
Abstract
The prevalence of obesity and related comorbidities is increasing worldwide. Furthermore, clinically meaningful body weight losses has proven difficult to achieve and especially to maintain through sustained lifestyle change in the form of diet and exercise. Pharmacotherapy against obesity is a non-invasive treatment as an adjunct to lifestyle changes, but approved anti-obesity drugs are currently few. This article reviews the major anti-obesity drugs and the benefit-risk profiles of the long-acting glucagon-like peptide-1 receptor agonists (GLP-1 RAs) liraglutide and semaglutide (a modified version of liraglutide with longer half-life and tripled receptor affinity). Generally, GLP-1 RAs are well tolerated and induce significant weight loss and lowering of comorbidities. Studies with liraglutide 3.0 mg/day have shown an average placebo-subtracted weight loss of 5.5 kg (range 4.6–5.9) in 1- to 3-year duration trials. One trial using semaglutide 0.4 mg once daily reported an average weight loss of 11.6% (~ 13.1 kg) after 1 year. Furthermore, semaglutide induced a ~ 6 percentage point larger placebo-subtracted body weight loss in a head-to-head comparison with liraglutide (11.6 vs. 5.5% weight loss, respectively). The safety profiles for both drugs were similar, with transient gastrointestinal disorders being the most commonly reported adverse events. The longest running trial and the most recent trials have not raised any new safety concerns. Long-term trials and post-marketing surveillance is warranted to fully assess both long-term efficacy and safety. Future combinational therapies of mimicked gut hormones involved in regulation of energy homeostasis and/or additional lifestyle change in the form of exercise might further improve efficacy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
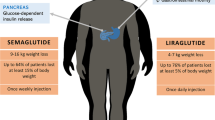
The glucagon-like peptide-1 receptor agonist liraglutide has been approved for the treatment of obesity, and studies demonstrate weight loss and a safety profile that is superior to those of other approved anti-obesity drugs. |
Semaglutide, a modified version of liraglutide, shows promise, with greater weight loss and a similar safety profile. |
1 Introduction
The number of individuals with obesity, defined as a body mass index (BMI) of > 30 kg/m2, has nearly tripled worldwide since 1975, increasing to more than 650 million or 13% of the world’s adult population as of 2016 [1]. In Europe the prevalence of adult obesity is 23% and around 36% in the USA [2]. Comorbidities associated with obesity include type 2 diabetes mellitus (T2DM), cardiovascular disease, arthritis, gallbladder disease, acute pancreatitis, non-alcoholic fatty liver disease and cancer [3, 4]. Obesity is also correlated with a decreased life expectancy, early mortality and increasing medical care costs [5, 6]. Studies have shown that a weight loss of > 5% significantly lowers comorbidities in patients with obesity [7,8,9]. The first line of treatment for obesity is lifestyle change in the form of diet and exercise; however, the success rate of maintaining a lifestyle-induced weight loss is < 10% [7, 8, 10]. To promote clinically meaningful weight loss (> 5%), pharmacotherapy is recommended as the second line of treatment and is indicated if BMI is ≥ 30 or ≥ 27 when related comorbidities to obesity are present [7, 11, 12]. Despite the increasing demand for new and improved medical options [1, 5, 6], the number of anti-obesity drugs has been reduced due to withdrawals and unfavourable benefit–risk profiles [13]. Several of these withdrawals have been subject to differences across regional medical agencies [13]. We present an overview of drug withdrawals and the anti-obesity drugs currently available.
Amphetamine-derivative compounds seem to decrease appetite and increase resting energy expenditure by increasing, in particular, norepinephrine release in the central nervous system, and became the primary drugs for obesity treatment in the 1940s and 1950s [14]. A group of these drugs (fenfluramine, dexfenfluramine, fenfluramine/phentermine) was withdrawn in the 1990s because of associated heart valve abnormalities and pulmonary hypertension [14, 15]. Phentermine as monotherapy is still approved by the US Food and Drug Administration (FDA) for short-term use only (a few weeks) [16, 17]. A new type of drug, rimonabant, which differed from these central-acting treatments by targeting the cannabinoid 1 receptor, emerged; trials reported weight loss of 4.7 kg compared with placebo groups, but the drug was never approved by the FDA and was withdrawn by the European Medicines Agency (EMA) in 2009 because of a series of adverse psychological reactions (depressed mood disorders and anxiety) [18,19,20,21]. In 2010, sibutramine, a serotonin-norepinephrine reuptake inhibitor that reduced appetite and increased energy expenditure, was withdrawn because of an increased risk of non-fatal myocardial infarction and non-fatal stroke [22,23,24,25,26]. Lorcaserin, a drug targeting the serotonin 5-hydroxytryptamine 2C receptor, which is involved in appetite control, reduced placebo-subtracted weight by 3.2 kg in trials and was approved by the FDA in 2012 [27]. However, in Europe, the approval of lorcaserin was cancelled in 2013 because of EMA concerns regarding potential carcinogenicity, psychiatric disorders and valvulopathy [28, 29]. A combination treatment consisting of phentermine and topiramate, an anti-seizure agent, was ultimately refused EMA authorisation in 2014 because of concern over long-term cardiovascular and psychiatric effects [30, 31].
Orlistat (a gastrointestinal lipase inhibitor [34]), naltrexone/bupropion (antagonist of pro-opiomelanocortin opioid auto-inhibition and stimulator of pro-opiomelanocortin neurons, respectively [35]) and liraglutide (glucagon-like peptide-1 receptor agonist [GLP-1 RA] [36]) are the only anti-obesity drugs currently approved by both the FDA and the EMA. Besides orlistat, liraglutide is the only approved anti-obesity drug that does not directly target the serotonin-noradrenaline-dopamine systems of the central nervous system, unlike many of the mentioned and withdrawn anti-obesity drugs (Table 1).
This article examines the GLP-1 RAs approved or pending approval for obesity treatment, namely liraglutide and semaglutide, from a benefit–risk perspective. We briefly describe the general mechanism of action of GLP-1 RAs and highlight the efficacy and safety data from trials of each drug.
2 Mechanism of Action of Glucagon-Like Peptide-1 Receptor Agonists in the Treatment of Obesity
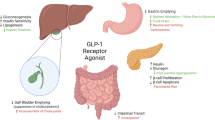
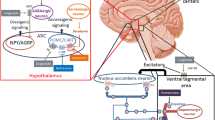
GLP-1 RAs mimic the effects of native GLP-1 hormone by activating the G protein-coupled GLP-1 receptor [37]. The GLP-1 receptor is found throughout the body, suggesting several physiological effects, with the anorexigenic (and insulinotropic) effects being the most important in the treatment of obesity [38, 39]. The anorexigenic effect mediated by the GLP-1 receptor is suggested to be due to both peripheral and central actions [40, 41]. Centrally, GLP-1 RAs may pass the blood–brain barrier and bind in hypothalamic regions, particularly the arcuate nucleus and paraventricular nucleus. In the arcuate nucleus, an indirect stimulatory effect is exerted on the orexigenic neuropeptide Y/agouti-related protein-expressing neuron group via γ-aminobutyric acid (GABA)-signalling, which may lead to appetite reduction [42,43,44]. Peripherally, it has been proposed that GLP-1 RAs also act on afferent vagal neurons [45, 46]. GLP-1 RAs also slow gastric emptying, but this effect does not seem to be the primary cause of the GLP-1 RA-induced weight loss and does not significantly change satiety or volume of fullness [47, 48]. Additionally, the weight reduction effect following GLP-1 RA treatment has been suggested to be influenced by a diminished reduction in circulating leptin following GLP-1 RA-induced weight loss, which may further increase the anorexigenic effect [49]. Evidence is currently inconsistent in demonstrating a change in energy expenditure by altered resting metabolic rate or diet-induced thermogenesis [50,51,52,53,54]. GLP-1 RAs ultimately lead to satiety, which can result in a decreased intake of food and a negative energy balance that leads to weight loss [41, 55]. Available GLP-1 RAs are exenatide, liraglutide, lixisenatide, albiglutide, dulaglutide and semaglutide, all of which are approved to treat T2DM [56, 57]. Only liraglutide is currently approved for the treatment of obesity, and semaglutide is under investigation as an obesity treatment [58, 59].
3 Methodology
We performed a literature search of PubMed (MEDLINE) on 9 November 2018 using the keywords liraglutide and obesity. Only double-blind randomised controlled trials (RCTs) designed to investigate the efficacy and safety of liraglutide 3.0 mg compared with a placebo group in overweight or obese participants aged 18–65 years were eligible for inclusion in the benefit–risk assessment. Subgroup analyses or reviews were excluded. The search yielded 217 results for liraglutide, 60 of which were clinical studies in humans. In total, 52 were discarded: 33 did not use liraglutide 3.0 mg (two did not use liraglutide), four were not RCTs, nine were subgroup analyses, five did not investigate efficacy and safety and one involved participants aged < 18 years. The only obesity RCT of semaglutide completed to date was included. Relevant RCTs and meta-analyses or reviews covering trials investigating liraglutide or semaglutide treatment compared with placebo groups in participants with T2DM were also included to assess safety and efficacy. Each risk–benefit profile was individually evaluated and discussed.
Liraglutide and semaglutide were examined using a standard benefit–risk assessment method comparing the efficacy data and safety probabilities from included trials [60]. Efficacy (placebo-subtracted weight loss in kg, rate of achieving > 5% weight loss, lowered probability of adverse events or comorbidities) was defined as the benefit component. The risk component included relevant adverse events (> 5% prevalence and between-group differences) and serious adverse events of particular interest associated with GLP-1-based therapies (pancreatitis, gallbladder-related events, cardiovascular risk, neoplasms and cancer risk, and hypoglycaemia) [61, 62]. This method was chosen to ensure the comparability of as many studies as possible.
4 Efficacy and Safety
4.1 Liraglutide
Liraglutide 3.0 mg is indicated for obesity treatment as an adjunct to diet and exercise lifestyle change [58, 70]. It is a subcutaneously administered analogue, sharing 97% homology of endogenously produced GLP-1 secreted by enteroendocrine L-cells in the gut [36, 38]. Native GLP-1 concentration typically increases postprandially but only exists in the human body for 1–2 min [38]. Liraglutide has thus been acylated to increase the plasma half-life to 12–13 h, requiring a daily dosing schedule, and up to 3.0 mg is approved for the treatment of obesity [36, 58].
Five of the included trials were phase III trials, the first being from 2009 with an extension published in 2012 [63,64,65,66,67,68,69]. Four are a part of the multicentre SCALE (Satiety and Clinical Adiposity—Liraglutide Evidence) programme, funded by Novo Nordisk A/S [65,66,67,68,69]. The most recent study, by O’Neil et al. [59], compared liraglutide and semaglutide, and the data are therefore presented in both the liraglutide and the semaglutide tables. Anthropometric and efficacy data are presented in Tables 2 and 3.
In all trials, participants followed a 4-week titration period followed by a constant dose period. From randomisation to completion, participants were instructed to follow a 500 kcal/day deficit of their individually calculated daily basal metabolic rate and were also counselled to maintain or increase physical activity to > 150 min/day. Wadden et al. [65] was the only trial with an initial low-calorie diet run-in period (1200–1400 kcal/day for 4–12 weeks), which had to lead to a weight loss of > 5% before randomisation. In the Davies et al. [66] trial, 88.8% of the participants randomised to liraglutide 3.0 mg and 90.5% of those in the placebo group were receiving one to three agents (metformin, sulfonylurea and glitazone), with the distribution of agents similar between the groups. Participants in Astrup et al. [64] were randomised into six trial arms for 1 year. All groups (except the orlistat group) were then pooled in a liraglutide 2.4 mg group that was later increased to 3.0 mg and completed after an additional year. The data presented in Table 2 are from the liraglutide 3.0 mg group before pooling (from week 0 to 52).
The placebo and liraglutide groups were anthropometrically similar within each trial in terms of baseline BMI and body weight. The placebo-subtracted weight losses were dependent on the length of the trial: the lowest weight loss, 4.4 kg, was seen in the 20-week trial, and the highest was seen in the trials that were longer than 6 months, demonstrating weight losses from 4.6 to 5.9 kg (p < 0.001 or lower). These placebo-subtracted weight losses are similar to those with naltrexone/bupropion (5.0 kg) and modest in comparison with EMA-withdrawn phentermine/topiramate (8.8 kg), see Table 1. A significantly higher mean weight loss of 3.8 kg (95% confidence interval [CI] 1.6–6.0; p < 0.0001) after 1 year of treatment was reported in a head-to-head comparison with orlistat [64]. In all studies, participants randomised to liraglutide treatment had a significantly higher chance of achieving a > 5% weight loss after randomisation than those in placebo groups, ranging from 46 to 76% and from 19 to 30%, respectively. In Pi-Sunyer et al. [67], 33.1% (liraglutide) versus 10.6% (placebo) had a weight loss of > 10% (p < 0.001), indicating that one-third of participants responded very well. Early responders have been defined as participants achieving a weight loss of > 5% at 12 weeks of treatment (not including titration period), and they are predicted to have a mean weight loss of 11.2% of their baseline body weight after 1 year of treatment [70]. In Pi-Sunyer et al. [67], 63.2% of participants were early responders. The studies of ≥ 1 year demonstrated a stabilisation of body weight in the liraglutide group after 35–40 weeks (except in Wadden et al. [65], where stabilisation occurred as early as after 20 weeks of treatment) until discontinuation of treatment where follow-up analyses found a regain of body weight in liraglutide groups [69].
Liraglutide also significantly improved other comorbidities compared with placebo in the Astrup et al. [64] trial; after 1 year of treatment with liraglutide 3.0 mg, the percentage of patients with pre-diabetes decreased from 31 to 10% compared with an increase from 36 to 37% with placebo. Liraglutide treatment also significantly lowered the prevalence of pre-diabetes compared with orlistat; 52–62% of participants with pre-diabetes treated with liraglutide returned to normal glucose tolerance after 2 years compared with 26% of those treated with orlistat [64].
4.1.1 General Observations and Adverse Events
The percentages of adverse events reported in > 5% of participants were lower in the placebo groups (range 47–79) than in the liraglutide groups (range 67–88) across all trials. Overall, withdrawal rates were comparable between liraglutide and placebo, with more withdrawals due to adverse events with liraglutide. Withdrawal rates due to adverse events in liraglutide groups were higher than with placebo in seven of eight trials. The event rate per 100 years of observation was higher with liraglutide in trials with an available value, indicating greater exposure to adverse events over time [59, 65, 68, 69]. The reports and distributions of adverse events in a review of the T2DM phase III trials using liraglutide 1.8 mg (LEAD trials) were similar [71].
The most common adverse events following liraglutide treatment were gastrointestinal disorders that were mild to moderate in severity and often presented in the titration period. Commonly reported events included nausea, vomiting, constipation, diarrhoea, dyspepsia and abdominal discomfort, of which nausea was the most common. In the Astrup et al. [64] trial, participants reporting nausea and/or vomiting had a higher mean weight loss than those without nausea and/or vomiting (10.0 vs. 7.1 kg, estimated difference 2.9 kg; 95% CI 0.5–5.3; p = 0.02). Gastrointestinal events reported in the first 1–6 weeks were often the cause for withdrawal [65, 67, 69]. In le Roux et al. [69], 8% of participants treated with liraglutide versus 2% with placebo withdrew because of gastrointestinal events. Two very commonly occurring events in both groups were nasopharyngitis and headache.
Several of the trials showed a significantly increased mean heart rate of approximately 2 beats/min during liraglutide treatment [66,67,68,69]. Blackman et al. [68] also reported an increased heart rate but a normal decrease in nocturnal heart rate. le Roux et al. [69] reported a decreased number of cardiovascular adverse events in the liraglutide group than in the placebo group (16 vs. 19%).
No apparent distribution or clustering between groups was seen in the few psychiatric disorders reported and according to mental health questionnaires administered in two of the trials [65, 69].
4.1.2 Serious Adverse Events of Interest
The longest running trial, 160 weeks [69], and the most recent trial [59] did not raise any new safety concerns. The percentage of participants experiencing any serious adverse events in the liraglutide groups was slightly higher than in the placebo groups in all trials, except the Astrup et al. [63] and O’Neil et al. [59] trials (in which the prevalence was higher in the placebo group). In Blackman et al. [68] and Davies et al. [66], the reported serious adverse events were single cases and with no apparent distribution within organ classes. O’Neil et al. [59] found a higher prevalence of allergic reactions (13% with liraglutide vs. 7% with placebo), but this was not found in other trials; the LEADER (Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes) trial found no significant difference in allergic reactions between groups (1.3% with liraglutide, 0.9% with placebo; p < 0.14) [72].
4.1.2.1 Pancreatitis
In le Roux et al. [69], the prevalence of pancreatitis was marginally higher in the liraglutide group (0.7%) than in the placebo group (0.3%) and was generally low in the shorter Pi-Sunyer et al. [67] trial (0.4 vs. < 0.1%, respectively). No reports of pancreatitis were found in the Wadden et al. [65], Blackman et al. [68] or Davies et al. [66] trials. Pi-Sunyer et al. [67] found that 2.5% of participants receiving liraglutide versus 1.1% of those receiving placebo had lipase levels three times higher than the upper limit of normal, but this finding had a low positive predictive value for pancreatitis in the trial (< 1%). Elevated lipase levels were also seen in Davies et al. [66]. An increase in serum lipase above three times the upper limit of normal is often used to guide the diagnosis of acute pancreatitis but is not a confirmation of the diagnosis [73]. Pooled data from the SCALE trials showed that increases in amylase/lipase activity did not predict acute pancreatitis onset, and FDA and EMA assessments have suggested that increases in lipase and amylase levels are not predictive of pancreatitis [74, 75]. The LEADER trial, which studied liraglutide 1.8 mg, found no significant difference in acute pancreatitis (0.4% with liraglutide vs. 0.5% with placebo; p = 0.44, total n = 9340, median follow-up 3.8 years) [72].
4.1.2.2 Gallbladder-Related Events
Gallbladder-related events, specifically acute cholecystitis and cholelithiasis, were also reported more frequently with liraglutide than with placebo by Pi-Sunyer et al. [67] (2.5 vs. 1.0%). These rates increased to 5 versus 2%, respectively, in the le Roux et al. [69] trial. Weight loss among participants with gallbladder-related events was larger than the overall mean weight loss and therefore might account for the differences, since rapid weight loss is associated with an increased risk of gallbladder-related events [67, 69, 76]. Furthermore, liraglutide has been shown to affect gallbladder motility, but a connection between GLP-1 RAs and gallbladder-related adverse events has not been fully established [77, 78]. Similarly, this discrepancy between events with liraglutide versus placebo was also seen in the LEADER trial, where liraglutide induced a 2.3 kg placebo-subtracted weight loss [72].
4.1.2.3 Hypoglycaemia
In Astrup et al. [64], 13 self-reported symptomatic hypoglycaemia events were recorded, 12 of which were from participants treated with liraglutide. In Pi-Sunyer et al. [67], the prevalence of hypoglycaemia was 1.3% with liraglutide and 1.0% with placebo, and none of the events were severe. In the Davies et al. [66] trial, more hypoglycaemic events were reported by patients with T2DM treated with liraglutide than by those with placebo, but the overall glycaemic control was significantly better, showing a reduction in participants’ net use of concomitant oral hypoglycaemic agents compared with placebo (13.1% with liraglutide vs. 5.7% with placebo decreased net use, odds ratio 5.63; 95% CI 3.62–8.76; p < 0.001). The severe cases of hypoglycaemia reported in the liraglutide group were all from participants receiving concomitant sulfonylurea treatment [66].
4.1.2.4 Neoplasms and Cancer
Trials of GLP-1 RAs have indicated that they increase calcitonin release and C-cell proliferation in the thyroid glands of mice, and C-cell tumours and medullary thyroid carcinomas in rodents are still mentioned in the FDA drug information for liraglutide 3.0 mg [58, 79]. None of the SCALE trials were powered sufficiently to determine carcinogenic effects. However, none [65,66,67,68] reported increased calcitonin levels or instances of medullary thyroid carcinoma. The LEADER trial, involving 9340 participants, was not powered to determine the effect of liraglutide on cancer risk, but it did not report any episodes of C-cell hyperplasia or medullary thyroid carcinoma or a significant difference in neoplasms in the liraglutide group [72]. Similar rates of neoplasms were generally reported or adjudicated between trial arms [59, 65, 66, 68], but Pi-Sunyer et al. [67] observed a higher incidence of malignant and pre-malignant breast neoplasms in the liraglutide groups (ten events in nine women with liraglutide vs. three events in three women with placebo; 56 weeks), as did le Roux et al. [69] (ten events in nine women with liraglutide vs. none with placebo; 160 weeks), with the greatest number of reports in women who had lost more body weight than the average compared with other women. This may suggest that some of the breast neoplasms could have been present before treatment but became visible with weight loss.
4.1.3 Cardiovascular Outcomes
Rates of serious adverse cardiovascular events were similar between groups in the le Roux et al. [69] trial. In the LEADER trial, the primary composite outcome was the first event (time-to-event analysis) of death from cardiovascular causes, non-fatal myocardial infarction or non-fatal stroke. The primary composite outcome occurred in significantly fewer participants treated with liraglutide (13.0 vs. 14.9% with placebo, hazard ratio [HR] 0.87; 95% CI 0.78–0.97; p < 0.001 for noninferiority, p = 0.01 for superiority) [72]. The LEADER findings count as a benefit in the assessment of the treatment and, in 2017, were deemed so significant that the EMA updated the liraglutide 3.0 mg prescription label with the 1.8 mg findings on lowered cardiovascular risk [80]. Systolic blood pressure was also reduced significantly more than in placebo groups, with Pi-Sunyer et al. [67] reporting a decrease of 2.80 mmHg (95% CI 2.09–3.56; p < 0.001). Results from trials have been inconsistent in demonstrating a significant decrease in diastolic blood pressure [59, 63,64,65,66,67,68,69].
4.2 Semaglutide
Semaglutide shares 94% homology with native GLP-1 and was developed from liraglutide in an attempt to increase the half-life while maintaining the physiological effects of liraglutide [81]. An amino acid substitution protects semaglutide from dipeptidyl-peptidase-4 degradation, the acylation has been modified and a more flexible linker has been used [81]. These changes have increased the affinity of semaglutide to the GLP-1 receptor threefold compared with liraglutide and increased its half-life to 165 h in humans [81]. The FDA (2017) and the EMA (2018) both approved subcutaneous semaglutide to a maximum of 1.0 mg for the treatment of T2DM [57, 82], but it is not yet approved for the treatment of obesity. Semaglutide trials have shown dose-dependent weight loss and adverse events [59, 83]. One dose-finding trial comparing semaglutide, liraglutide and placebo has been completed [59]. Four phase III studies (STEP 1–4) are set for completion in 2020: a weight loss study, a non-insulin T2DM study, a maximising weight loss study and a weight loss maintenance study [84]. Furthermore, as required by FDA guidelines for pre-approval and post-approval for drugs managing glycaemic regulation, Novo Nordisk A/S plans to conduct a landmark cardiovascular trial that is estimated for completion in 2023 (SELECT [Semaglutide Effects on Cardiovascular Outcomes in People with Overweight or Obesity]) [85]. The trial is set to include 17,500 participants and will compare daily subcutaneous injections of semaglutide versus placebo for 31–59 months in non-diabetic individuals with prior cardiovascular disease [85].
Table 4 presents the anthropometric and efficacy data for the highest dose of semaglutide from the completed phase II trial with non-diabetic participants with obesity. Additionally, trials using semaglutide as monotherapy or as an add-on to other standard hypoglycaemic agents in participants with T2DM are included: two phase III trials (one monotherapy trial [SUSTAIN-1] and one add-on trial [SUSTAIN-6]), and a trial comparing the subcutaneous and oral analogue.
All trials had a titration period; in the diabetes trials by Marso et al. [87] and Davies et al. [88], background treatment with standard hypoglycaemic agents was continued, whereas O’Neil et al. [59] and Sorli et al. [86] used semaglutide as monotherapy. The variations in background treatment, combined with the varying doses and once-daily/once-weekly administration, may explain the differences in efficacy between trials: weight loss of 11.6% with semaglutide 0.4 mg once daily [59], 3.6 kg with subcutaneous semaglutide 1.0 mg once weekly [86] and 5.7 kg with oral semaglutide 40 mg once daily [88].
The trials involving patients with T2DM were anthropometrically similar but the results differed from those reported in the obesity trial by O’Neil et al. [59]. The diabetes trials demonstrated a placebo-subtracted weight loss of 3.6–5.7 kg, whereas the obesity trial demonstrated a 11.6% weight loss, which was noticeably higher than the 5.5% weight loss seen with liraglutide 3.0 mg in the same trial. The participants receiving oral semaglutide 40 mg in the Davies et al. [88] trial lost numerically more placebo-subtracted weight than the participants receiving subcutaneous semaglutide 1.0 mg (6.2% with oral semaglutide 40 mg vs. 5.9% with subcutaneous semaglutide 1.0 mg), but no significant difference between the two groups was seen. Overall, significantly more participants treated with semaglutide achieved a > 5% weight loss versus placebo.
The estimated treatment difference between semaglutide 0.4 mg and liraglutide 3.0 mg was − 6.1% body weight (95% CI − 8.4 to − 3.8; p < 0.0001), strongly favouring semaglutide [59]. Furthermore, the group receiving semaglutide 0.4 mg experienced a significant reduction in glycated haemoglobin (HbA1c: − 0.29 ± 0.03% standard deviation; p < 0.0001), but no change was seen in the placebo group [59]. Marso et al. [87] treated patients with T2DM and reported an HbA1c of 8.7 ± 1.5% for both the semaglutide 1.0 mg and the placebo groups; semaglutide 1.0 mg significantly lowered HbA1c (1.1%; 95% CI 0.9–1.2; p < 0.0001) compared with placebo. Significantly more participants receiving placebo than those receiving semaglutide 1.0 mg needed additional treatment with hypoglycaemic agents (39 vs. 20%, respectively) [87]. In Davies et al. [88], more than 90% of participants treated with oral semaglutide 40 mg or subcutaneous semaglutide 1.0 mg achieved an HbA1c of < 7% (p < 0.001) versus 28% with placebo.
4.2.1 General Observations and Adverse Events
Overall withdrawal rates were similar in all trials except for Davies et al. [88]. This might be because of the relatively low number of participants in the randomised groups, making this study more prone to greater percentage point shifts. Withdrawals due to adverse events were more frequent with semaglutide (range 5–23% vs. 1–8% with placebo) in all trials (see Table 5). The overall withdrawal rate in O’Neil et al. [59] did not show an association with different semaglutide doses. The adverse event rate per 100 years of observation in O’Neil et al. [59] was 743 with semaglutide and 485 with placebo, indicating greater exposure to adverse events over time with semaglutide. These general observations were similar to those with liraglutide.
In both obesity and T2DM trials, the distribution of adverse events was similar between liraglutide and semaglutide [59, 65,66,67,68,69, 71]. The most common adverse events were gastrointestinal disorders, which were largely mild to moderate (range 15–38% with placebo, 38–82% with semaglutide). In Marso et al. [87], Sorli et al. [86] and O’Neil et al. [59], most adverse event-related withdrawals were due to gastrointestinal events, commonly reported as nausea, vomiting, and constipation or diarrhoea, with nausea the most common. For semaglutide, similarly to liraglutide, nausea was mostly present in the titration period and diminished over time [59, 86, 88]. Treatment with semaglutide also induced an increase in mean resting heart rate of approximately 2–3 beats/min compared with placebo [86, 87].
4.2.2 Serious Adverse Events of Interest
No unexpected safety concerns were identified in any of the trials [59, 86,87,88]. The percentage of participants reporting any serious adverse event was higher with semaglutide than with placebo in Sorli et al. [86] and O’Neil et al. [59] but lower in Marso et al. [87] and Davies et al. [88]. In particular, compared with the other trials, the rate of serious adverse events was unusually high for both placebo and semaglutide in Marso et al. [87] (36 vs. 34%, respectively).
4.2.2.1 Pancreatitis and Gallbladder-Related Events
Only O’Neil et al. [59] reported a difference in gallbladder-related events (cholelithiasis and cholecystitis; concurrent with five adjudicated pancreatitis events) with semaglutide 0.4 mg versus placebo (6 vs. 4%); no apparent relationship was seen between the groups. Marso et al. [87] found no imbalance of pancreatitis and gallbladder-related events between semaglutide and placebo.
4.2.2.2 Hypoglycaemia and Retinopathy
Hypoglycaemia events were generally few across trials and evenly distributed between groups, as seen in Marso et al. [87] in patients with T2DM (21.7% with semaglutide vs. 21.0% with placebo). In Marso et al. [87], 83.5% of participants had pre-existing retinopathy complications, evenly distributed between groups; however, retinopathy complications occurred in 3.0% of participants treated with semaglutide and in 1.8% of those treated with placebo (HR 1.76; 95% CI 1.11–2.78; p = 0.02). These findings are dissimilar to those from other human and animal trials. A meta-analysis by Dicembrini et al. [89] found that treatment with GLP-1 RAs (pooled data from exenatide, liraglutide, lixisenatide, albiglutide, dulaglutide and semaglutide trials) in patients with diabetes did not significantly increase the incidence of retinopathy (Mantel–Haenszel odds ratio 0.92; 95% CI 0.74–1.16; p = 0.49) [89]. GLP-1 receptor activation has also been shown to prevent retinal neurodegeneration in diabetic mice [90].
4.2.2.3 Neoplasms and Cancer
Calcitonin levels were low with both semaglutide and placebo in Sorli et al. [86], and none of the trials reported instances of thyroid carcinoma. Nevertheless, the FDA still cautions against the same adverse events as they do for liraglutide 3.0 mg [82]. In Sorli et al. [86], singular cases of malignant neoplasms were seen in the semaglutide 1.0 mg group, and even distributions of neoplasms were seen in O’Neil et al. [59]. In the larger Marso et al. [87] trial, the distribution of malignant neoplasms was similar between the semaglutide and placebo groups (HR 0.94; 95% CI 0.67–1.32; data from 1648 participants treated with semaglutide 0.5 mg or 1.0 mg and 1649 participants receiving placebo), whereas the rate of pancreatic cancer was higher with placebo [87].
4.2.3 Cardiovascular Outcomes
In the Marso et al. [87] trial, which investigated cardiovascular risks, mean systolic pressure was lowered by 2.6 mmHg with semaglutide compared with placebo (p < 0.001), and the primary composite outcome (first occurrence of cardiovascular death, non-fatal myocardial infarction or non-fatal stroke) was significantly lower in participants receiving semaglutide 0.5 mg or 1.0 mg versus placebo (6.6% with semaglutide vs. 8.9% with placebo; HR 0.74; 95% CI 0.58–0.95; p < 0.001 for non-inferiority of semaglutide). This was a drug benefit similar to that reported in the LEADER trial [72].
5 Discussion
Semaglutide induced a larger weight loss than liraglutide (− 6.1%; 95% CI − 8.4 to − 3.8; p < 0.0001) and unlike other obesity drugs, the weight loss with higher doses of semaglutide seemed to continue throughout the 52-week trial by O’Neil et al. [59] instead of plateauing. Therefore it will be interesting to see whether these results are similar in the STEP trials. The prospect of improving drug delivery with once-weekly oral administration with semaglutide instead of daily subcutaneous injections is also encouraging, as it may improve drug compliance. Furthermore, additional exercise combined with GLP-1 analogue treatment may have beneficial effects on weight loss and health. Mensberg et al. [91] showed that, in patients with T2DM, liraglutide 1.8 mg plus exercise for 16 weeks compared with placebo plus exercise significantly improved fasting glucose (− 3.4 vs. − 0.3 mM; p < 0.001) and systolic blood pressure (− 5.4 vs. − 0.6 mmHg; p < 0.01) [91]. Thus, investigating the benefit of longer term controlled exercise with liraglutide treatment versus liraglutide or exercise alone in weight loss management is warranted.
When compared with orlistat, liraglutide induced a significantly greater weight loss (3.8 kg; 95% CI 1.6–6.0; p < 0.0001) [64]. Naltrexone/bupropion trials compared with liraglutide trials have shown similar weight loss; however, the beneficial effects on reversal of pre-diabetes and lowering of cardiovascular risk factors favour liraglutide [33, 72, 92]. A trial investigating the cardiovascular risks of naltrexone/bupropion, set to enrol 8910 participants, was terminated prematurely after a breach of confidentiality, so no solid assessment of its safety can yet be made [92]. The cardiovascular outcomes trial investigating lorcaserin reported a placebo-subtracted weight loss of 2.8 kg (95% CI 2.6–3.0) and no significant increase in major cardiovascular events (HR 0.99; 95% CI 0.85–1.14; p < 0.001 for noninferiority) after 1 year [93]. While no equivalent cardiovascular risk trial has investigated liraglutide 3.0 mg in non-diabetic participants, both efficacy and safety results for lorcaserin seem inferior to those for liraglutide 3.0 mg. Head-to-head trials have not been performed between liraglutide and naltrexone/bupropion or lorcaserin to compare their efficacy and safety.
Overall, the liraglutide and semaglutide safety outcomes reviewed in this article were similar. Trials of both drugs have shown safety similar to that of other GLP-1 RAs in the treatment of T2DM: In a meta-analysis by Bethel et al. [94] of the cardiovascular outcome trials ELIXA (lixisenatide), LEADER (liraglutide), SUSTAIN-6 (semaglutide) and EXSCEL (extended-release exenatide), GLP-1 RAs compared with placebo showed a significant 10% reduction in the primary composite outcome (death from cardiovascular causes, non-fatal myocardial infarction or non-fatal stroke; HR 0.90; 95% CI 0.82–0.99; p = 0.033), a 13% reduction in cardiovascular death (HR 0.87; 95% CI 0.79–0.96; p = 0.007) and a 12% reduction in all-cause mortality (HR 0.88; 95% CI 0.81–0.95; p = 0.002) [94]. Furthermore, the meta-analysis found no significant differences in hypoglycaemia, pancreatitis, pancreatic cancer or medullary thyroid cancer [94]. Another study of the cardiovascular outcome trials using restricted mean survival time instead of HRs also found a reduction in cardiovascular risk with liraglutide and semaglutide [95]. These lowered risks count as benefits in the evaluation of GLP-1 RAs. Nevertheless, these positive findings are still to be established in larger, sufficiently powered long-term safety trials in non-diabetic individuals and with doses suitable for the treatment of obesity, alongside post-marketing surveillance [13].
In the Look AHEAD trial, which randomised 5145 obese participants with T2DM into an intervention group (intensive lifestyle counselling and increased physical activity) and a control group, a significant difference in percentage body weight loss was seen throughout the trial (8.5 vs. 0.6% after 1 year and 4.7 vs. 2.1% after 8 years, respectively), but no significant reduction in the rate of cardiovascular events was seen (intervention group HR 0.95; 95% CI 0.83–1.09, p = 0.51; 1.83 and 1.92 events per 100 person-years in the intervention and control groups, respectively) [96]. Thus, the cardiovascular risk reduction induced by liraglutide treatment seen in, for example, Pi-Sunyer et al. [67], le Roux et al. [69] or Marso et al. [87] seems to exceed the risk reduction potential from weight loss alone [8, 67, 69, 72]. Therefore, the complications associated with successful therapy seem to be outweighed by the potential for a decrease in cardiac events that exceeds the potential for weight loss alone.
In a clinical setting, the current prices for liraglutide in obesity treatment are substantially high, estimated at around $US1200 per month of treatment at the highest dosage [97]. While some insurance and other savings plans exist (depending on country), it must be expected that the prices will discourage some potential patients. A cost-effectiveness meta-analysis of RCTs has been performed on various non-surgical weight loss strategies, including liraglutide 3.0 mg [97]. Average cost-effectiveness ratios were calculated from 12 months of treatment in US dollars and weight loss in kilograms; 1 kg of weight loss cost an average of $US2102 with liraglutide 3.0 mg, $US823 with lorcaserin, $US541 with naltrexone/bupropion, $US327 with phentermine/topiramate, $US251 with orlistat, and $US134 with Weight Watchers (weekly lifestyle counselling), (perscription prices obtained in 2017 and Weight Watchers-program in 2018) [97]. A real-life, open-label cost-effectiveness RCT of liraglutide 3.0 mg (STRIVE [Saxenda in Obesity Services] study) is currently recruiting participants with complex obesity (BMI ≥ 35 and one or more comorbidities; prediabetes, T2DM, hypertension or sleep apnoea) and is set for completion in 2021. The trial is planned to investigate cost-effectiveness and budget implications by targeting early responders by investigating standard care consisting of obesity-specialist care compared with obesity specialist care plus liraglutide treatment and pre-specified stopping rules for the medication [98]. Targeting early responders and strict stopping rules might be a method to decrease the average cost-effectiveness ratio and avoid unneccesary exposure of liraglutide treatment. Currently, the EMA recommends discontinueing treatment if not ≥ 5% of initial body weight has been lost after 12 weeks [70].
GLP-1 RAs are also thought to have effects on other organ systems [41]. A direct cardioprotective effect has been seen in mice via remote ischaemic conditioning, and a neuroprotective effect has also been proposed but has yet to be established in humans [99, 100]. Another study has shown a direct anti-inflammatory effect in humans treated with lixisenatide via macrophage phenotype changes [101]. The effects are not yet fully established and remain to be determined on a larger scale in humans but are potentially beneficial.
Results from gut hormonal changes after bariatric surgery have provided insight into several gut hormones and their relevance in energy homeostasis and metabolism, e.g. GLP-1, gastric inhibitory peptide (GIP), cholecystokinin, pancreatic polypeptide, peptide YY, oxyntomodulin, and the orexigenic hormone ghrelin [102, 103]. Future treatments mimicking endogenous gut hormones and potentially combining them (i.e. dual agonism by GLP-1/GIP or triple agonism by GLP-1/oxyntomodulin/peptide YY) might augment efficacy [104, 105].
Note that all of the trials included in Tables 2 and 4 were funded by Novo Nordisk A/S, which has patent and commercial interests in liraglutide and semaglutide.
6 Conclusion
Obesity is an increasing problem worldwide and requires new and improved treatment options. Mimicking gut hormones marks a new era of anti-obesity drugs. So far, GLP-1-based therapy in the form of liraglutide has overall tolerable risks and mainly mild and transient adverse events. The efficacy of liraglutide is comparable to that of other anti-obesity drugs and significantly increases the chance of achieving and maintaining clinically meaningful weight loss. Overall, the benefit–risk profile of liraglutide in the treatment of obesity is favourable and superior to other approved anti-obesity drugs. The completed phase II obesity trial of semaglutide demonstrates more promising weight loss than liraglutide with a similar safety profile, which will be reassessed in upcoming phase III trials and a large cardiovascular risk trial. The potential for a decrease in cardiac events seems to outweigh the complications of successful therapy and therefore exceed the potential for weight loss alone. Finally, long-term trials and post-marketing surveillance are required to determine the long-term safety of the new anti-obesity drugs.
Change history
30 April 2019
In the original publication of this article, the following correction should be noted in Table 1.
References
World Health Organization. Obesity and overweight. 2018. http://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 28 Mar 2019.
World Health Organization. Prevalence of obesity among adults, BMI ≥ 30, age-standardized Estimates by WHO region. 2017. http://apps.who.int/gho/data/view.main.REGION2480A?lang=en. Accessed 28 Mar 2019.
Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9:88. https://doi.org/10.1186/1471-2458-9-88.
Pi-Sunyer X. The medical risks of obesity. Postgrad Med. 2009;121(6):21–33. https://doi.org/10.3810/pgm.2009.11.2074.
Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Affairs (Project Hope). 2009;28(5):w822–31. https://doi.org/10.1377/hlthaff.28.5.w822.
Peeters A, Barendregt JJ, Willekens F, Mackenbach JP, Al Mamun A, Bonneux L. Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Ann Intern Med. 2003;138(1):24–32.
Jensen MD, Ryan DH, Apovian CM, Ard JD, Comuzzie AG, Donato KA et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol. 2014;63(25 Pt B):2985–3023. https://doi.org/10.1016/j.jacc.2013.11.004.
The Look AHEAD Research Group. Eight-year weight losses with an intensive lifestyle intervention: the look AHEAD study. Obesity (Silver Spring, Md). 2014;22(1):5–13. https://doi.org/10.1002/oby.20662.
Wing RR, Lang W, Wadden TA, Safford M, Knowler WC, Bertoni AG, et al. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care. 2011;34(7):1481–6. https://doi.org/10.2337/dc10-2415.
Fildes A, Charlton J, Rudisill C, Littlejohns P, Prevost AT, Gulliford MC. Probability of an obese person attaining normal body weight: cohort study using electronic health records. Am J Public Health. 2015;105(9):e54–9. https://doi.org/10.2105/AJPH.2015.302773.
Fried M, Yumuk V, Oppert JM, Scopinaro N, Torres A, Weiner R, et al. Interdisciplinary European guidelines on metabolic and bariatric surgery. Obes Surg. 2014;24(1):42–55. https://doi.org/10.1007/s11695-013-1079-8.
Garvey WT, Mechanick JI, Brett EM, Garber AJ, Hurley DL, Jastreboff AM, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Comprehensive Clinical Practice Guidelines for medical care of patients with obesity. Endocr Pract. 2016;22(Suppl 3):1–203. https://doi.org/10.4158/EP161365.GL.
Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of anti-obesity medicinal products because of adverse drug reactions: a systematic review. BMC Med. 2016;14(1):191. https://doi.org/10.1186/s12916-016-0735-y.
Haslam D. Weight management in obesity—past and present. Int J Clin Pract. 2016;70(3):206–17. https://doi.org/10.1111/ijcp.12771.
Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337(9):581–8. https://doi.org/10.1056/nejm199708283370901.
US Food and Drug Administration. Full prescribing information Suprenza (phentermine hydrochloride) orally disintegrating tablet. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202088s005lbl.pdf. Accesssed 28 Mar 2019.
European Medicines Agency. The European Agency for the Evaluation of Medicinal Products. 1999. https://www.ema.europa.eu/documents/press-release/extraordinary-meeting-finalise-review-anorectic-agents_en.pdf. Accessed 28 Mar 2019.
Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365(9468):1389–97. https://doi.org/10.1016/s0140-6736(05)66374-x.
Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370(9600):1706–13. https://doi.org/10.1016/s0140-6736(07)61721-8.
European Medicines Agency. Acomplia. https://www.ema.europa.eu/en/medicines/human/EPAR/acomplia. Accessed 28 Mar 2019.
European Medicines Agency. Zimulti. https://www.ema.europa.eu/medicines/human/EPAR/Zimulti. Accessed 28 Mar 2019.
Rolls BJ, Shide DJ, Thorwart ML, Ulbrecht JS. Sibutramine reduces food intake in non-dieting women with obesity. Obes Res. 1998;6(1):1–11.
Luque CA, Rey JA. The discovery and status of sibutramine as an anti-obesity drug. Eur J Pharmacol. 2002;440(2):119–28. https://doi.org/10.1016/S0014-2999(02)01423-1.
European Medicines Agency. European Medicines Agency recommends suspension of marketing authorisation for sibutramine. 2010. https://www.ema.europa.eu/news/european-medicines-agency-recommends-suspension-marketing-authorisation-sibutramine?fbclid=IwAR0j-JZEdrJ3qbW2iDOGiVJoiIPZVKuJSZkV5j9a438Lt9h6O6lMP0c34qA. Accessed 28 Mar 2019.
US Food and Drug Administration. FDA Drug Safety Communication: FDA Recommends Against the Continued Use of Meridia (sibutramine). 2010. https://www.fda.gov/Drugs/DrugSafety/ucm228746.htm. Accessed 28 Mar 2019.
James WP, Caterson ID, Coutinho W, Finer N, Van Gaal LF, Maggioni AP, et al. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med. 2010;363(10):905–17. https://doi.org/10.1056/NEJMoa1003114.
US Food and Drug Administration. Center for Drug Evaluation and Research 208524Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208524Orig1s000Approv.pdf. Accessed 28 Mar 2019.
European Medicines Agency. Withdrawal assessment report for Belviq. 2013. https://www.ema.europa.eu/documents/withdrawal-report/withdrawal-assessment-report-belviq_en.pdf. Accessed 28 Mar 2019.
Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374(6522):542–6. https://doi.org/10.1038/374542a0.
Porter RJ, Dhir A, Macdonald RL, Rogawski MA. Chapter 39—mechanisms of action of antiseizure drugs. In: Stefan H, Theodore WH, editors. Handbook of clinical neurology. Amsterdam: Elsevier; 2012. p. 663–81.
European Medicines Agency. Refusal of the marketing authorisation for Qsiva (phentermine / topiramate). 2013. https://www.ema.europa.eu/documents/smop-initial/questions-answers-refusal-marketing-authorisation-qsiva-phentermine/topiramate_en.pdf. Accessed 28 Mar 2019.
Arterburn DE, Crane PK, Veenstra DL. The efficacy and safety of sibutramine for weight loss: a systematic review. Arch Intern Med. 2004;164(9):994–1003. https://doi.org/10.1001/archinte.164.9.994.
Khera R, Murad MH, Chandar AK, Dulai PS, Wang Z, Prokop LJ, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA. 2016;315(22):2424–34. https://doi.org/10.1001/jama.2016.7602.
Davidson MH, Hauptman J, DiGirolamo M, et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat: a randomized controlled trial. JAMA. 1999;281(3):235–42. https://doi.org/10.1001/jama.281.3.235.
Apovian CM, Aronne L, Rubino D, Still C, Wyatt H, Burns C, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring, Md). 2013;21(5):935–43. https://doi.org/10.1002/oby.20309.
Agerso H, Jensen LB, Elbrond B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45(2):195–202. https://doi.org/10.1007/s00125-001-0719-z.
Madsbad S, Holst JJ. Treatment with GLP-1 receptor agonists. In: Bonora E, DeFronzo R, editors. Diabetes. Epidemiology, genetics, pathogenesis, diagnosis, prevention, and treatment. Cham: Springer International Publishing; 2018. p. 1–45.
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–39. https://doi.org/10.1152/physrev.00034.2006.
Torekov SS, Madsbad S, Holst JJ. Obesity—an indication for GLP-1 treatment? Obesity pathophysiology and GLP-1 treatment potential. Obes Rev. 2011;12(8):593–601. https://doi.org/10.1111/j.1467-789X.2011.00860.x.
Madsbad S. The role of glucagon-like peptide-1 impairment in obesity and potential therapeutic implications. Diabetes Obes Metab. 2014;16(1):9–21. https://doi.org/10.1111/dom.12119.
Muscogiuri G, DeFronzo RA, Gastaldelli A, Holst JJ. Glucagon-like Peptide-1 and the Central/Peripheral Nervous System: Crosstalk in Diabetes. Trends Endocrinol Metab. 2017;28(2):88–103. https://doi.org/10.1016/j.tem.2016.10.001.
Iepsen EW, Zhang J, Thomsen HS, Hansen EL, Hollensted M, Madsbad S, et al. Patients with obesity caused by melanocortin-4 receptor mutations can be treated with a glucagon-like peptide-1 receptor agonist. Cell Metab. 2018;28(1):23–32. https://doi.org/10.1016/j.cmet.2018.05.008.
Knudsen LB, Secher A, Hecksher-Sørensen J, Pyke C. Long-acting glucagon-like peptide-1 receptor agonists have direct access to and effects on pro-opiomelanocortin/cocaine- and amphetamine-stimulated transcript neurons in the mouse hypothalamus. Journal of diabetes investigation. 2016;7 Suppl 1(Suppl Suppl 1):56–63. https://doi.org/10.1111/jdi.12463.
Secher A, Jelsing J, Baquero AF, Hecksher-Sorensen J, Cowley MA, Dalboge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Investig. 2014;124(10):4473–88. https://doi.org/10.1172/JCI75276.
Wettergren A, Wojdemann M, Meisner S, Stadil F, Holst JJ. The inhibitory effect of glucagon-like peptide-1 (GLP-1) 7-36 amide on gastric acid secretion in humans depends on an intact vagal innervation. Gut. 1997;40(5):597–601.
Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology. 2011;152(8):3103–12. https://doi.org/10.1210/en.2011-0174.
Halawi H, Khemani D, Eckert D, O’Neill J, Kadouh H, Grothe K, et al. Effects of liraglutide on weight, satiation, and gastric functions in obesity: a randomised, placebo-controlled pilot trial. Lancet Gastroenterol Hepatol. 2017;2(12):890–9. https://doi.org/10.1016/s2468-1253(17)30285-6.
Jelsing J, Vrang N, Hansen G, Raun K, Tang-Christensen M, Knudsen LB. Liraglutide: short-lived effect on gastric emptying—long lasting effects on body weight. Diabetes Obes Metab. 2012;14(6):531–8. https://doi.org/10.1111/j.1463-1326.2012.01557.x.
Iepsen EW, Lundgren J, Dirksen C, Jensen JE, Pedersen O, Hansen T et al. Treatment with a GLP-1 receptor agonist diminishes the decrease in free plasma leptin during maintenance of weight loss. Int J Obes (2005). 2015;39(5):834–41. https://doi.org/10.1038/ijo.2014.177.
Bradley DP, Kulstad R, Racine N, Shenker Y, Meredith M, Schoeller DA. Alterations in energy balance following exenatide administration. Applied physiology, nutrition, and metabolism = Physiologie appliquee, nutrition et metabolisme. 2012;37(5):893–9. https://doi.org/10.1139/h2012-068.
van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WH. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int J Obes (2005). 2014;38(6):784–93. https://doi.org/10.1038/ijo.2013.162.
Pannacciulli N, Bunt JC, Koska J, Bogardus C, Krakoff J. Higher fasting plasma concentrations of glucagon-like peptide 1 are associated with higher resting energy expenditure and fat oxidation rates in humans. Am J Clin Nutr. 2006;84(3):556–60. https://doi.org/10.1093/ajcn/84.3.556.
Harder H, Nielsen L, Tu DT, Astrup A. The effect of liraglutide, a long-acting glucagon-like peptide 1 derivative, on glycemic control, body composition, and 24-h energy expenditure in patients with type 2 diabetes. Diabetes Care. 2004;27(8):1915–21.
Shalev A, Holst JJ, Keller U. Effects of glucagon-like peptide 1 (7-36 amide) on whole-body protein metabolism in healthy man. Eur J Clin Invest. 1997;27(1):10–6.
Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology. 2009;150(4):1680–7. https://doi.org/10.1210/en.2008-1045.
Tran KL, Park YI, Pandya S, Muliyil NJ, Jensen BD, Huynh K, et al. Overview of glucagon-like peptide-1 receptor agonists for the treatment of patients with type 2 diabetes. Am Health Drug Benefits. 2017;10(4):178–88.
European Medicines Agency. Ozempic. https://www.ema.europa.eu/en/medicines/human/EPAR/ozempic. Accessed 28 Mar 2019.
US Food and Drug Administration. Full prescribing information saxenda (liraglutide [rDNA origin] injection), solution for subcutaneous use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206321Orig1s000lbl.pdf. Accessed 28 Mar 2019.
O’Neil PM, Birkenfeld AL, McGowan B, Mosenzon O, Pedersen SD, Wharton S, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392(10148):637–49. https://doi.org/10.1016/s0140-6736(18)31773-2.
Guo JJ, Pandey S, Doyle J, Bian B, Lis Y, Raisch DW. A review of quantitative risk-benefit methodologies for assessing drug safety and efficacy-report of the ISPOR risk-benefit management working group. Value Health. 2010;13(5):657–66. https://doi.org/10.1111/j.1524-4733.2010.00725.x.
European Medicines Agency. Assessment report for GLP-1 based therapies. 2013. https://www.ema.europa.eu/documents/report/assessment-report-article-53-procedure-glp-1-based-therapies_en.pdf. Accessed 28 Mar 2019.
Filippatos TD, Panagiotopoulou TV, Elisaf MS. Adverse Effects of GLP-1 Receptor Agonists. Rev Diabetic Stud. 2014;11(3–4):202–30. https://doi.org/10.1900/RDS.2014.11.202.
Astrup A, Rossner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374(9701):1606–16. https://doi.org/10.1016/s0140-6736(09)61375-1.
Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (2005). 2012;36(6):843–54. https://doi.org/10.1038/ijo.2011.158.
Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (2005). 2013;37(11):1443–51. https://doi.org/10.1038/ijo.2013.120.
Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjoth TV, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE diabetes randomized clinical trial. JAMA. 2015;314(7):687–99. https://doi.org/10.1001/jama.2015.9676.
Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11–22. https://doi.org/10.1056/nejmoa1411892.
Blackman A, Foster GD, Zammit G, Rosenberg R, Aronne L, Wadden T et al. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: the SCALE Sleep Apnea randomized clinical trial. Int J Obes (2005). 2016;40(8):1310–9. https://doi.org/10.1038/ijo.2016.52.
le Roux CW, Astrup A, Fujioka K, Greenway F, Lau DCW, Van Gaal L, et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double-blind trial. Lancet. 2017;389(10077):1399–409. https://doi.org/10.1016/s0140-6736(17)30069-7.
European Medicines Agency. Summary of produce characteristics. https://www.ema.europa.eu/documents/product-information/saxenda-epar-product-information_en.pdf. Accessed 28 Mar 2019.
Rigato M, Fadini GP. Comparative effectiveness of liraglutide in the treatment of type 2 diabetes. Diab Metabol Syndr Obesi. 2014;7:107–20. https://doi.org/10.2147/DMSO.S37644.
Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22. https://doi.org/10.1056/nejmoa1603827.
Rompianesi G, Hann A, Komolafe O, Pereira SP, Davidson BR, Gurusamy KS. Serum amylase and lipase and urinary trypsinogen and amylase for diagnosis of acute pancreatitis. Cochrane Database Syst Rev. 2017. https://doi.org/10.1002/14651858.cd012010.pub2.
Egan AG, Blind E, Dunder K, de Graeff PA, Hummer BT, Bourcier T et al. Pancreatic safety of incretin-based drugs—FDA and EMA assessment. N Engl J Med. 2014;370(9):794–7. https://doi.org/10.1056/nejmp1314078.
Steinberg WM, Rosenstock J, Wadden TA, Donsmark M, Jensen CB, DeVries JH. Impact of liraglutide on amylase, lipase, and acute pancreatitis in participants with overweight/obesity and normoglycemia, prediabetes, or type 2 diabetes: secondary analyses of pooled data from the SCALE Clinical Development Program. Diabetes Care. 2017;40(7):839–48. https://doi.org/10.2337/dc16-2684.
Portincasa P, Moschetta A, Palasciano G. Cholesterol gallstone disease. Lancet. 2006;368(9531):230–9. https://doi.org/10.1016/s0140-6736(06)69044-2.
Nexøe-Larsen CC, Sørensen PH, Hausner H, Agersnap M, Baekdal M, Brønden A, et al. Effects of liraglutide on gallbladder emptying: a randomized, placebo-controlled trial in adults with overweight or obesity. Diabetes Obes Metab. 2018;20(11):2557–64. https://doi.org/10.1111/dom.13420.
Smits MM, Tonneijck L, Muskiet MHA, Hoekstra T, Kramer MHH, Diamant M, et al. Biliary effects of liraglutide and sitagliptin, a 12-week randomized placebo-controlled trial in type 2 diabetes patients. Diabetes Obes Metab. 2016;18(12):1217–25. https://doi.org/10.1111/dom.12748.
Madsen LW, Knauf JA, Gotfredsen C, Pilling A, Sjogren I, Andersen S, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology. 2012;153(3):1538–47. https://doi.org/10.1210/en.2011-1864.
Novo Nordisk. CHMP endorses EU label update of Saxenda® based on the LEADER trial. 2017. https://www.novonordisk.com/bin/getPDF.2115243.pdf. Accessed 28 Mar 2019.
Lau J, Bloch P, Schaffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–80. https://doi.org/10.1021/acs.jmedchem.5b00726.
US Food and Drug Administration. Center for drug evaluation and research 209637Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209637Orig1s000lbl.pdf. Accessed 28 Mar 2019.
Nauck MA, Petrie JR, Sesti G, Mannucci E, Courreges JP, Lindegaard ML, et al. A phase 2, randomized, dose-finding study of the novel once-weekly human GLP-1 analog, semaglutide, compared with placebo and open-label liraglutide in patients with type 2 diabetes. Diabetes Care. 2016;39(2):231–41. https://doi.org/10.2337/dc15-0165.
Novo Nordisk. Strengthen leadership in obesity. 2017. https://www.novonordisk.com/content/dam/Denmark/HQ/investors/irmaterial/cmd/2017/05_Strengthen%20leadership%20in%20obesity.pdf. Accessed 28 Mar 2019.
ClinicalTrials.gov. Semaglutide Effects on Heart Disease and Stroke in Patients With Overweight or Obesity (SELECT). NCT03574597. 2018.https://clinicaltrials.gov/ct2/show/NCT03574597. Accessed 28 Mar 2019.
Sorli C, Harashima SI, Tsoukas GM, Unger J, Karsbol JD, Hansen T, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5(4):251–60. https://doi.org/10.1016/s2213-8587(17)30013-x.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–44. https://doi.org/10.1056/NEJMoa1607141.
Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial. JAMA. 2017;318(15):1460–70. https://doi.org/10.1001/jama.2017.14752.
Dicembrini I, Nreu B, Scatena A, Andreozzi F, Sesti G, Mannucci E, et al. Microvascular effects of glucagon-like peptide-1 receptor agonists in type 2 diabetes: a meta-analysis of randomized controlled trials. Acta Diabetol. 2017;54(10):933–41. https://doi.org/10.1007/s00592-017-1031-9.
Hernandez C, Bogdanov P, Corraliza L, Garcia-Ramirez M, Sola-Adell C, Arranz JA, et al. Topical administration of GLP-1 receptor agonists prevents retinal neurodegeneration in experimental diabetes. Diabetes. 2016;65(1):172–87. https://doi.org/10.2337/db15-0443.
Mensberg P, Nyby S, Jorgensen PG, Storgaard H, Jensen MT, Sivertsen J, et al. Near-normalization of glycaemic control with glucagon-like peptide-1 receptor agonist treatment combined with exercise in patients with type 2 diabetes. Diabetes Obes Metab. 2017;19(2):172–80. https://doi.org/10.1111/dom.12797.
Nissen SE, Wolski KE, Prcela L, Wadden T, Buse JB, Bakris G, et al. Effect of Naltrexone-Bupropion on major adverse cardiovascular events in overweight and obese patients with cardiovascular risk factors: a randomized clinical trial. JAMA. 2016;315(10):990–1004. https://doi.org/10.1001/jama.2016.1558.
Bohula EA, Wiviott SD, McGuire DK, Inzucchi SE, Kuder J, Im K, et al. Cardiovascular safety of Lorcaserin in overweight or obese patients. N Engl J Med. 2018;379(12):1107–17. https://doi.org/10.1056/NEJMoa1808721.
Bethel MA, Patel RA, Merrill P, Lokhnygina Y, Buse JB, Mentz RJ, et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol. 2018;6(2):105–13. https://doi.org/10.1016/s2213-8587(17)30412-6.
Kaneko M, Narukawa M. Assessment of cardiovascular risk with glucagon-like peptide 1 receptor agonists in patients with type 2 diabetes using an alternative measure to the hazard ratio. Ann Pharmacother. 2018;52(7):632–8. https://doi.org/10.1177/1060028018757407.
The Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369(2):145–54. https://doi.org/10.1056/NEJMoa1212914.
Finkelstein EA, Verghese NR. Incremental cost-effectiveness of evidence-based non-surgical weight loss strategies. Clin Obes. 2019;0(0):e12294. https://doi.org/10.1111/cob.12294.
ClinicalTrials.gov. Saxenda in obesity services (STRIVE Study) (STRIVE). NCT03036800. 2017. https://clinicaltrials.gov/ct2/show/NCT03036800. Accessed 28 Mar 2019.
Basalay MV, Mastitskaya S, Mrochek A, Ackland GL, Del Arroyo AG, Sanchez J, et al. Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res. 2016;112(3):669–76. https://doi.org/10.1093/cvr/cvw216.
Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. 2012;166(5):1586–99. https://doi.org/10.1111/j.1476-5381.2012.01971.x.
Vinue A, Navarro J, Herrero-Cervera A, Garcia-Cubas M, Andres-Blasco I, Martinez-Hervas S, et al. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia. 2017;60(9):1801–12. https://doi.org/10.1007/s00125-017-4330-3.
Holst JJ, Madsbad S, Bojsen-Møller KN, Svane MS, Jørgensen NB, Dirksen C, et al. Mechanisms in bariatric surgery: gut hormones, diabetes resolution, and weight loss. Surg Obes Relat Dis. 2018;14(5):708–14. https://doi.org/10.1016/j.soard.2018.03.003.
Meek CL, Lewis HB, Reimann F, Gribble FM, Park AJ. The effect of bariatric surgery on gastrointestinal and pancreatic peptide hormones. Peptides. 2016;77:28–37. https://doi.org/10.1016/j.peptides.2015.08.013.
Sadry SA, Drucker DJ. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat Rev Endocrinol. 2013;9(7):425–33. https://doi.org/10.1038/nrendo.2013.47.
Alexiadou K, Anyiam O, Tan T. Cracking the combination: gut hormones for the treatment of obesity and diabetes. J Neuroendocrinol. 2018:e12664. https://doi.org/10.1111/jne.12664.
Author information
Authors and Affiliations
Contributions
Conceptualisation and methodology: SST. Writing first draft: RMC. Critically editing, re-writing, analysing and reviewing the manuscript: SST, RMC, and CRJ.
Corresponding author
Ethics declarations
Funding
This work was supported by the Tripartite Immunometabolism Consortium [TrIC], Novo Nordisk Foundation; grant number NNF15CC0018486.
Conflict of interest
Rasmus Michael Christensen was supported by the Novo Nordisk Foundation, grant number NNF15CC0018486. Christian Rimer Juhl has no conflicts of interest that are directly relevant to the content of this article. Signe Sørensen Torekov has received research grants from the Novo Nordisk Foundation and Novo Nordisk A/S.
Additional information
The original version of this article was revised: The drug “Orlistat” was incorrectly changed to “Gastrointestinal lipase inhibitor” in Table 1 and the same has been corrected.
Rights and permissions
About this article

Cite this article
Christensen, R.M., Juhl, C.R. & Torekov, S.S. Benefit-Risk Assessment of Obesity Drugs: Focus on Glucagon-like Peptide-1 Receptor Agonists. Drug Saf 42, 957–971 (2019). https://doi.org/10.1007/s40264-019-00812-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-019-00812-7