clinically silent); however, when complicated by superimposed thrombosis (in a process known as atherothrombosis), the resulting clinical manifestations include, among others, acute coronary syndrome [ACS; e.g. myocardial infarction (MI)], cerebrovascular disease [CBVD; stroke and transient ischaemic attack (TIA)] and peripheral artery disease (PAD; e.g. critical limb ischaemia) [1, 2]. Atherothrombotic complications of atherosclerosis continue to be leading cause of morbidity and mortality in industrialized regions of the world, including Europe [3].
Despite the use of standard-of-care antiplatelet therapies, patients with ACS remain at high risk for recurrent thrombotic cardiovascular (CV) events [4, 5], as do clinically stable patients with established atherosclerotic disease receiving additional secondary prevention drug therapies, including antihypertensives and lipid-lowering agents [6]. The rationale for antiplatelet therapy in patients with atherothrombotic disease rests on the pivotal role played by platelet activation in atherothrombosis [7, 8]. In this respect, existing oral antiplatelet agents target two of the three major pathways involved in platelet activation, namely those mediated by thromboxane A2 (TxA2) and adenosine diphosphate (ADP) [9–11]. In the setting of ACS, dual antiplatelet therapy (DAPT) with aspirin (a cyclo-oxygenase-1 inhibitor) plus clopidogrel (a thienopyridine ADP P2Y12 receptor antagonist) significantly reduces the rate of major CV events compared with aspirin alone, albeit at the expense of a higher bleeding risk [12]. The antithrombotic benefits with more potent thienopyridine and non-thienopyridine ADP P2Y12 receptor antagonists (e.g. prasugrel [4] and ticagrelor [5]) are more pronounced, although they are still associated with bleeding risks. There is also evidence, albeit inconclusive, to suggest that DAPT with aspirin plus clopidogrel significantly reduces the risk of major CV events in patients with established atherosclerosis; however, this apparent benefit is again accompanied by an increased risk of bleeding [13, 14]. Overall, benefits and risks with these agents may be because the TxA2- and ADP-mediated pathways are important not only in pathological thrombosis, but also in normal (protective) haemostasis [8]; the need to both enhance antithrombotic benefit and, in as far as it is possible, uncouple it from bleeding risk, has led to the investigation of therapies that target the third—and most potent—major pathway involved in platelet activation, namely that mediated by thrombin [9–11, 15].
Thrombin, a multifunctional serine protease generated at sites of vascular injury, plays a key role in the control of thrombus formation. In terms of normal (protective) haemostasis, thrombin-mediated cleavage of fibrinogen into fibrin is more important than thrombin-mediated platelet activation. However, thrombin activates platelets at concentrations lower than those required for activation of the coagulation cascade, and thrombin-mediated platelet activation contributes to pathological thrombosis through the formation of an occlusive platelet-rich thrombus [16, 17]. Thrombin activates platelets through stimulation of the two protease-activated receptor (PAR) subtypes that are expressed on human platelets, namely PAR-1 and PAR-4, with the former being the principal platelet thrombin receptor (half maximal effective concentrations of 50 and 5000 pmol/L, respectively) [11].
Vorapaxar (Zontivity®) is a first-in-class, potent and orally-active PAR-1 antagonist that blocks thrombin-mediated platelet activation without interfering with thrombin-mediated fibrin deposition [18]. In the EU, vorapaxar is indicated as an adjunct to antiplatelet therapy with aspirin and, where appropriate, clopidogrel, for the reduction of atherothrombotic events in adult patients with a history of MI [19].
This article briefly summarizes the pharmacological properties of vorapaxar and reviews, mainly from an EU perspective, its efficacy and tolerability in preventing atherothrombotic events in clinically stable patients with established atherosclerotic disease. Vorapaxar has also been evaluated for the long-term prevention of recurrent ischaemic CV events in patients presenting with a non-ST-segment elevation (NSTE) ACS [20]; however, detailed discussion of the data pertaining to this separate clinical situation—and unapproved indication—is beyond the scope of this article.
2 Pharmacological Properties
The pharmacodynamics and pharmacokinetic properties of vorapaxar are briefly summarized in Tables 1 and 2, respectively.
2.1 Pharmacodynamic Properties
Vorapaxar is a synthetic tricyclic 3-phenylpyridine analogue of himbacine (an alkaloid isolated from the bark of the Australian magnolia), but lacks the muscarinic M2 antagonist activity characteristic of himbacine [21, 22]. It produces selective and reversible PAR-1 inhibition [19]; however, its effects are irreversible for the life span of platelets because of its long elimination half-life (≈8 days; see Table 2).
As part of the largest phase I study to be performed, once-daily administration of vorapaxar 2.5 mg to 16 healthy Caucasian and Japanese volunteers for 4 weeks resulted in complete (i.e. ≥80 %) inhibition of PAR-1-mediated platelet aggregation in 75 % of the subjects on day 7, and in all of the subjects on days 14, 21 and 28. Inhibition of PAR-1-mediated platelet aggregation was evaluated by the inhibition of thrombin receptor agonist peptide (TRAP)-induced platelet aggregation. Platelet function gradually recovered following discontinuation of vorapaxar, with levels of ≥50 % reached 2–3 weeks after treatment withdrawal [24]. However, platelet aggregation at levels of ≥80 % could last for 2–4 weeks after treatment discontinuation [19]. The slow reversal of platelet inhibition after stopping treatment with vorapaxar is consistent with the long-half life of the drug [23, 24].
2.2 Pharmacokinetic Properties
Vorapaxar is eliminated primarily by metabolism and excretion (mainly of metabolites) in the faeces (Table 2).
As regards special patient populations, vorapaxar exposure is not altered to the extent that dosage adjustment is necessary, based on age, gender, ethnicity, renal impairment or mild hepatic impairment [19]. The drug should, however, be used with caution in patients with severe renal impairment or end-stage renal disease, due to a lack of therapeutic experience [19]. Furthermore, despite vorapaxar exposure being similar between healthy patients with those with moderate hepatic impairment, the drug should be used with caution in the latter. Vorapaxar is contraindicated in patients with severe hepatic impairment [19].
Concomitant treatment with vorapaxar and strong cytochrome P450 (CYP) 3A inhibitors or inducers should be avoided) (Table 2). However, no increase in bleeding risk or reduction in the efficacy of vorapaxar was observed in patients taking weak or moderate CYP3A inhibitors in the TRA 2°P-TIMI 50 and TRACER trials, and dosage adjustment is not considered necessary for concomitant administration of these drugs with vorapaxar [19].
In vitro metabolism studies indicate that neither vorapaxar nor M20 (the major active circulating metabolite; Table 2) is likely to cause clinically significant inhibition or induction of major CYP isoforms or inhibition of the following efflux or uptake transporters: breast cancer resistance protein, organic anion transporter (OAT) 1, OAT3, organic anion transporter polypeptide (OATP) 1B1, OATP1B3 and organic cation transporter 2 [19, 29]. Vorapaxar is a weak inhibitor of the intestinal P-glycoprotein (P-gp) transporter; however, coadministration of vorapaxar with digoxin (a P-gp substrate) does not necessitate a change of the dosage of either drug [19].
3 Therapeutic Efficacy
The use of vorapaxar to reduce the risk of recurrent atherothrombotic events in patients with established atherosclerosis receiving standard therapy (including oral antiplatelet agents) has been examined in a large, randomized, double-blind, placebo-controlled, multinational trial, namely TRA 2°P-TIMI 50 [18]. Information about and results from this trial are available from full publications [31–36], abstracts [37–39], the EU summary of product characteristics (SPC) [19], the US prescribing information [29] and an FDA draft briefing document [40].
3.1 TRA 2°P-TIMI 50 Trial
Eligible patients had a history of: (1) spontaneous MI or ischaemic stroke within the previous 2 weeks to 12 months or (2) PAD associated with a history of intermittent claudication in conjunction with either an ankle brachial index <0.85 or previous revascularization for limb ischaemia. Patients enrolled were randomized to receive vorapaxar 2.5 mg/day (n = 13,225) or placebo (n = 13,224) in addition to standard of care until the end of the study. Randomization was stratified by the qualifying disease (in a hierarchical order) and the intention to administer a thienopyridine [18]. As an example of the hierarchical enrolment, patients with a history of stroke who also had a history of MI or PAD were recruited to the MI and PAD cohorts, respectively; this approach resulted in some overlap of atherosclerotic disease conditions between the strata (based on the qualifying event and the timing of the event in history). By design, enrollment in both the PAD- and stroke-qualifying cohorts was completed after reaching ≈15 % of the planned overall trial population [31–33].
Of note, the Data and Safety Monitoring Board (DSMB) reported an increased risk of intracranial haemorrhage (ICH) with vorapaxar in patients with prior stroke after a median of 2 years of follow-up; they recommended that the study medication be discontinued in all patients with a history of ischaemic stroke before or during the trial [32–34].
In addition, before the database was locked and while blinded treatment was ongoing, the TIMI Study Group reordered the hierarchy of the efficacy endpoints, such that the composite of CV death, MI, stroke or urgent coronary revascularization (UCR)—originally the primary endpoint—became the major secondary outcome, while the composite of CV death, MI or stroke—originally the major secondary outcome—became the primary endpoint [31]. This change was made following a review by the TIMI Study Group of data from another large, randomized, double-blind, placebo-controlled, multinational trial, namely TRACER [20], which examined the use of oral vorapaxar to reduce the risk of recurrent ischaemic CV events in patients with NSTE ACS receiving standard therapy (see Sect. 6 for a brief discussion of this trial). Nevertheless, data submitted for regulatory approval retained the original primary and secondary composite outcomes [19, 29].
Bleeding was assessed according to Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO) and Thrombolysis in Myocardial Infarction (TIMI) criteria; the main safety endpoint was GUSTO moderate or severe bleeding [31]. Results for net clinical outcome analyses that take into account both efficacy and bleeding endpoints are presented in this section; results for bleeding endpoints are discussed in detail in Sect. 4. All event rates presented in this section (and Sect. 4) are 3-year Kaplan-Meier estimates.
The TRA 2°P-TIMI 50 trial was not designed to evaluate the efficacy of vorapaxar in individual patient subgroups. This notwithstanding, subgroup analyses based on qualifying atherosclerotic disease were prespecified [18]. Of note, subgroup analyses of patients with no history of CBVD (i.e. all patients who qualified for the trial with a diagnosis of either MI or PAD without a history of stroke or TIA [29]) and post-MI patients with no history of CBVD (i.e. all patients who qualified for the trial with a diagnosis of MI without a history of stroke or TIA [19]) were performed subsequent to the DSMB decision described above.
In terms of concomitant antiplatelet medication, nearly all trial participants were receiving aspirin [93.5 % (including 98.1, 80.9 and 87.9 % of post-MI, post-stroke and PAD patients, respectively)], nearly two-thirds were receiving a thienopyridine [62.2 % (78.1, 23.6 and 36.8 %, respectively)] and more than one-half were receiving aspirin plus a thienopyridine [57.0 % (76.8, 7.3 and 28.2 %, respectively)] [31–34]. The thienopyridine used in this trial was almost always clopidogrel [34]; <1 % of participants received prasugrel during the study [31].
3.1.1 Overall Patient Population
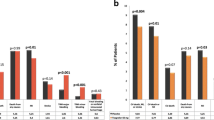
In the TRA 2°P-TIMI 50 trial, vorapaxar was more effective than placebo in preventing major CV events in patients with established atherosclerotic disease who were receiving standard therapy [31] (Table 3). In the overall trial population, vorapaxar significantly reduced the risk of the composite endpoint of CV death, MI or stroke by 13 %, and that of the composite endpoint of CV death, MI, stroke or UCR by 12 %, compared with placebo [31] (Table 3). In terms of the individual components of these composite endpoints, however, only the rate of MI was significantly reduced with vorapaxar [31] (Table 3). The overall stroke rate was the same in the two treatment groups [31] (Table 3)
Regarding the composite outcome of CV death, MI or stroke, no significant heterogeneity of the treatment effect was seen across all but one of the major subgroups of patients defined by demographic (e.g. age, gender, region) or disease (e.g. history of stroke, diabetes, current smoker, aspirin use, renal function) characteristics at baseline [31]. The sole exception was bodyweight; the hazard ratio (HR) for the aforementioned composite outcome was 0.85 (95 % CI 0.78–0.92) and 1.22 (95 % CI 0.88–1.69) in patients weighing ≥60 and <60 kg, respectively (p = 0.03 for interaction) [31]. Notably, vorapaxar significantly reduced the risk of CV death, MI or stroke relative to placebo, regardless of whether or not thienopyridine therapy was planned at randomization or whether or not patients were already receiving a thienopyridine at randomization or at 18 months after randomization [31, 38]. The effect of vorapaxar on the risk of CV death, MI or stroke and that of CV death, MI, stroke or UCR in the predefined subgroups based on qualifying atherosclerotic disease is discussed later in this section.
Vorapaxar was associated with a significant reduction in arterial revascularization rates compared with placebo [13.6 vs. 15.5 %; HR 0.89 (95 % CI 0.83–0.95); p < 0.001]. Significant reductions were seen in both peripheral and coronary revascularizations, including coronary artery bypass graft (CABG) surgery, as well as in elective revascularizations (p ≤ 0.044 for all) [37]. Vorapaxar was also associated with a significant reduction in the risk of definite stent thrombosis, according to Academic Research Consortium criteria (1.1 vs. 1.4 %; HR 0.71; p = 0.04) [39].
In net clinical outcome analyses, there was a significant difference in favour of vorapaxar over placebo for the combined endpoint of all-cause mortality, MI, stroke or GUSTO severe bleeding [11.9 vs. 12.8 %; HR 0.92 (95 % CI 0.85–0.99); p = 0.02], but not for the combined endpoint of CV death, MI, stroke or GUSTO moderate or severe bleeding [11.7 vs. 12.1 %; HR 0.97 (95 % CI 0.90–1.04)] or the combined endpoint of CV death, MI, stroke, UCR or GUSTO moderate or severe bleeding [13.4 vs. 14.0 %; HR 0.96 (95 % CI 0.89–1.02)] [31].
3.1.2 Patients with No History of Cerebrovascular Disease
In the large subgroup of post-MI or PAD patients with no history of CBVD (n = 20,170), vorapaxar significantly reduced the risk of the composite endpoint of CV death, MI or stroke by 20 % [7.9 vs. 9.5 %; HR 0.80 (95 % CI 0.73–0.89); p < 0.001], the composite endpoint of CV death, MI, stroke or UCR by 17 % [10.1 vs. 11.8 %; HR 0.83 (95 % CI 0.76–0.90); p < 0.001], MI by 18 % [5.4 vs. 6.4 %; HR 0.82 (95 % CI 0.73–0.93); p = 0.002] and stroke by 33 % [1.2 vs. 1.6 %; HR 0.67 (95 % CI 0.52–0.87); p = 0.002], compared with placebo [35].
3.1.3 Patients Qualifying with Myocardial Infarction
Post-MI patients (n = 17,779) represented the largest qualifying atherosclerotic disease cohort in the TRA 2°P-TIMI 50 trial. The lack of a sample size calculation notwithstanding (Sect. 3.1), there was (based on the number of events) an estimated power in excess of 85 % to detect a treatment difference of at least 16 % in this subgroup [34].
Among post-MI patients, vorapaxar significantly reduced the risk of the composite endpoint of CV death, MI or stroke by 20 %, the composite endpoint of CV death, MI, stroke or UCR by 17 %, MI by 21 % and ischaemic stroke by 34 %, compared with placebo [34] (Table 3).
Regarding the composite outcome of CV death, MI or stroke, no significant heterogeneity of treatment effect was found across key subgroups in the post-MI cohort. For example, vorapaxar significantly reduced the risk of CV death, MI or stroke, irrespective of the timing of the qualifying MI relative to randomization [34], whether or not thienopyridine therapy was planned at randomization [34], or whether or not patients had diabetes mellitus [34, 36].
In net clinical outcome analyses, there was a significant difference in favour of vorapaxar over placebo for the combined endpoint of all-cause mortality, MI, stroke or GUSTO severe bleeding [10.1 vs. 11.4 %; HR 0.86 (95 % CI 0.78–0.95); p = 0.003] and the combined endpoint of CV death, MI, stroke, UCR or GUSTO moderate or severe bleeding [12.5 vs. 13.4 %; HR 0.91 (95 % CI 0.84–0.99); p = 0.038], but not for the combined endpoint of CV death, MI, stroke or GUSTO moderate or severe bleeding [10.2 vs. 11.0 %; HR 0.91 (95 % CI 0.83–1.01)] [34].
3.1.3.1 Patients Qualifying with Myocardial Infarction with no History of Cerebrovascular Disease
In the subgroup of post-MI patients with no history of CBVD (n = 16,897), vorapaxar significantly reduced the risk of CV death, MI or stroke by 22 % and that of CV death, MI, stroke or UCR by 18 % compared with placebo (Table 3). A beneficial effect of vorapaxar was consistently observed across many subgroups of this cohort [19].
In addition, a significant difference in favour of vorapaxar over placebo for the net clinical outcome based on multiple occurrences of CV death, MI, stroke or GUSTO severe bleeding was consistently observed at all timepoints assessed (i.e. 12, 18, 24, 30 and 36 months; p ≤ 0.033 for all) [19].
Vorapaxar also appeared to reduce the rate of definite stent thrombosis compared with placebo in patients receiving any stent before or during the study [HR 0.70 (95 % CI 0.50–0.98)] [19].
3.1.4 Patients Qualifying with Peripheral Artery Disease
In the underpowered subgroup of patients who qualified for the trial with a diagnosis of PAD (n = 3787), vorapaxar was associated with statistically nonsignificant reductions in the composite endpoints of CV death, MI or stroke, and CV death, MI, stroke or UCR; similar results were seen for the individual outcomes of CV death and ischaemic stroke (Table 3) [32].
Regarding manifestations of PAD, however, vorapaxar significantly reduced the risk of limb ischaemic events, including hospitalization for acute limb ischaemia [by 42 %; 2.3 vs. 3.9 %; HR 0.58 (95 % CI 0.39–0.86); p = 0.006], any peripheral revascularization [by 16 %; 18.4 vs. 22.2 %; HR 0.84 (95 % CI 0.73–0.97); p = 0.017], urgent peripheral revascularization [by 35 %; 3.1 vs. 4.7 %; HR 0.65 (95 % CI 0.46–0.91); p = 0.012], and elective peripheral revascularization [by 14 %; 16.5 vs. 19.5 %; HR 0.86 (95 % CI 0.74–0.9995); p = 0.049], compared with placebo [32].
Furthermore, with respect to broader vascular endpoints that not only included events involving the peripheral circulation, but also those involving the coronary and cerebral circulations, vorapaxar significantly reduced the risk of urgent vascular hospitalization [5.8 vs. 8.0 %; HR 0.72 (95 % CI 0.56–0.93); p = 0.011] and any revascularization [26.2 vs. 30.3 %; HR 0.88 (95 % CI 0.78–0.99); p = 0.036] compared with placebo [32]. Vorapaxar also significantly reduced the risk of the composite endpoints of CV death, MI, stroke or urgent vascular hospitalization [15.9 vs. 18.6 %; HR 0.85 (95 % CI 0.73–0.998); p = 0.047] and CV death, MI, stroke, urgent vascular hospitalization and revascularization [32.7 vs. 38.0 %; HR 0.87 (95 % CI 0.78–0.97); p = 0.009] [32].
3.1.4.1 Patients Qualifying with Peripheral Artery Disease with no History of Cerebrovascular Disease
Vorapaxar was associated with statistically nonsignificant reductions in the composite endpoints of CV death, MI or stroke [HR 0.87 (95 % CI 0.71–1.06)] and CV death, MI, stroke or UCR [HR 0.88 (95 % CI 0.71–1.09)] in the subgroup of PAD patients with no history of CBVD (n = 3273) [35, 41].
3.1.5 Patients Qualifying with Stroke
Vorapaxar, as compared with placebo, did not significantly reduce the risk of the composite endpoint of CV death, MI or stroke, the composite endpoint of CV death, MI, stroke or UCR, or any of the individual components of these composites in the underpowered subgroup of patients who qualified for the trial with a diagnosis of stroke (n = 4883) [33] (Table 3).
Likewise, vorapaxar was associated with statistically nonsignificant reductions in CV death, MI or stroke [HR 0.95 (95 % CI 0.80–1.11)] and CV death, MI, stroke or UCR [HR 0.94 (95 % CI 0.80–1.10)] compared with placebo in the larger cohort of patients with a history of stroke (i.e. all trial participants with a history of stroke, regardless of qualifying atherosclerosis; n = 5746) [31].
4 Tolerability
This section focuses primarily on haemorrhagic adverse events that occurred during the conduct of the TRA 2°P-TIMI 50 trial in patients with established atherosclerosis in which vorapaxar was used as an adjunct to standard therapy, including oral antiplatelet agents (see Sect. 3.1 for study design details). The discussion on non-haemorrhagic adverse events (Sect. 4.2) derives data from the TRA 2°P-TIMI 50 trial [41] and a combined analysis of the TRA 2°P-TIMI 50 and TRACER trials [29, 42].
4.1 Haemorrhagic Adverse Events
Adding vorapaxar to standard antiplatelet therapy in the TRA 2°P-TIMI 50 trial (Sect. 3) was associated with increased rates of bleeding, including GUSTO moderate to severe bleeding (main safety endpoint) and TIMI clinically significant bleeding (Table 4).
In the overall trial population, vorapaxar significantly increased the risk of GUSTO moderate or severe bleeding by 66 % compared with placebo (Table 4); no significant heterogeneity of the treatment effect was seen across any of the major subgroups of patients defined by demographic or disease characteristics at baseline [31]. Vorapaxar significantly increased the risk of GUSTO moderate or severe bleeding by 55 % in the subgroup of patients with no history of CBVD [3.7 vs. 2.4 %; HR 1.55 (95 % CI 1.30–1.86); p < 0.001][35], by 61, 62 and 93 % in the MI-, PAD- and stroke-qualifying cohorts, respectively (Table 4), and by 74 % in the cohort of patients with a history of stroke [4.7 vs. 2.8 %; HR 1.74 (95 % CI 1.26–2.39); p < 0.001] [31]. Among post-MI patients with no history of CBVD, vorapaxar significantly increased the risk of GUSTO moderate or severe bleeding and that of GUSTO moderate bleeding, but not that of GUSTO severe bleeding, compared with placebo (Table 4). The effect of vorapaxar on GUSTO moderate or severe bleeding relative to placebo was consistent across the subgroups examined within this cohort [19].
Vorapaxar, as compared with placebo, significantly increased the risk of TIMI clinically significant bleeding in the overall trial population (by 46 %; Table 4), the cohort of patients with no history of CBVD [by 47 %; 15.5 vs. 10.9 %; HR 1.47 (95 % CI 1.35–1.60); p value not reported] [29], the MI-qualifying cohort (by 49 %; Table 4), the subgroup of post-MI patients with no history of CBVD (by 46 %; Table 4) and the stroke-qualifying cohort (by 37 %; Table 4). The corresponding result for the PAD-qualifying cohort was not reported [32]. Vorapaxar also significantly increased the risk of TIMI clinically significant bleeding in the cohort of patients with a history of stroke [by 38 %; 15.2 vs. 10.8 %; HR 1.38 (95 % CI 1.17–1.63); p < 0.001] [31]. Among post-MI patients with no history of CBVD, vorapaxar did not significantly increase the risk of TIMI (non-CABG) major bleeding compared with placebo (Table 4). Vorapaxar did, however, significantly increase the risk of TIMI (non-CABG) minor bleeding relative to placebo [1.4 vs. 0.6 %; HR 2.23 (95 % CI 1.58–3.15); p < 0.001] [19].
There was a statistically significant, ≈2- to 2.5-fold increase in the risk of ICH, inclusive of intracerebral and subdural bleeding, with vorapaxar relative to placebo in analyses of the overall trial population (Table 4), the stroke-qualifying cohort (Table 4) and the cohort of patients with a history of stroke [2.4 vs. 0.9 %; HR 2.55 (95 % CI 1.52–4.28); p < 0.001] [31]. An analysis of the stroke-qualifying cohort indicated that the increased risk of ICH with vorapaxar emerged early and persisted during treatment [33]. In comparison, the increase in the risk of ICH with vorapaxar relative to placebo was generally smaller (≈1.5- to 2-fold) and not statistically significant in the cohort of patients with no history of CBVD [0.6 vs. 0.4 %; HR 1.46 (95 % CI 0.92–2.31); p = 0.1] [35], the MI- and PAD-qualifying cohorts (Table 4), and the cohort of post-MI patients with no history of CBVD (Table 4).
There were no significant between-group differences in the (very low) rates of fatal bleeding in the overall trial population or in any of the key subgroups examined, including the cohort of post-MI patients with no history of CBVD (Table 4).
4.2 Non-Haemorrhagic Adverse Events
Similar proportions of vorapaxar (n = 13,186) and placebo (n = 13,166) recipients reported treatment-related non-haemorrhagic adverse events (16.1 vs. 15.7 %), treatment-related non-haemorrhagic serious adverse events (0.6 vs. 0.7 %) and treatment-related non-haemorrhagic adverse events that led to study discontinuation (2.5 vs. 2.4 %), based on the as-treated population in the TRA 2°P-TIMI 50 study [41].
Anaemia was the most common non-haemorrhagic adverse event among vorapaxar recipients, according to a combined analysis of the TRA 2°P-TIMI 50 and TRACER studies; it occurred in 5.0 % of the 19,632 vorapaxar-treated patients versus 4.0 % of the 19,607 placebo-treated patients. Other adverse events occurring in >2 % of patients treated with vorapaxar in these trials were depression (2.4 vs. 2.1 % for placebo), and rashes, eruptions and exanthemas (2.2 vs. 2.0 %). Diplopia and related oculomotor disturbances occurred in 0.2 % of vorapaxar recipients (30 patients), compared with 0.06 % of placebo recipients (10 patients) [29].
The incidences of commonly occurring serious non-haemorrhagic adverse events were, with few exceptions, markedly similar in vorapaxar and placebo recipients in the combined analysis of the TRA 2°P-TIMI 50 and TRACER studies. One notable exception was anaemia (which occurred in 0.3 % of vorapaxar-treated patients versus 0.1 % of placebo-treated patients); another was pulmonary embolism (0.1 vs. 0.3 %) [42].
5 Dosage and Administration
In the EU, oral vorapaxar, coadministered with aspirin and, where appropriate, clopidogrel, is indicated for the reduction of atherothrombotic events in adult patients with a history of MI [19]. The recommended dosage is one 2 mg film-coated tablet [which contains 2.08 mg of vorapaxar (as vorapaxar sulphate)] taken once daily, with or without food. Treatment should be initiated at least 2 weeks after a MI and preferably within the first 12 months from the acute event [19].
Vorapaxar increases the risk of bleeding, including ICH and sometimes fatal bleeding [19]. It is contraindicated in patients with a history of stroke, TIA, or ICH; treatment should be discontinued in patients who experience a stroke, TIA, or ICH while receiving vorapaxar [19]. Vorapaxar is also contraindicated in patients with any active pathological bleeding as well as those with severe hepatic impairment. There is limited clinical experience with prasugrel and no experience with ticagrelor in phase 3 studies; hence, vorapaxar should not be used in combination with these ADP P2Y12 receptor antagonists [19].
Of note, withholding vorapaxar for a brief period will not be useful in managing an acute bleeding event because of its long half-life (Sect. 2). Furthermore, there is no known treatment to reverse the antiplatelet effect of vorapaxar [19].
Local prescribing information should be consulted for full details of contraindications, warnings and precautions.
6 Current Status of Vorapaxar for the Secondary Prevention of Atherothrombotic Events
Prior to the inception of the TRA 2°P TIMI 50 trial, it had not been conclusively demonstrated that DAPT (with aspirin plus clopidogrel) was effective in reducing the risk of thrombotic events in patients with established atherosclerotic disease [18] (see Sect. 1).
Against this background, TRA 2°P TIMI 50 evaluated the efficacy of adding vorapaxar to aspirin (with or without clopidogrel) in the secondary prevention of atherothrombotic events in a broad group of 26,449 patients with established atherosclerosis (prior MI, stroke or PAD). Compared with placebo, vorapaxar significantly reduced the risk of the major composite endpoints of CV death, MI or stroke, and CV death, MI, stroke or UCR in the overall study population (Sect. 3.1.1). However, consistent with other strategies aimed at intensifying antithrombotic therapy, the addition of vorapaxar to standard therapy was associated with increased bleeding complications. Compared with placebo, vorapaxar significantly increased the risk of GUSTO moderate or severe bleeding (primary safety outcome), TIMI clinically significant bleeding and ICH in the overall trial population (Sect. 4.1).
Subgroup analyses of TRA 2°P TIMI 50 have been performed with the aim of identifying those individuals with established atherosclerosis who, on balance, may benefit from receiving vorapaxar in addition to standard antiplatelet therapy. Among the notable findings are the following:
-
In the large cohorts of post-MI-qualifying patients (≈67 % of the study population) and post-MI-qualifying patients with no history of stroke or TIA (≈64 %), vorapaxar reduced the risk of major CV events (Sect. 3.1.3). There was an accompanying increase in the risk of major bleeding events (GUSTO moderate and/or severe and/or TIMI clinically significant), but not that of ICH (Sect. 4.1);
-
In the underpowered cohort of patients with PAD-qualifying patients (≈14 %), vorapaxar did not reduce the risk of major CV events, although it did reduce the risk of limb ischaemic events (Sect. 3.1.4). There was an accompanying increase in the risk of major bleeding events, but not that of ICH (Sect. 4.1);
-
In the large cohort of post-MI- or PAD-qualifying patients with no history of CBVD (≈76 %), vorapaxar reduced the risk of major CV events (Sect. 3.1.2). There was an accompanying increase in the risk of major bleeding events, but not that of ICH (Sect. 4.1);
-
In the underpowered cohort of prior stroke-qualifying patients (≈19 %), vorapaxar did not reduce the risk of major CV events (Sect. 3.1.5). Moreover, there was an accompanying increase in the risk of major bleeding events and ICH; the latter led to the early termination of treatment in patients with a history of stroke before (or during) the trial (Sects. 3.1, 4.1).
On this basis of these results, vorapaxar has been approved as an adjunctive antiplatelet therapy for use both in the EU and USA, albeit in different patient populations: post-MI patients in the EU [19] and compared with post-MI patients or patients with PAD in the USA [29]. In both regions, however, vorapaxar is contraindicated for use in patients with a history of stroke, TIA or ICH [19, 29]. According to both drug labels [19, 29], vorapaxar increases the risk of bleeding in proportion to the patient’s underlying bleeding risk; the known risk factors for bleeding include older age, lower body weight, reduced renal or hepatic function, history of bleeding disorders and concomitant use of certain medications (including anticoagulants and fibrinolytics).
Vorapaxar is the first PAR-1 antagonist to become commercially available for the long-term secondary prevention of major CV events; it has a long half-life (Sect. 2) and only needs to be administered (orally) once daily (Sect. 5), which may be advantageous from the perspective of patient adherence to treatment in this setting. To date, only one other PAR-1 antagonist has completed phase II clinical development, namely atopaxar; phase III studies of this agent are needed to further evaluate its efficacy and tolerability (as an adjunct to standard antiplatelet therapy) in preventing recurrent ischaemic events in high-risk patients [44].
Results generally consistent to those seen in TRA 2°P TIMI 50 were also observed in TRACER, a parallel study to TRA 2°P TIMI 50, which compared vorapaxar (40 mg then 2.5 mg/day) with placebo in patients presenting with NSTE ACS receiving standard therapy (including DAPT with aspirin plus a thienopyridine) [20] (see Sect. 3.1). Vorapaxar did not significantly reduce the risk of the 2-year primary outcome (a composite of CV death, MI, stroke, recurrent ischaemia with rehospitalization, or UCR); although it did significantly reduce the risk of the major secondary endpoint (a composite of CV death, MI or stroke), superiority could not be declared, as the primary and secondary endpoints were tested in sequence. Vorapaxar also significantly increased the risk of major bleeding, including ICH; follow-up was stopped early because of bleeding [20]. Overall, the results of this trial do not support a role for adjunctive antithrombotic therapy with vorapaxar in patients presenting with NSTE ACS; the drug is not approved for use in this clinical situation [29] (Sect. 1). Whether adjunctive antithrombotic therapy with vorapaxar will improve outcomes in patients with ST-segment elevation ACS remains to be investigated [15].
In conclusion, vorapaxar can be considered for use in carefully selected patients with established atherosclerosis, i.e. those for whom the potential antithrombotic benefit outweighs the risk of bleeding. On the basis of the TRA 2°P TIMI 50 results, vorapaxar has been approved in the EU as an adjunctive treatment for the secondary prevention of atherothrombotic events in patients with prior MI who do not have a history of stroke, TIA or ICH.
Data selection sources:
Relevant medical literature (including published and unpublished data) on Vorapaxar was identified by searching databases including MEDLINE (from 1946), PubMed (from 1946) and EMBASE (from 1996) [searches last updated 31 March 2015], bibliographies from published literature, clinical trial registries/databases and websites. Additional information was also requested from the company developing the drug.
Search terms: Vorapaxar, Zontivity, MK-5348, SCH 530348.
Study selection: Studies in patients with established atherosclerotic disease who received Vorapaxar. When available, large, well designed, comparative trials with appropriate statistical methodology were preferred. Relevant pharmacodynamic and pharmacokinetic data are also included.
References
Leys D. Atherothrombosis: a major health burden. Cerebrovasc Dis. 2001;11(Suppl 2):1–4.
Steg PG, Dorman SH, Amarenco P. Atherothrombosis and the role of antiplatelet therapy. J Thromb Haemost. 2011;9(Suppl 1):325–32.
Perk J, De Backer G, Gohlke H, et al. European Guidelines on cardiovascular, disease prevention in clinical practice (version 2012). The fifth joint task force of the European society of cardiology and other societies on cardiovascular disease prevention in clinical practice. Eur Heart J. 2012;33:1635–701.
Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15.
Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–57.
Steg PG, Bhatt DL, Wilson PWF, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA. 2007;297(11):1197–206.
Domandy JA. Rationale for antiplatelet therapy in patients with atherothrombotic disease. Vasc Med. 1998;3(3):253–5.
Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–94.
Leonardi S, Tricoci P, Becker RC. Thrombin receptor antagonists for the treatment of atherosclerosis: therapeutic potential of vorapaxar and E-5555. Drugs. 2010;70(14):1771–83.
Cho JR, Rollini F, Franchi F, et al. Unmet needs in the management of acute myocardial infarction: role of novel protease-activated receptor-1 antagonist vorapaxar. Vasc Health Risk Manag. 2014;10:177–88.
Tello-Montoliu A, Tomasello SD, Ueno M, et al. Antiplatelet therapy: thrombin receptor antagonists. Br J Clin Pharmacol. 2011;72(4):658–71.
Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494–502.
Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354(16):1706–17.
Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol. 2007;49(19):1982–8.
Duerschmied D, Bode C. Vorapaxar expands antiplatelet options. Which patients may benefit from thrombin receptor antagonism? Hamostaseologie. 2012;32(3):221–7.
Martorell L, Martinez-Gonzalez J, Rodrigues C, et al. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb Haemost. 2008;99(2):305–15.
Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74(4):597–607.
Morrow DA, Scirica BM, Fox KA, et al. Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2°P)-TIMI 50 trial. Am Heart J. 2009;158(3):335–41.e3.
Merck Sharp & Dohme Ltd. Zontivity 2 mg film-coated tablets: EU summary of product characteristics. 2015. http://www.ema.europa.eu/ema. Accessed 5 Mar 2015.
Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366(1):20–33.
Chackalamannil SDR, Asberom T, Doller D, et al. A highly efficient total synthesis of (+)-himbacine. J Am Chem Soc. 1996;118:9812–3.
Doller DCS, Czarniecki M, McQuade R, et al. Design, synthesis, and structure-activity relationship studies of himbacine derived muscarinic receptor antagonists. Bioorg Med Chem Lett. 1999;9:901–6.
Kosoglou T, Reyderman L, Kasserra C, et al. No differences in the pharmacodynamics and pharmacokinetics of the thrombin receptor antagonist vorapaxar between healthy Japanese and Caucasian subjects. Eur J Clin Pharmacol. 2012;68(3):291–300.
Kosoglou T, Reyderman L, Tiessen RG, et al. Pharmacodynamics and pharmacokinetics of the novel PAR-1 antagonist vorapaxar (formerly SCH 530348) in healthy subjects. Eur J Clin Pharmacol. 2012;68(3):249–58.
Chackalamannil SDR, Wang Y, Greenlee WJ, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51(11):3061–4.
Chintala M, Kurowski S, Zhai Y, et al. SCH 530348 is a novel oral antiplatelet agent selective for protease-activated receptor-1 (PAR-1) receptor subtype without partial agonist activity. FASEB J. 2009;23(Meeting Abstract Suppl):757.6.
Storey RF, Kotha J, Smyth SS, et al. Effects of vorapaxar on platelet reactivity and biomarker expression in non-ST-elevation acute coronary syndromes. The TRACER Pharmacodynamic Substudy. Thromb Haemost. 2014;111(5):883–91.
Kosoglou T, Hunt TL, Xuan F, et al. Effect of the thrombin receptor antagonist (PAR-1) vorapaxar on QT/QTc interval in healthy volunteers: a randomized, placebo- and positive-controlled, parallel group trial. Clin Pharmacol Drug Dev. 2014;3(1):18–24.
Merck & Co. Inc. Zontivity™ (vorapaxar) tablets 2.08 mg (equivalent to 2.5 mg vorapaxar sulfate), for oral use: US prescribing information. 2014. http://www.merck.com. Accessed 11 Aug 2014.
Ghosal A, Lu X, Penner N, et al. Identification of human liver cytochrome P450 enzymes involved in the metabolism of SCH 530348 (Vorapaxar), a potent oral thrombin protease-activated receptor 1 antagonist. Drug Metab Dispos. 2011;39(1):30–8.
Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404–13.
Bonaca MP, Scirica BM, Creager MA, et al. Vorapaxar in patients with peripheral artery disease: results from TRA2°P-TIMI 50. Circulation. 2013;127(14):1522–9.
Morrow DA, Alberts MJ, Mohr JP, et al. Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke. 2013;44(3):691–8.
Scirica BM, Bonaca MP, Braunwald E, et al. Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2°P-TIMI 50 trial. Lancet. 2012;380(9850):1317–24.
Magnani G, Bonaca MP, Braunwald E, et al. Efficacy and safety of vorapaxar as approved for clinical use in the United States. J Am Heart Assoc. 2015;4:e001505.
Cavender MA, Scirica BM, Bonaca MP, et al. Vorapaxar in patients with diabetes and prior MI: findings from the TRA 2°P-TIMI 50 trial. Circulation. 2015;131:1047–53.
Bohula May EA, Bonaca MP, Scirica BM, et al. Rates of arterial revascularization in patients treated with vorapaxar vs. placebo in the TRA 2°P-TIMI 50 trial. Circulation. 2012;126(21 Suppl 1):A19221.
Bonaca MP, Scirica BM, Braunwald E, et al. Efficacy of vorapaxar is not modified by thienopyridine therapy: results from TRA 2°P-TIMI 50 trial. Circulation. 2012;126(21 Suppl 1):A18595.
Bonaca MP, Scirica BM, Braunwald E, et al. Vorapaxar reduces coronary stent thrombosis: results from the TRA2P-TIMI 50 trial. Circulation. 2013;128(22 Suppl 1):A18928.
Food and Drug Administration. FDA draft briefing document for the Cardiovascular and Renal Drugs Advisory Committee (CRDAC). Sponsor: Merck Sharp & Dohme Corp, Inc. Drug: Zontivity (vorapaxar sulfate) 2.5 mg tablets. 2014. http://www.fda.gov. Accessed 25 Mar 2015.
Kalidas C. Vorapaxar in secondary prevention of atherothrombosis. http://www.fda.gov. Accessed 14 Oct 2014.
Cardiovascular and Renal Drugs Advisory Committee Meeting. Vorapaxar advisory committee briefing document. Vorapaxar sulfate: secondary prevention of atherothrombosis in patients with a history of myocardial infarction. NDA 204886. 2014. http://www.fda.gov. Accessed 13 Oct 2014.
European Medicines Agency. Zontivity. International non-proprietary name: vorapaxar. European public assessment report. 2015. http://www.ema.europa.eu. Accessed 25 Mar 2015.
Wurster T, May AE. Atopaxar. A novel player in antiplatelet therapy? Hamostaseologie. 2012;32(3):228–33.
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process, the manufacturer of the agent under review was offered an opportunity to comment on this article. Changes resulting from comments received were made by the author on the basis of scientific and editorial merit. James Frampton is a salaried employee of Adis/Springer.
Author information
Authors and Affiliations
Corresponding author
Additional information
The manuscript was reviewed by: D. Duerschmied, Department of Cardiology and Angiology I Heart Center, University of Freiburg, Freiburg, Germany; M. Paciaroni, Stroke Unit and Division of Internal and Cardiovascular Medicine, Santa Maria della Misericordia Hospital, University of Perugia, Perugia, Italy; A Tello-Montoliu, Department of Cardiology, Hospital Clinico Universitario Virgen de la Arrixaca, Murcia, Spain.
Rights and permissions
About this article

Cite this article
Frampton, J.E. Vorapaxar: A Review of Its Use in the Long-Term Secondary Prevention of Atherothrombotic Events. Drugs 75, 797–808 (2015). https://doi.org/10.1007/s40265-015-0387-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0387-9