
Abstract
Four “third-generation” antiepileptic drugs (AEDs) were approved for adjunctive treatment of refractory focal onset seizures during the past 10 years. Long-term efficacy and safety of the drugs were demonstrated in large extension studies and in reports of subgroups of patients not studied in pivotal trials. Reviewing extension study and post-marketing outcome series for the four newer AEDs—lacosamide, perampanel, eslicarbazepine acetate and brivaracetam—can guide clinicians in treating and monitoring patients. AED extension studies evaluate treatment retention, drug tolerability, and drug safety during individualized treatment with flexible dosing and thus provide information not available in rigid pivotal trials. Patient retention in the studies ranged from 75 to 80% at 1 year and from 36 to 68% at 2-year treatment intervals. Safety findings were generally similar to those of pivotal trials, with no major safety risks identified and with several specific adverse drug effects, such as hyponatremia, reported. The third-generation AEDs, some through new mechanisms and others with improved tolerability compared to related AEDs, provide new options in efficacy and tolerability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Safety and efficacy of new antiepileptic drugs (AEDs) was evaluated in long-term extension studies and post-marketing series. |
Lacosamide, perampanel, eslicarbazepine acetate, and brivaracetam have high retention during 1 year of open treatment, with gradual reductions over several years. |
At high doses, treatment with the new AEDs was associated with similar central nervous system-related adverse drug effects, with some specific drug effects. |
Comparing long-term treatment outcomes for newer AEDs aids clinicians in monitoring patients. |
1 Introduction
Drug manufacturers must prove efficacy and safety of their new products in preclinical and phase I, II, and III trials for US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval. By the time a new antiepileptic drug (AED) is approved for market, usually several hundred patients have been exposed to the drug and followed for several years.
However, post-marketing and long-term follow-up is vital because of the possibility of rare effects or side effects that only arise after long exposures. Randomized pivotal trials are relatively brief—typically providing 8–12 weeks of exposure to investigational AEDs. European and US regulatory agencies require safety assessments of AEDs over longer exposure periods, and this is provided in extension studies. These studies monitor patients during 1–8 years of “open” treatment. Patients completing pivotal trials often are pooled into larger extension studies, improving detection of potential infrequent adverse drug effects. These studies evaluate safety during chronic exposure and demonstrate patient retention—a general index of treatment success over several year periods [1].
This narrative review discusses and compares the extension study results for four “third-generation” AEDs: lacosamide, perampanel, eslicarbazepine acetate, and brivaracetam. These studies evaluate tolerability and safety across a broader range of doses than are typically used in pivotal trials (e.g., lacosamide is indicated for 400 mg/day maximum dosage, but there is substantial safety information during treatment with dosages up to 800 mg/day) [2]. Initial pivotal trials also require rigid treatment with randomly assigned fixed AED doses and do not permit adjustments of concomitant AEDs. Extension studies provide some insight into outcomes with flexible dosing, when doses of concomitant AEDs can be adjusted. For example, during an extension study, pharmacodynamic interactions between lacosamide and other sodium channel modulators causing dizziness and sedation usually could be resolved by down-titrating the concomitant AEDs [3].
Patient retention in extension studies also provides a reasonable index of long-term treatment outcome. Patients often discontinue treatment in extension studies if they develop adverse drug effects or do not benefit from reduced seizures. Efficacy data were reported by median seizure frequency reduction and ≥ 50% responder rates; for extension trials, efficacy (seizure control) is influenced by the aforementioned flexible dosing of concomitant AEDs. In addition, responder rates are often enriched because of study dropouts and so may best represent seizure outcomes after 1–2 years of treatment rather than longer periods. Patients typically enroll in safety studies over several-year periods; those entering extension study treatment near the completion of a study program may have limited 1- or 2-year exposures because of a study closing, rather than because of a favorable or unfavorable treatment outcome. Consequently, it is usually most accurate to represent study retention and responder outcomes based on enrollment dates. Unfortunately, not all extension studies reported these data.
Extension studies provide important safety information on new AEDs but occasionally do not detect all unexpected or infrequent adverse drug effects. Vigabatrin, for example, was approved in many countries while US extension studies were still ongoing. Visual dysfunction was detected in a small number of patients in the UK receiving post-marketed treatment [4] and was then shown in the US extension study to be due to inner retinal dysfunction causing peripheral visual field loss [5]. Only later was the drug released in the US with an orphan drug treatment indication for infantile spasms and refractory complex partial seizures, with careful visual safety monitoring [6].
Confidence in the sensitivity of extension studies to exclude major safety risks can be approximated by Hanley's law which states the maximum risk (> 95% confidence interval) equals 3/n. Many of the pooled AED extension studies enrolled approximately 1,000 patients, and thus the corresponding risk for undetected major safety risks for these studies is less than 3 in 1000. Consistent with this, no new serious safety problems were detected in the AED extension studies or in post-marketing series. This suggests extension studies and post-marketing series provide reasonable screening for most common serious safety risks. Rare safety risks (e.g., < 1 per 1000 exposures) may be missed. Felbamate, for example, the first new AED approved in 15 years in 1993, was approved after fewer than 300 patients were monitored in extension studies. 1 year later, 110,000 patients were exposed to felbamate and 31 cases of aplastic anemia and 18 cases of hepatic failure were reported [8]. This highlights the importance of evaluating safety in the large extension study pools reviewed here and the need to be vigilant for very rare or unanticipated adverse drug effects [9].
This long-term tolerability and efficacy data for third-generation AEDs provide a helpful comparison with older AEDs used in the adjunctive treatment trials. The review highlights major differences in mechanisms and tolerability between the newer third-generation AEDs and previous therapies, which helps show how they might be matched with individual patient treatment needs.
2 Lacosamide
Lacosamide is approved for adjunctive and monotherapy treatment of partial-onset seizures for patients ≥ 17 years old in the USA and ≥ 16 years old in Europe, based on several successful phase II and phase III, placebo-controlled, randomized trials [10,11,12]. Lacosamide is a functionalized amino acid molecule that selectively enhances the slow inactivation of voltage-gated sodium channels [10]. Lacosamide is approved at total oral daily doses of 200–400 mg; an intravenous solution was approved as a temporary substitute for oral treatment [13].
2.1 Clinical Trials
One phase II and two phase III, randomized, placebo-controlled trials evaluated lacosamide treatment in 1294 patients with resistant partial-onset seizures at US and global sites [10,11,12]. Monthly seizure frequencies during the studies decreased 26–39% across a dose range of 200–400 mg (total daily dose). Less than 10% of patients became seizure free, but 20–30% had a > 75% reduction in seizures [13]. The most common treatment-related adverse events for lacosamide were dizziness, nausea, blurred vision, and imbalance, though these symptoms were most common during dose titration [13]. Lacosamide 600 mg/day was effective compared to placebo, but efficacy was similar to 400 mg/day, and since adverse events were increased, the 600 mg/day dosage was not approved in the USA and EU. In a later post hoc analysis, this seemed due to a pharmacodynamic interaction between lacosamide and other sodium channel-modulating AEDs—patients taking lacosamide with AEDs with other mechanisms often tolerated and benefitted from the 600 mg/day dosage [14].
2.2 Long-Term Efficacy Data
Patients completing phase II and phase III trials enrolled in one of four open-label extension trials. All four trials allowed lacosamide dosage adjustments of 100–800 mg/day, and the first three trials allowed treatment with up to three concomitant AEDs, with dosage adjustments permitted. The first study, SP756, enrolled 308 patients for up to 5 years of exposure [15]. The median duration of treatment was 2.9 years; the median dosage was 500 mg/day. Seventy-five percent of patients had > 1 year of exposure, and 29% had > 4 years of exposure. 55.2% of patients discontinued lacosamide treatment, primarily because of lack of efficacy (26%) and adverse events (11%). The median percent seizure frequency reduction was 53.4–62.5% across 1-, 2-, 3-, and 4-year cohorts; corresponding ≥ 50% responder rates were 52.8–62.5%. For each completer cohort, the initial reduction in seizure frequency during the first 6 months was maintained through further time points. The increasing seizure reductions and ≥ 50% responder rates with long treatment periods partially reflect enrichment due to dropouts in these extension studies.
The second study, SP774, enrolled 376 patients for up to 5.5 years of exposure [16]. The median duration of treatment was 3.2 years; the median dosage was 400 mg/day. Patient retention in the study (based on Kaplan–Meier methods) was 74.5% at 1 year, 52.9% at 3 years, and 40.6% at 5 years. 57.4% of patients discontinued, primarily because of lack of efficacy (24.5%), withdrawn consent (17.6%), and adverse events (9.0%). The median percent seizure frequency reduction was 55.4% for the 1-year cohort and 62.3% for the 3-year cohort. Corresponding ≥ 50% responder rates were overall 55.9% for the 1-year cohort and 63.0% for the 3-year cohort; the 1-year cohort saw an increase from 55.2 to 58.8% from 6 months to 1 year, and the 3-year cohort saw an increase from 55.5 to 66.0% from 6 months to 3 years.
The third extension study, SP615, enrolled 370 patients for up to 8 years of treatment [2]. The median duration of treatment was 2.8 years; the median lacosamide dosage was 400 mg/day. Patient retention in the study according to Kaplan–Meier analysis was 76.8% at 1 year, 50.8% at 3 years, and 38.7% at 5 years. 67.6% of patients discontinued lacosamide treatment, most commonly because of lack of efficacy (33.8%), adverse events (13.2%), and withdrawn consent (13.0%). The median percent seizure frequency reduction was 47.3–65.2% across 1-, 3-, and 5-year cohorts; corresponding ≥ 50% responder rates were 48.8–63.4%; the seizure frequency reduction by each yearly completer cohort was generally maintained over time.
The fourth study, SP902, was a 2-year lacosamide monotherapy study that enrolled patients from study SP904, a conversion-to-monotherapy study [17]. The median duration was 729 days (~ 2 years); the median dosage was 500 mg/day. Most patients (292 of 322 patients; 90.7%) achieved lacosamide monotherapy at some point during the study; however, only 151 patients had lacosamide monotherapy throughout the entire study. A total of 112 (34.8%) discontinued treatment, most commonly because of withdrawn consent (9.3%). For the 292 patients on monotherapy at some point, responder rates were favorable, with 62.7–74.2% having ≥ 50% seizure reduction and 39.2–57.3% having ≥ 75% seizure reduction. 5.6% were seizure free at 24 months of open treatment. Percentages of seizure frequency reduction over time were maintained both for those who were on lacosamide monotherapy consistently throughout the study and for those who had lacosamide monotherapy at any point during the study.
Several open treatment trials examined the tolerability and efficacy of lacosamide after marketing release. One multicenter, prospective study in Italy and Germany included 118 patients with uncontrolled generalized and focal epilepsy [18]. A total of 81 patients (68.6%) reached the 1-year follow-up, with discontinuation due to lack of efficacy (15.2%), an increase in seizure frequency (10.2%), and adverse events (5.9%). The ≥ 50% responder rate was 43.2% at 1 year of open treatment.
A study in Italy evaluated patients (n = 58) who had become seizure free with adjunctive lacosamide treatment and showed 63.8% were able to convert to monotherapy, while 36% required continued polytherapy. 55.2% remained seizure free [19]. Patients’ history of fewer than three lifetime trials of AEDs was a predictor of seizure freedom.
A third multicenter, prospective study in Italy studied 18 infants and young children with refractory focal epilepsy [20]. The ≥ 50% responder rate after 3 months of treatment was 44% (n = 8), which included 17% (n = 3) of patients who became seizure free. At 1 year, however, half of the initial responders had loss of efficacy, and only two patients were still seizure free. Due to adverse events, lacosamide was stopped in one patient (imbalance) and reduced in three (drowsiness, vomiting).
Multiple small, long-term, retrospective studies followed outcome of lacosamide treatment in patients with focal epilepsy. A study of 21 children in Korea, treated for 6–13 months, found 67% were ≥ 50% responders, with 19% becoming seizure free [21]. Two patients (10%) discontinued treatment because of adverse events (aggressive behaviors and depression). A study in Spain monitored 60 patients with refractory focal epilepsy during 13–24 months of treatment; 47% of patients had a ≥ 50% reduction in seizure frequency, with only two patients achieving seizure freedom [22]. Thirteen percent of patients discontinued treatment because of adverse events (n = 6) and seizure worsening (n = 2). In a separate retrospective study in Spain (n = 66), 64% were seizure free during an average of 15.5 months of lacosamide treatment [23]. Fifteen percent of patients discontinued treatment, mostly because of adverse events (n = 3) or lack of efficacy (n = 6). In a retrospective study of 22 children in Canada, 45% were > 50% responders during an average period of treatment of 12 months [24]. One patient became seizure free and 41% (n = 9) of patients discontinued treatment because of adverse events (n = 6) and lack of efficacy (n = 3).
A small, 18-month study of adults (n = 19) with Lennox-Gastaut syndrome did not show efficacy with lacosamide treatment; two patients had a > 50% reduction in seizures, and 47.7% discontinued treatment because of seizure worsening and other adverse events [25].
2.3 Long-Term Safety Data
Safety data are available from the four long-term (2–8 years of exposure) extension trials as well as from nine 1-year post-marketing studies. Adverse events were generally mild to moderate in severity and were similar to those reported in pivotal trials. Adverse events were most commonly nervous system-related (i.e., dizziness, headache). Studies noted, however, that dose adjustments and addition of concomitant AEDs may have affected treatment emergent adverse event (TEAE) occurrence.
For the 5-year open-label extension study SP756, including 308 patients, 93.5% experienced TEAEs, most commonly (≥ 15%) dizziness (50.0%), headache (21.8%), contusion (18.5%), nausea (18.5%), convulsion (17.2%), nasopharyngitis (17.2%), fall (15.9%), vomiting (15.9%), and diplopia (15.3%) [15]. Dizziness and convulsion were the only TEAEs that led to discontinuation in ≥ 1.0% of patients. 23.1% of patients experienced a treatment emergent serious adverse event (SAE). Of these, four patients each experienced convulsion and dizziness, and two patients each experienced atrial fibrillation and ventricular extrasystoles.
For the 5.5-year open-label extension study SP774, including 376 patients, 82.7% of patients reported TEAEs, most commonly dizziness (24.2%), headache (14.4%), diplopia (13.8%), and nasopharyngitis (13.8%) [16]. Nine percent of patients discontinued because of adverse events, most commonly dizziness (1.3%). 23.1% of patients experienced treatment emergent SAEs, with three types occurring with an incidence ≥ 1%: convulsion (4.0%), epilepsy (1.9%), and status epilepticus (1.3%).
For the 8-year open-label extension study SP615, including 307 patients, 71.4% reported TEAEs, most commonly dizziness (39.7%), headache (20.8%), nausea (17.3%), diplopia (17.0%), fatigue (16.5%), upper respiratory tract infection (16.5%), nasopharyngitis (16.2%), and contusion (15.4%) [2]. 12.7% of patients discontinued treatment because of TEAEs; dizziness and convulsion were the only TEAEs that resulted in discontinuation in > 1% of patients. 33.8% of patients experienced treatment emergent SAEs; convulsion (6.2%) was the only treatment emergent SAE that occurred in > 2% of patients.
A 2-year monotherapy extension study, SP904, helped identify adverse events linked to lacosamide treatment alone [17]. Out of 292 patients on monotherapy, 83.9% had TEAEs, most commonly dizziness (17.5%), headache (15.1%), upper respiratory tract infection (10.6%), nausea (10.3%), and convulsion (10.3%). 4.1% of patients reported TEAEs leading to discontinuation, most commonly convulsion (1.8%) and postictal state (1.2%). Twelve percent reported SAEs that were seizure related, most commonly convulsion (2.7%) and syncope (0.7%). Serious TEAEs had a mild trend of being more common above the dosage of 300 mg/day.
A multicenter, prospective study in Italy and Germany included 118 patients with uncontrolled generalized and partial-onset seizures [18]. 29.7% experienced side effects, most commonly dizziness (6.8%), headache (5.9%), somnolence (4.3%), dyspepsia (4.3%), vomiting (4.3%), irritability (3.4%), and nausea (3.4%). Seven subjects withdrew because of adverse events.
A second multicenter, prospective study in Italy evaluated 18 infants and young children with refractory focal epilepsy; 33% experienced at least one adverse event with lacosamide treatment, most commonly drowsiness (21%), nervousness (12.5%), vomiting (8%), and instability and difficulty walking (4%) [20]. All side effects were tolerable or resolved through dose reduction or discontinuation.
A third prospective study in Italy, including 58 adult patients receiving lacosamide monotherapy, found 20.8% reported mild to moderate adverse events, most commonly drowsiness (n = 7), dizziness (n = 3), and headache (n = 2) [19]. No SAEs were reported.
Several retrospective studies of patients with focal epilepsy reported similar patterns of mild to moderate adverse events. The Korean study of 21 pediatric patients found 38% had mild TEAEs: somnolence (n = 3), dizziness (n = 2), personality change (n = 2), sleep disturbance (n = 1), and nausea (n = 1) [21]. No patient experienced seizure worsening. The study in Spain of 60 patients with refractory focal epilepsy and nocturnal seizures found 33% experienced TEAEs, most commonly dizziness (n = 16) [22]. No SAEs were reported. In the second study in Spain, including 66 patients treated with lacosamide monotherapy, 22.7% experienced mild to moderate adverse events, most commonly fatigue (7.5%), dizziness (6.1%). 4.5% of patients discontinued lacosamide treatment because of adverse events, such as severe fatigue (n = 1), dizziness (n = 1), and pruritus and insomnia (n = 1) [23]. In the study of 22 pediatric patients in Canada, 50% of patients had adverse events, most commonly dizziness (23%) and drowsiness (23%) [24]. No SAEs were reported. An observational, post-authorization safety study in Germany compared the incidence of cardiovascular and psychiatric-related TEAEs during 1 year of treatment with adjunctive lacosamide versus another AED; both groups had a low incidence of these TEAEs [26].
In the retrospective Colombia study of 19 adult patients with Lennox-Gastaut syndrome, 47.7% (n = 9) patients reported adverse events, most commonly seizure worsening (47.7%), aggressive behavior (47.7%), irritability (47.7%), and somnolence (31.6%) [25].
2.4 Discussion
A large range of lacosamide dosages (200–800 mg/day) were evaluated in long-term extension studies, with no new safety abnormalities detected compared to phase II/III randomized studies. The ≥ 50% responder rates were comparable across the open-label, regulatory extension studies and the prospective and retrospective, single- and multicenter monitoring studies. The > 50% responder rates ranged from 42 to 67% across these studies. Some of the increases in efficacy during the extension studies reflect enrichment due to dropouts of non-responders. However, patient retention was high (74–76%) at 1-year follow-up across the adjunctive therapy extension trials. Discontinuation in these trials was most commonly due to lack of efficacy, adverse events, and withdrawn consent. Lacosamide was similarly efficacious in these larger extension studies and the phase II/phase III pivotal trial results. Patients in monotherapy extension studies had particularly strong treatment responses. Studies of pediatric patients also found treatment efficacious. Central nervous system (CNS)-related adverse events were the most commonly reported signs and symptoms during extension treatment.
3 Perampanel
Perampanel is a non-competitive AMPA glutamate receptor antagonist approved in the USA and Europe for adjunctive treatment of partial-onset seizures with or without secondary generalization in patients aged 12 years and older [27]. It was also effective and approved for treating primary generalized epilepsy with tonic–clonic seizures [28].
3.1 Clinical Trials
In 2012, the FDA and EMA approved perampanel for adjunctive treatment of partial-onset seizures with or without secondary generalization after three phase III, double-blind, randomized, controlled trials [29,30,31]. Dosages of 4 mg/day, 8 mg/day, and 12 mg/day were effective. The trials included 1478 patients (≥ 12 years old) with refractory partial-onset seizures in more than 40 countries at 262 centers. During the 13-week treatment periods (following a 6-week titration period), the mean relative reduction in seizure frequency was 23.3–28.8% for perampanel versus 12.8% for placebo. The pooled ≥ 50% responder rate was 28.5–35.0%, compared to 19.3% in the placebo groups [32]. Most TEAEs were mild/moderate; the most common TEAEs were dizziness, somnolence, and headache [32].
Two placebo-controlled, dose-escalation, phase II trials preceded these trials, and found perampanel was tolerated across a dosage range of 2–12 mg/day in adult patients [33].
3.2 Long-Term Efficacy Data
Long-term safety data on perampanel are available from two long-term extension studies, from one small, 1-year prospective, observational study, and from four retrospective studies. The 3-year open-label extension study 307 enrolled 96.4% (1216/1264) of patients who had completed phase III studies [27]. Seizure outcomes were analyzed in patients completing ≥ 6 months (n = 1090), ≥ 9 months (n = 980), ≥ 1 year (n = 874), and ≥ 2 years (n = 337) of treatment. The Kaplan–Meier curve estimated 75.9% retention of patients at 1 year and 34.2% at 2 years. Patients discontinued treatment overall most frequently because of subject choice (13.8%), adverse events (12.9%), and inadequate therapeutic effect (11.6%). At 1 year, 1.8% discontinued because of adverse events, 2.6% because of lack of efficacy, 3.9% because of subject choice, and 0.4% because of other reasons, while at the 2-year time point, 0.4% discontinued because of adverse events, 1.3% because of lack of efficacy, 1.3% because of subject choice, and 1.1% because of other reasons. Changes in seizure frequency and responder rate were comparable at similar time points; seizure frequency reduction remained stable for each yearly completer cohort over time. The overall ≥ 50% responder rate was 31.1% in the first 3 months and 62.7% in the last 3 months of follow-up, partially reflecting enrichment due to dropouts of non-responders. The corresponding median percent seizure frequency reductions were 29.1 and 58.1%. The seizure-free rate over the first 6 months was 4.9% (45/918 patients), and over the last 6 months (for patients with ≥ 2 years of data), it was 10.6% (15/141).
The 8-year open-label extension study 207 enrolled 138 adult patients out of 180 who had completed the phase II studies [34]. In a 4-year interim analysis, over a third of the patients (n = 53, 38.4%) remained on perampanel; 41.3% had ≥ 3 years of exposure and 13.0% of patients had ≥ 4 years. Patients discontinued treatment most commonly because of withdrawal of patient consent (23.2%), lack of efficacy (18.1%), and adverse events (13.0%). The median percent change in seizure frequency was 43.7–52.0% for 1- to 4-year cohorts, and the corresponding ≥ 50% responder rates were 43.8–51.5%.
Five post-marketing studies were done: one prospective and four retrospective. A 1-year, prospective, observational study in Denmark followed 22 adult patients with drug-resistant focal epilepsy. At 1 year, 54.5% of patients remained on perampanel treatment; 45.5% (n = 10) of patients discontinued, with 31.8% (n = 7) withdrawing because of adverse events. 27.2% were ≥ 50% responders, and 9.1% were seizure free [35].
Four retrospective studies evaluated responses from patients who had been excluded from pivotal trials (e.g., because of medical and psychiatric comorbidities). A 1-year observational study, FYDATA, in Spain examined 464 patients (≥ 12 years of age, with focal epilepsy) [36]. FYDATA had a 60.6% retention rate; patients discontinued most frequently because of adverse events (17.9%) and lack of efficacy (17.5%). The median percentage seizure frequency reduction was 33.3% at 1 year, with 7.2% of patients achieving seizure freedom. The ≥ 50% responder rate at 1 year was 26.8%.
A single-center, retrospective study in the USA examined outcomes of perampanel treatment in 85 adult and 16 pediatric patients with epilepsy over approximately 1 year [37]. Twenty-seven percent of patients discontinued treatment: 23% discontinued because of adverse events and 4% because of lack of efficacy. The median seizure frequency reduction was 50% overall, 50% in children, and 33% in adults. The ≥ 50% responder rate was 51% (63% in children and 49% in adults), and 6% reached seizure freedom.
Two retrospective studies included many patients with intellectual disability, behavioral problems, or psychiatric problems. A 1-year study in the Netherlands examined 62 adult and pediatric patients with intellectual disabilities and refractory epilepsy [38]. 53.2% achieved seizure reduction, with none reaching seizure freedom; 58.1% completed 1 year of treatment. Patients discontinued most frequently because of adverse events (44.4%), lack of efficacy (14.8%), or a combination of both (40.7%). The second study, conducted in France, pooled retrospective data from 101 adult patients with refractory epilepsy who were prescribed treatment over the course of 13 months [39]. The study included patients with learning disabilities (37.6%) and psychiatric comorbidities (49.5%). Mean retention was 8.1 months (range 14 days to 17 months). 49.5% of patients discontinued, most commonly because of side effects (n = 20), lack of efficacy (n = 2), or a combination of both (n = 25). Seizure reduction was > 50% in 41.6% of patients; 6.9% of patients became seizure free.
3.3 Long-Term Safety Data
Safety data beyond 1 year of exposure were available from the two regulatory, open-label extension studies; adverse events were mostly mild or moderate in severity and were similar to those in pivotal trials, although dose adjustments and concomitant AEDs may have affected adverse event occurrence. The 3-year extension study (study 307) of 1216 patients found 81.7% (n = 993) of patients had TEAEs, most commonly dizziness (46.8%), somnolence (21.2%), headache (18.3%), and fatigue (13.1%) [27]. Adverse events occurred most frequently during dose titration. Only two adverse events, dizziness (3.9%) and irritability (1.3%) led to discontinuation in ≥ 1% of patients. 18.7% (n = 227) of patients had SAEs; the only SAEs occurring in > 1% of patients were those related to seizures, although 3.9% had at least one psychiatric SAE. Three deaths occurred [car accident, sudden unexpected death in epilepsy (SUDEP), cerebral hemorrhage]; none were considered to be related to study treatment.
In the phase II, open-label extension study (study 207) of 138 patients, 93.5% (n = 129) experienced at least one TEAE [34]. Most were mild or moderate in severity, most commonly dizziness (41.3%), headache (21.0%), and somnolence (19.6). Frequencies of these three TEAEs were halved during both the second and third years of treatment. 12.3% of patients discontinued treatment because of TEAEs: dizziness (n = 3), vertigo (n = 2), upper abdominal pain (n = 2), fatigue (n = 2), headache (n = 2), and status epilepticus (n = 2). 15.2% of patients experienced SAEs, and these were considered possibly related to treatment in four patients and probably related in one.
Safety data were also available from five 1-year observational studies. In the 1-year observational study conducted in Denmark, including 22 adult patients with focal epilepsy, 59.1% reported side effects, most commonly tiredness (36.4%), behavioral changes (primarily aggression) (22.7%), and dizziness (18.2%), leading to discontinuation in 31.8% of subjects [35].
In the FYDATA study from Spain, including 464 patients, 62.9% experienced adverse events, most commonly dizziness (23.2%), somnolence (19.8%), and irritability (17.9%); most of the adverse events appeared in the first 6 months of treatment [36].
In the US retrospective study of 85 adult and 16 pediatric patients, 47% of patients experienced adverse events, most commonly sleepiness/fatigue (18%), dizziness/falls (18%), and behavioral changes (15%) [37]. Twenty-three percent of patients discontinued treatment because of adverse events, of which 35% experienced sleepiness/fatigue, 30% experienced behavioral problems, and 22% experienced dizziness. Frequency of adverse events was correlated with dose (7.3 mg average dose for patients with adverse events vs 5.5 mg for patients without).
Behavioral problems (i.e., aggression, irritability) were recognized as a potential serious adverse effect of perampanel. The retrospective study conducted in the Netherlands followed 62 patients with intellectual disabilities and refractory epilepsy [38]. 58.1% of patients experienced adverse events; 45.2% of patients experienced somatic adverse events (somnolence, motor problems and unsteadiness, gastrointestinal problems), and 40.3% experienced behavioral adverse events (aggression, agitation, disruptive behavior, mood problems). 80.6% experienced adverse events within 3 months of starting perampanel, and 12.9% discontinued treatment because of adverse events. The retrospective study conducted in France followed 101 patients with refractory epilepsy as well as learning disabilities and psychiatric comorbidities [39]. 62.4% experienced adverse events, the most common of which were irritability (33.7%), asthenia (18.8%), aggression (15.8%), sedation (13.9%), and dizziness (10.9%).
3.4 Discussion
The large, phase III, open-label extension study demonstrated similar efficacy in patients converting from placebo to perampanel and those receiving initial treatment. Retention rates and ≥ 50% responder rates were comparable across all extension studies. Retention rates at 1 year ranged from 50.5 to 73%; ≥ 50% responder rates ranged from 26.8 to 51%. Patients discontinued because of lack of efficacy (2–18.1%), withdrawal of consent (13.8–23.2%), and adverse events (12.9–44.4%). The study conducted in the Netherlands with patients with intellectual disability had no patients reaching seizure freedom [38]; all the other long-term studies reported patients (6–7%) achieving seizure freedom. Studies unanimously concluded that perampanel treatment can provide clinically meaningful improvement, with most studies demonstrating results comparable to those of pivotal trials, despite high numbers of patients with highly refractory seizures.
The tolerability profile of perampanel in the long-term, phase II and phase III extension trials was consistent with the safety data from the pivotal trials. The median dose was approximately 10 mg in the forced-titration extension study 307 and 6–8 mg in the other trials. Up-titration occurred every 2 weeks for extension studies and varied every 2–4 weeks for the other trials. Patterns of adverse event drug effects were similar across the extension studies. Rates of discontinuation due to adverse events were comparable in six of the seven studies, ranging from 12.3 to 23%. In the small (n = 22), prospective study in Denmark, the rate was slightly higher (31.8%) [35]. This study also reported a higher rate (22%) of behavioral abnormalities (primarily aggressive behavior). The retrospective study in France, which included patients with psychiatric comorbidities and learning disabilities, also found high rates of irritability (34.7%) and aggressive behavior (15.8%), but specifically screened for these symptoms [39]. This study also found that having psychiatric comorbidity did not increase patient risk for these symptoms. Similarly, the study conducted in the Netherlands examined patients with epilepsy as well as behavioral disorders and found that pre-existing behavioral problems do not predict the occurrence of additional behavioral adverse effects [38]. In contrast, the observational study FYDATA, conducted in Spain and including 464 patients, found that patients with psychiatric comorbidities were more likely to experience psychiatric adverse events [36]. FYDATA also suggested that weekly (2 mg/week) titration increased the likelihood of experiencing an adverse event. The US retrospective study similarly suggested that slower rates of dose titration were responsible for lower adverse event frequency and severity [37]. The study conducted in Denmark also reported the fewest side effects with a slow titration rate (2 mg/4 weeks). For all trials, no new safety abnormalities were detected.
4 Eslicarbazepine Acetate
Eslicarbazepine acetate (ESL) was approved in 2009 by the EMA, and later in 2013 by the US FDA. ESL was initially approved for adjunctive therapy in adults with refractory partial-onset seizures, and in 2015, it was approved for monotherapy by the FDA [40, 41]. Like carbamazepine and oxcarbazepine, ESL has a dibenzazepine nucleus with a 5-carboxamide substitution; the 10, 11 position is structurally different from carbamazepine and oxcarbazepine. ESL is converted to (S)-licarbazepine (also called eslicarbazepine); while oxcarbazepine is converted to both racemic forms of (l)- and (S)-licarbazepine.
Results from two 1-year open-label extension studies; one clinical observational, 2-year follow-up study; and one 1-year clinical observational study are available, as is a recent review of post-marketing experience [42].
4.1 Clinical Trials
Four phase III studies (pivotal trials) of adjunctive eslicarbazepine acetate treatment showed efficacy at dosages of 800–1200 mg/day [43,44,45,46,47]. These studies included over 1700 patients in 23 countries at 125 centers [42]. ESL was approved by the EMA in 2009 on the basis of the results of three pivotal trials. An FDA review, however, criticized the data quality in study 2093–303 and of seizure diary data collection in the studies (diaries did not all require the absence of seizures be logged), and questioned whether the 800 mg/day dosage was effective. They required an additional pivotal trial (trial 304) be performed with patients in North America, and subsequently approved ESL in 2013 [47,48,49]. In the initial three trials, the pooled 50% responder rate was 36–44%, compared to a responder rate of 22% in the placebo groups [43]. During the 12-week treatment periods (following a 2-week titration period), the median relative reduction in seizure frequency was 35–39%, versus 15% for placebo [43]. In the fourth trial, required by the FDA, the ≥ 50% responder rate was 42.6% in the 1200-mg/day group, but was not significantly different from placebo in the 800-mg/day group [47]. Most TEAEs occurred during the first 6 weeks of treatment and were dose dependent; in weeks 6–12, there was no difference in adverse events between the treatment groups and the placebo groups. The most common TEAEs were dizziness, somnolence, headache, and nausea (experienced by > 10% of patients taking ESL), and TEAEs were dose dependent [43].
For monotherapy approval, two studies compared withdrawal of concomitant AEDs to ESL monotherapy (at 1200 or 1600 mg ESL) to a historical, pseudo-placebo control group [50, 51]. In one study, 19 patients (12.3%) met exit criteria of seizure control worsening over the 18-week study period, well below the 65.3% historical control exit rate [50]. In the other, the exit rates for the 1200- and 1600-mg groups were 28.7% and 44.4%, respectively, also significantly below the historical control exit rate. The most common TEAEs were headache (25.0%) and dizziness (17.4%); TEAEs were dose dependent, with severe TEAEs more common in the 1600-mg group [seizures; three patients with hyponatremia; one patient each with drug reaction with eosinophilia and systemic symptoms (DRESS), pruritic rash, depression, anxiety, dyspnea, hypertension, cardiogenic shock, hypokalemia, car accident, pancreatic neoplasm, pulmonary edema, ankle fracture, post-concussive syndrome, tibia fracture, syncope, and spontaneous abortion] [50]. TEAEs were more common during the titration period and AED conversion period than during the monotherapy period.
4.2 Long-Term Efficacy Data
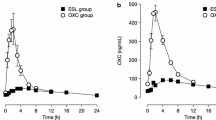
Little long-term safety and efficacy data beyond 1 year of follow-up have been published. Efficacy data up to 1 year post-trial are available from two open-label extension studies, the phase III trials 2093–301 and 2093–302, which were performed in Europe, South America, and Africa. Extension data from trial 304, which included patients in North America, and was designed to meet FDA requirements [47, 48], have not been published. The extension trial 2093–301 included 314 patients who entered the open-label extension with a median dosage of 800 mg daily; during the trial, the dosage could be individualized to 400–1200 mg daily. Of the 312 intent-to-treat population, 239 (76.6%) completed 1 year of treatment. Of the 73 (23.4%) patients who stopped treatment before 1 year, 39 (12.5%) withdrew consent, 11 (3.5%) had unacceptable adverse events, eight (2.6%) had protocol violations, eight (2.6%) were withdrawn at investigators’ discretion, two (0.6%) had seizure exacerbation, and 13 (4.2%) had another unspecified reason. Compared to baseline, there was a median seizure reduction of 39% during the first 4 weeks and 56.3% in the last 12 weeks of the trial (weeks 41–52). The mean seizure reduction was 37.5% in the first 4 weeks of the extension, and 40.6% in the last 12 weeks. The 50% responder rate was 41% in the first 16 weeks of the trial and increased gradually over time to 53% during weeks 41–52. Quality of Life in Epilepsy Inventory (QOLIE-31) scores and depressive symptoms also improved significantly [52].
The extension trial 2093–302 enrolled 325 patients, of whom 267 (82.2%) completed 6 months and 223 (68.6%) completed 1 year of treatment. Of the 102 patients (31.4%) who discontinued prior to 1 year of exposure, 34 (10.5%) did so for lack of efficacy, 32 (9.8%) did so for adverse events, 20 (6.2%) withdrew consent, 12 (3.7%) withdrew at investigators’ discretion, one (0.3%) withdrew because of pregnancy, and three (0.9%) withdrew for other unspecified reasons. Similar to trial 301, efficacy was stable or increased during 1 year of treatment; the median seizure frequency reduction was 32% in the first 4 weeks of treatment, increasing each 12-week interval to 39.3% in the 41- to 52-week time period. The ≥ 50% responder rate was 37% during the first month of the extension study, increasing to 41.5% during the last interval (41–52 weeks). The proportion of patients who were seizure free was 5% initially and increased during each 12-week block. Eight patients (2.5%) were seizure free for the entire 12 months of follow-up. Median number of Days per 4 weeks with seizures also decreased gradually during the follow-up period to 3.7 during weeks 41–52; 43 patients (10.8%) were seizure free during weeks 41–52 [53].
Two-year follow-up was reported for 152 consecutive patients in Portugal starting ESL after EMA approval; retention rates were 71.3% at 12 months of treatment and 62.8% at 24 months [54]. The ≥ 50% responder rates were 25.7% at 12 months and 17.1% at 24 months of treatment. The median seizure frequency reduction was 62.5% at 12 months and 60% at 24 months, while seizure-free rates were 7.9% at 12 months and 5.3% at 24 months (which included three patients on ESL monotherapy) [54]. Dose changes of concomitant AEDs and other medication changes were possible during this observation period.
The ESLIBASE study reported 1-year follow-up data for patients started on ESL after EMA approval in Spain [55]. Of 327 patients started on ESL, 237 (72.5%) continued taking ESL after 1 year; the ≥ 50% responder rate was 52.5% at 12 months, and the seizure-free rate was 25.3% at 12 months.
4.3 Long-Term Safety Data
A pooled analysis of the four pivotal trials and post-marketing surveillance from October 2009 to October 2015 was recently published [42]. In this analysis, the most common TEAEs (affecting > 10% of patients) were dizziness, somnolence, headache, and nausea, which were dose dependent. In post-marketing reporting, the most commonly reported TEAEs were hyponatremia (n = 206 patients, 10.2% of TEAEs reported), seizure (n = 118 patients, 5.8% of TEAEs reported), dizziness (n = 82 patients, 4.1% of TEAEs reported), rash (n = 76 patients, 2.6% of TEAEs reported), fatigue (n = 42 patients, 2.1% of TEAEs), and nausea (n = 37 patients, 1.8% of TEAEs) [42]. Psychiatric disorders including confusional state, depression, suicidal ideation, and aggression comprised 7.4% of TEAEs; there were two suicide attempts and two completed suicides [42]. There were 21 cases of cardiac abnormalities [42].
The 2-year clinical observational study (n = 152) found dizziness, somnolence, and nausea to be the most common TEAEs, similar to shorter study periods. Four patients had serious TEAEs: three with rash and one with bicytopenia (thrombocytopenia and lymphocytopenia) [54]. In this longer study, the majority (56%) of TEAEs occurred within the first 6 months of treatment. TEAEs were more likely in patients who were also taking carbamazepine [54].
Detailed safety data for up to 1 year of exposure are available from the two published open-label extension studies enrolling patients completing two phase III trials [2093-301 (n = 314) and 2093-302 (n = 325)] performed in Europe, South America, and Africa. Extension data from trial 304, which included patients in North America [47,48,49] have not been published separately. In all studies, adverse events, laboratory tests (hematology, biochemistry, thyroid, coagulation, urinalysis), vital signs, weight, and electrocardiogram (ECG) results were tracked. While the majority of TEAEs occurred in the first few weeks of the phase III and monotherapy trials [43, 50, 51], the TEAEs continued to arise during the open-label extension studies (patients from the placebo arms of the studies who entered the extensions received ESL for the first time). Sixty-seven percent of patients reported an adverse event at least once, mostly mild or moderate, with dizziness, somnolence, and headache being most common [52, 53]. A total of 48 patients (7.5%) discontinued the extension studies early, 11 from 301 and 39 from 302. Trial 301 found that TEAE and dizziness incidence decreased over time, from 35% during the first 3 months to 12.7% during the last 3 months [52]. Serious TEAEs occurred in 19 patients, with those occurring in more than one patient being tonic–clonic seizures (three patients) and drug toxicity (two patients) [52]. There were two cases of DRESS in pivotal trial 301 and two in post-marketing surveillance [42].
Due to the similarity of ESL and oxcarbazepine, risks for hyponatremia were assessed; it was of particular interest whether patients without hyponatremia after initial weeks of ESL treatment were at risk during longer treatment periods. In post-marketing surveillance, the most commonly reported TEAE was hyponatremia, accounting for 10.2% of TEAEs [42]. However, whether these patients had clinically diagnosed hyponatremia with serum sodium < 125 mEq/L or had a reduction in sodium was not described. The risks for hyponatremia (< 125 mEq/L) in pivotal trials was 0.5 with the 800-mg/day dosage and 1.5% with the 1200-mg/day dosage. The extension of study 301 found ten patients (3.2%) with non-significant decreases in sodium, and sodium decreased for one patient (0.3%) from normal to significantly “low” [52]. The extension study 302 found “no relevant laboratory results raising concerns” with a mean sodium of 139.4 (± 3.7) mmol/L at time of entry and 140.3 (± 4.4) mmol/L at the end of week 52, though four participants (1.2%) reported a sodium decrease and two participants (0.6%) reported a sodium increase [53]. Patients converting from placebo to ESL in the extension study and developing hyponatremia are not described. Trial 304 (with a 12-week treatment period) found that serum sodium tended to stabilize after 2 months of treatment with ESL [47]. No other clinically significant changes in laboratory values, vital signs, weight, or ECGs were reported in the extension studies [52, 53].
There were three deaths due to drowning and one due to severe coronary atherosclerosis during the extension periods. These deaths were considered unrelated to the study medication [52, 53].
In the 1-year observational study from Spain (n = 327), 40.7% of patients reported TEAEs by 12 months. Dizziness, somnolence, and nausea were the most common TEAEs; rash/pruritus occurred in 12 patients (3.6%), cognitive or memory changes in 11 (3.3%), hyponatremia in nine (2.7%), visual hallucinations in one (0.3%), and hepatic enzyme elevation in 1 (0.3%) [55].
One observational study of 32 patients starting ESL after new-onset, post-stroke seizures found hyponatremia in four patients (12.5%) followed up to 2 years, of which three patients had symptomatic hyponatremia [56].
4.4 Concomitant AEDs
The four pivotal trials excluded patients taking oxcarbazepine [because of a shared active (l)-licarbazepine metabolite] or felbamate [43]. More than 20% of patients took carbamazepine, lamotrigine, and valproic acid, which allowed additional analysis of subgroups of patients taking those AEDs. The three pivotal trials showed no difference in efficacy regarding whether or not patients were also taking carbamazepine, lamotrigine, or valproic acid [43]. These subgroups were not examined in the currently available data from the open-label extension studies. However, the observational, 1-year ESLIBASE study found a significant difference in responder rates between patients taking a sodium channel AED at baseline (47.7%) and those not taking a sodium channel AED at baseline (66.7%) [55].
4.5 Discussion
Responder rates were similar in the clinical trial and extension studies of ESL; in the pivotal trials of adjunctive ESL treatment, the pooled ≥ 50% responder rate was 36–44% [43,44,45,46], while the ≥ 50% responder rate was 37–53% in the extension studies [52], increasing over time (which may be expected as patients who did not have efficacy stopped the medication). Retention rates at 1 year were 68–76% [52, 53]. The median relative reduction in seizure frequency was similar: 35–39% [43] in the clinical trials and 32–40% in the extension studies [52, 53]. Long-term efficacy data from the additional North American trial requested by the FDA to satisfy concerns over data completeness [47, 49] are not available.
The tolerability and safety profile of ESL in the limited available long-term safety data was consistent with that of the pivotal clinical trials; nausea, somnolence, headache, and dizziness were the most common TEAEs in both short- and long-term studies. While most TEAEs occurred in the first few weeks of the phase III and monotherapy trials [43, 50, 51], new instances of TEAEs occurred during the open-label extension studies, and the majority (67%) of patients reported at least one TEAE [52, 53]. The proportion of patients with hyponatremia in the extension studies was low, with four patients (1.2%) and 11 patients (3.5%) in the extension studies experiencing a decrease in sodium, compared to the 5.1% of patients experiencing at least a 10-point drop in sodium reported on the product insert. In the small (n = 32), 2-year observational study of post-stroke seizure patients, 12.5% experienced hyponatremia. Initial trials reported that serum sodium tended to stabilize after the first 2 months [47]. Measuring plasma sodium after approximately a month of therapy has been recommended. The first serious hematologic abnormality, thrombocytopenia and lymphocytopenia, occurred during long-term follow-up [54]. Hypersensitivity reactions (rash and DRESS syndrome), while rare, are a larger concern with ESL than with the other new AEDs studied in this review.
5 Brivaracetam
Brivaracetam is a high-affinity ligand for synaptic vesicle protein 2A (SV2A), with a tenfold higher affinity than levetiracetam [57]. Long-term data on brivaracetam are available from post-trial extension studies for 6- to 60-month patient exposures.
5.1 Clinical Trials
Brivaracetam was approved by the EMA and FDA after two phase II [57, 58] and four phase III, double-blind, randomized, controlled trials, with 12- to 16-week treatment periods [59,60,61,62]. Doses of 20–200 mg were found to be effective, though dosages of 50 mg/day were not effective across trials. The ≥ 50% responder rate during the trials was 27.3–32.7% for 50 mg/day [59, 60], 36.0–38.9% for brivaracetam 100 mg/day [59, 62] and 37.8% for brivaracetam 200 mg/day [62], and 34.1% in a flexible dose study [61].
Retention rates were 90.6–92.2% in the phase III trials. The most common TEAEs reported were headache, somnolence, dizziness, and fatigue [59,60,61,62].
5.2 Long-Term Efficacy Data
Pooled data from follow-up of phase II and phase III studies found that the 12-, 24-, and 60-month retention rates were 79.8, 68.1 and 54.4%, respectively [63]. Of the 2186 patients who received brivaracetam in one of the phase II or III trials, the majority (93.86%) completed the studies and continued in open-label extension studies [63]. Of the 4140 patients in the combined safety and efficacy populations who entered long-term follow-up, 1813 (43.8%) were still in long-term follow-up after 60 months. Reasons for discontinuing were lack of efficacy in 1041 (25.1%), adverse events in 553 (13.3%), patient choice in 341 (8.0%), loss to follow-up in 143 (3.5%), and other unspecified reasons in 219 (5.3%).
In the long-term follow-up of patients from the clinical trials, the ≥ 50% responder rate increased from 43.5% from months 1 to 3 of follow-up to 71.0% at 58–60 months of follow-up. Of the 4.9% of patients who were seizure free at 6 months, there was a slight decrease in seizure freedom over time, with 4.2, 3.0 and 3.3% remaining seizure free at 12, 24 and 60 months, respectively [63]. 16.9% of patients were seizure free for any 6-month interval during follow-up, 10.4% for any 12-month interval, and 5.5% for any 24-month interval.
Post-marketing follow-up of 262 patients for up to 12 months found a median retention time of 6.1 months and a relatively stable rate of withdrawal over 12 months in patients without prior levetiracetam exposure [64].
5.3 Long-Term Safety Data
In pooled data at 12, 24, and 60 months of extension study treatment for 2186 patients, 84.5% reported at least one TEAE [63]. Headache and dizziness were the most common TEAEs, followed by somnolence, nasopharyngitis, fatigue, and seizure. A total of 264 patients (12.1%) had significant TEAEs leading to discontinuation of brivaracetam, most commonly seizure (31 patients, 1.4%), pregnancy (19 patients, 0.9%), somnolence (16 patients, 0.7%), depression (14 patients, 0.6%), dizziness (14 patients, 0.6%), fatigue (12 patients, 0.5%), suicidal ideation (11 patients, 0.5%) and suicide attempt (10 patients, 0.5%) [63].
Psychiatric and behavioral effects are of particular concern in brivaracetam, because of the risks associated with levetiracetam treatment. In the long-term follow-up data, 156 patients (7.1%) reported depression, 114 (5.2%) reported irritability, and 107 (4.9%) reported anxiety. The investigators did not find a dose-dependent effect for psychiatric TEAEs. Forty-three patients (2.0%) reported suicidal ideations, and two patients completed suicide [63].
Thirty-three patients taking brivaracetam died, with six deaths due to cancer, four due to drowning, four due to SUDEP, three due to myocardial infarction, two to accident, and two to suicide [63]. Three cases of SUDEP and one suicide were considered possibly treatment related.
The pooled data reports that “other safety and tolerability assessments, including laboratory tests, did not reveal any issues of clinical concern, with no dosage-related effects or increases in incidence over time,” and reports no significant ECG changes [63].
5.4 Concomitant AEDs
Levetiracetam use was allowed in some phase III trials [59, 61] and excluded in another [62]. Klein et al. found a significant difference in the seizure reduction rate between patients who had previously taken levetiracetam and in those who were levetiracetam naïve [62], with the response rates being higher in those who had not previously tried levetiracetam. Those who stopped levetiracetam because of adverse effects had a higher response than did those who stopped because of inefficacy [62]. The long-term follow-up studies did not examine the efficacy or side effects by specific concomitant AEDs.
5.5 Discussion
Efficacy of brivaracetam in long-term studies was similar or higher than that during clinical trials, with ≥ 50% responder rates of 27–38% during clinical trials [59,60,61,62] and ≥ 50% responder rates of 43.5% increasing to 71% during long-term follow-up (likely reflecting the tendency for patients who do not receive efficacy to discontinue the medication) [63]. The retention rates were compared to those completing blinded study periods; 90–92% of patients completed the assigned phase III trial, and retention rates fell from 79.8% at 12 months to 54.4% at 60 months [63]. The discontinuation rate for patients on brivaracetam was stable during the first 12 months of surveillance during one post-marketing study of 262 patients, with a median retention time of 6.1 months [64].
The majority of patients (84.5%) reported at least one TEAE in long-term brivaracetam follow-up, with no dose-dependent effect reported [63]. The most common TEAEs in long-term follow-up were similar to those seen in clinical trials (dizziness, headache, fatigue) [63]. However, depression, irritability, and anxiety were seen at higher rates in long-term data than during clinical trials, and suicidal ideation, ten suicide attempts, and two completed suicides (one thought possibly related to brivaracetam treatment) were reported in the long-term follow-up data, underscoring the importance of long-term safety monitoring, especially for mood disorders.
6 Comparing Extension Study Results for “Third-Generation” AEDs
The extension studies for lacosamide, perampanel, eslicarbazepine acetate, and brivaracetam contain some heterogeneity in patient recruitment that influenced study retention and tolerability results. Many patients treated with perampanel, for example, received treatment with three concomitant AEDs, while eslicarbazepine acetate restricted treatment to two other AEDs for most patients. Patient retention during extension studies, mean seizure responder rates, and occurrence of common adverse drug effects can be compared, though, with the caution that studies varied in the severity of epilepsy in the populations they enrolled. As shown in Figs. 1, 2, study retention rates during 1–2 years of open study treatment were comparable for the four AEDs; approximately 48–59% of patients reported long-term improvements (≥ 50% responder rates) at 1 year and 57–61% at 2 years. No unusual safety concerns were detected in extension studies that were not recognized during the controlled pivotal trials (Table 1, Fig. 3). Hypersensitivity, while rare, was more common in patients on ESL than the other new AEDs studied. Psychiatric side effects such as depression and irritability were lowest in lacosamide patients, and ranged from 7.1 to 10.9% in the other AEDs in this review. A major finding is that a large subset of patients completing each of the pivotal trials and those who tolerated and continued into “open” extension treatment appeared to benefit from the new AED treatment. Large proportions of patients continued treatment for 1–4 years, with stable responses and with no new adverse events reported. A clinical conclusion from these comparisons is that a large proportion of patients tolerate each of these AEDs well and choose to continue long-term treatment, with continued reductions in seizures. Careful monitoring for the most common and serious TEAEs reported in the long-term extension studies would permit most patients with medically refractory epilepsy to try each of these new treatments.

Retention rates at 1 and 2 years of exposure during adjunctive treatment of partial-onset seizures: pooled extension studies for four new AEDs. AED antiepileptic drug, BRV brivaracetam, ESL eslicarbazepine acetate, LCM lacosamide, PER perampanel. †No 2-year extension study outcome data available for ESL. *Retention data for BIA-2093-301 and BIA-2093-302 only [2, 15, 16, 27, 34, 52, 53, 63]

≥50% responder rates at 1 and 2 years of exposure during adjunctive treatment of partial-onset seizures: pooled extension studies for four new AEDs. AED antiepileptic drug, BRV brivaracetam, ESL eslicarbazepine acetate, LCM lacosamide, PER perampanel. †No 2-year extension study outcome data available for ESL. *Responder rate data for BIA-2093-301 and BIA-2093-302 only [2, 15, 16, 27, 34, 52, 53, 63]

Incidence of common† TEAEs during adjunctive treatment of partial-onset seizures: pooled extension studies for four new AEDs. AED antiepileptic drug, BRV brivaracetam, ESL eslicarbazepine acetate, LCM lacosamide, PER perampanel, TEAE treatment emergent adverse event. †TEAEs > 10%; also included other specific TEAEs for comparison. Data is unreported or unavailable for < 5% incidence [2, 15, 16, 27, 34, 42, 52, 53, 63]
7 Comparing “Third-Generation” AEDs with Previous AEDs: Contributions of Extensions Studies
Third-generation AEDs provide treatment options for the 30–40% of patients with drug resistant epilepsy and for additional patients who fail to tolerate previous AEDs. There are limited data directly comparing the efficacy of new and older AEDs; however, tolerability, safety, and pharmacologic data can be compared using extension study results.
-
1.
Lacosamide has a slow sodium channel inactivation mechanism that differentiates it from traditional “fast” sodium channel modulators, such as carbamazepine and phenytoin. Consequently, it may be effective in treating seizures in patients failing treatment with older sodium channel modulators. The most common adverse drug effects of lacosamide—dizziness, imbalance, sedation—are similar to traditional sodium modulators, and it is common to see pharmacodynamic interactions with dizziness, imbalance, sedation, etc. when combining lacosamide with high doses of traditional sodium channel modulators. Extension studies and post-marketing series show patients tolerate high dosages of lacosamide (400–600 mg daily) if dosages of other sodium channel agents are decreased or not used [3, 14]. In extension studies, many patients tolerated and appeared to benefit from lacosamide 600 mg/day (300 mg twice daily) dosages, even though pharmacodynamic interactions limited responses in adjuvant use with other sodium channel modulators.
-
2.
Perampanel has a 105-h half-life and is the first selective AMPA receptor antagonist to be approved to treat epilepsy with an indication for focal onset and generalized onset seizures. Due to perampanel’s long half-life and receptor mechanisms, it was tolerated well in extension studies when started slowly—2-mg increases every 2 weeks to 8-mg daily dosage [27]. Perampanel is dosed at bedtime to avoid transient post-dose drowsiness. Perampanel efficacy is correlated with increasing mean plasma concentrations [65]; carbamazepine induces perampanel metabolism and reduces concentrations. Clinicians may need to titrate perampanel to high dosages (e.g., 10 or 12 mg daily) when combined with carbamazepine (or discontinue carbamazepine).
-
3.
ESL is chemically related to oxcarbazepine, with an improved tolerability profile in pivotal and extension trials, particularly when comparing tolerability at high doses [65,66,67]. This appears to be due to ESL being a pro-drug that is converted predominantly to S-licarbazepine (also called eslicarbazepine). Oxcarbazepine is converted into S-licarbazepine and higher amounts of l-licarbazepine than ESL. The l-licarbazepine enantiomer has a shorter half-life and apparently more adverse CNS-related effects than S-licarbazepine. Although there is limited published ESL extension study data, the drug was well tolerated, with a low incidence of CNS-related drug effects [66]. Due to the drugs’ common active metabolites, however, patients with persisting seizures on high doses of oxcarbazepine may not respond to ESL treatment. Extension study treatment shows patients who previously did not tolerate oxcarbazepine may tolerate and benefit from ESL therapy.
-
4.
Like levetiracetam, brivaracetam is a ligand of SV2A and has a similar tolerability profile [68, 69]. Some patients, however, with behavioral problems during levetiracetam treatment have improved behavior when converted to brivaracetam [70]. Similarly, some patients with somnolence or irritability with levetiracetam therapy tolerate and respond to brivaracetam [64]. Brivaracetam treatment reduced seizure frequency in patients previously failing levetiracetam therapy; however, responses were reduced [62].
8 Conclusion
A large proportion of patients continued treatment with third-generation AEDs over 1–4 years. While most had stable responses with no new adverse events reported, the trials enrolled patients with drug-resistant epilepsy and responses to the newer AEDs were variable. Some patients not tolerating older AEDs benefit from treatment with new AEDs, including related compounds, e.g., oxcarbazepine–eslicarbazepine [69] and levetiracetam–brivaracetam [64]. Perampanel offers treatment with a new AMPA receptor mechanism and has broad spectrum efficacy against focal onset seizures and generalized onset tonic–clonic seizures. Lacosamide through a new slow sodium inactivation mechanism often provides benefit for patients who fail treatment with traditional sodium channel agents (carbamazepine, phenytoin), though, these may have pharmacodynamic interactions when used together. These treatment options often benefit individual patients, particularly those who have not tolerated older AEDs. Overall, extension studies show that a large proportion of patients tolerated third-generation AEDs, with most reporting no new adverse events.
References
US Food and Drug Administration. Step 3: clinical research. US Food Drug Administration. [Internet]. 2015; pp 1–4. http://www.fda.gov/ForPatients/Approvals/Drugs/ucm405622.htm#Clinical_Research_Phase_Studies. Accessed 9 Aug 2017.
Rosenfeld W, Fountain NB, Kaubrys G, Ben-Menachem E, Mcshea C, Isojarvi J, et al. Safety and efficacy of adjunctive lacosamide among patients with partial-onset seizures in a long-term open-label extension trial of up to 8 years. Epilepsy Behav. 2014;41:164–70. https://doi.org/10.1016/j.yebeh.2014.09.074.
Edwards HB, Cole AG, Griffiths AS, Lin B, Bean A, Krauss GL. Minimizing pharmacodynamic interactions of high doses of lacosamide. Acta Neurol Scand. 2012;125:228–33.
Eke T, Talbot JF, Lawden MC. Severe persistent visual field constriction associated with vigabatrin. Br Med J. 1997;314:180–1.
Krauss GL, Johnson MA, Miller NR. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings. Neurology [Internet]. 1998;50:614–8. Available from: http://search.ebscohost.com/login.aspx?direct=true&db=cmedm&AN=9521245&site=ehost-live. Accessed 9 Aug 2017.
Krauss G, Faught E, Foroozan R, Pellock JM, Sergott RC, Shields WD, et al. Sabril ® registry 5-year results: characteristics of adult patients treated with vigabatrin. Epilepsy Behav [Internet]. 2016;56:15–9. https://doi.org/10.1016/j.yebeh.2015.12.004.
Eypasch E, Lefering R, Kum CK, Troidl H. Probability of adverse events that have not yet occurred: a statistical reminder. BMJ [Internet]. 1995; 311:619–20. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2550668&tool=pmcentrez&rendertype=abstract. Accessed 9 Aug 2017.
Thakkar K, Billa G, Rane J, Chudasama H, Goswami S, Shah R. The rise and fall of felbamate as a treatment for partial epilepsy—aplastic anemia and hepatic failure to blame? Expert Rev Neurother Inform Healthc. 2015;15:1373–5.
Berlin JA, Glasser SC, Ellenberg SS. Adverse event detection in drug development: recommendations and obligations beyond phase 3. Am J Public Health. 2008;98:1366–71 (American Public Health Association).
Ben-Menachem E, Biton V, Jatuzis D, Abou-Khalil B, Doty P, Rudd GD. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308–17.
Chung S, Sperling MR, Biton V, Krauss G, Hebert D, Rudd GD, et al. Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia. 2010;51:958–67.
Halász P, Kälviäinen R, Mazurkiewicz-Beldzińska M, Rosenow F, Doty P, Hebert D, et al. Adjunctive lacosamide for partial-onset seizures: efficacy and safety results from a randomized controlled trial. Epilepsia. 2009;50:443–53.
Biton V. Lacosamide for the treatment of partial-onset seizures. Expert Rev. Neurother. [Internet]. 2012;12: 645–55. http://www.expert-reviews.com/doi/abs/10.1586/ern.12.50?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub=pubmed. Accessed 9 Aug 2017.
Sake JK, Hebert D, Isojrvi J, Doty P, De Backer M, Davies K, et al. A pooled analysis of lacosamide clinical trial data grouped by mechanism of action of concomitant antiepileptic drugs. CNS Drugs. 2010;24:1055–68.
Husain A, Chung S, Faught E, Isojarvi J, McShea C, Doty P. Long-term safety and efficacy in patients with uncontrolled partial-onset seizures treated with adjunctive lacosamide: results from a phase III open-label extension trial. Epilepsia. 2012;53:521–8.
Rosenow F, Kelemen A, Ben-Menachem E, Mcshea C, Isojarvi J, Doty P, et al. Long-term adjunctive lacosamide treatment in patients with partial-onset seizures. Acta Neurol Scand. 2016;133:136–44.
Vossler DG, Wechsler RT, Williams P, Byrnes W, Therriault S. Long-term exposure and safety of lacosamide monotherapy for the treatment of partial-onset (focal) seizures: results from a multicenter, open-label trial. Epilepsia. 2016;57:1625–33.
Verrotti A, Loiacono G, Pizzolorusso A, Parisi P, Bruni O, Luchetti A, et al. Lacosamide in pediatric and adult patients: comparison of efficacy and safety. Seizure. 2013;22:210–6.
Lattanzi S, Cagnetti C, Foschi N, Provinciali L, Silvestrini M. Lacosamide monotherapy for partial onset seizures. Seizure. 2015;27:71–4.
Grosso S, Parisi P, Spalice A, Verrotti A, Balestri P. Efficacy and safety of lacosamide in infants and young children with refractory focal epilepsy. Eur J Paediatr Neurol [Internet]. 2014; 18:55–9. http://www.scopus.com/inward/record.url?eid=2-s2.0-84892507265&partnerID=40&md5=5f28e91dc70822f386224ea1ccc93091. Accessed 9 Aug 2017.
Kim JS, Kim H, Lim BC, Chae J-H, Choi J, Kim KJ, et al. Lacosamide as an adjunctive therapy in pediatric patients with refractory focal epilepsy. Brain Dev. 2014;36:510–5.
García-Morales I, Delgado RT, Falip M, Campos D, García ME, Gil-Nagel A. Early clinical experience with lacosamide as adjunctive therapy in patients with refractory focal epilepsy and nocturnal seizures. Seizure. 2011;20:801–4.
Giraldez BG, Toledano R, Garcia-Morales I, Gil-Nagel A, Lopez-Gonzalez FJ, Tortosa D, et al. Long-term efficacy and safety of lacosamide monotherapy in the treatment of partial-onset seizures: a multicenter evaluation. Seizure [Internet]. 2015; 29:119–22. http://www.elsevier.com/inca/publications/store/6/2/3/0/7/1/index.htt%5Cnhttp://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=emed17&AN=604863003%5Cnhttp://www.tdnet.com/AalSy/resolver?sid=OVID:embase&id=pmid:26076854&id=doi:10.1016%2Fj.se. Accessed 9 Aug 2017.
Toupin J-F, Lortie A, Major P, Diadori P, Vanasse M, Rossignol E, et al. Efficacy and safety of lacosamide as an adjunctive therapy for refractory focal epilepsy in paediatric patients: a retrospective single-centre. Epileptic Disord. 2015;17:436–43.
Andrade-Machado R, Luque-Navarro-De Los Reyes J, Benjumea-Cuartas V, Restrepo JFA, Jaramillo-Jiménez E, Andrade-Gutierrez G, et al. Efficacy and tolerability of add-on lacosamide treatment in adults with Lennox-Gastaut syndrome: an observational study. Seizure. 2015;33:81–7.
Steinhoff BJ, Eckhardt K, Doty P, De Backer M, Brunnert M, Schulze-Bonhage A. A long-term noninterventional safety study of adjunctive lacosamide therapy in patients with epilepsy and uncontrolled partial-onset seizures. Epilepsy Behav. [Internet]. 2017; 58:35–43. https://doi.org/10.1016/j.yebeh.2016.02.041.
Krauss GL, Perucca E, Ben-Menachem E, Kwan P, Shih JJ, Clément JF, et al. Long-term safety of perampanel and seizure outcomes in refractory partial-onset seizures and secondarily generalized seizures: results from phase III extension study 307. Epilepsia. 2014;55:1058–68.
French JA, Krauss GL, Wechsler RT, Wang X-F, DiVentura B, Brandt C, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy: a randomized trial. Neurology [Internet]. 2015; 85:950–7. http://www.ncbi.nlm.nih.gov/pubmed/26296511%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4567458. Accessed 9 Aug 2017.
French JA, Krauss GL, Biton V, Squillacote D, Yang H, Laurenza A, et al. Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79:589–96.
French JA, Krauss GL, Steinhoff BJ, Squillacote D, Yang H, Kumar D, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. 2013;54:117–25.
Krauss GL, Serratosa JM, Villanueva V, Endziniene M, Hong Z, French J, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurol. [Internet]. 2012; 78:1408–15. http://www.neurology.org/content/78/18/1408.abstract. Accessed 9 Aug 2017.
Steinhoff BJ, Ben-Menachem E, Ryvlin P, Shorvon S, Kramer L, Satlin A, et al. Efficacy and safety of adjunctive perampanel for the treatment of refractory partial seizures: a pooled analysis of three phase III studies. Epilepsia. 2013;54:1481–9.
Krauss GL, Bar M, Biton V, Klapper JA, Rektor I, Vaiciene-Magistris N, et al. Tolerability and safety of perampanel: two randomized dose-escalation studies. Acta Neurol Scand. 2012;125:8–15.
Rektor I, Krauss GL, Bar M, Biton V, Klapper JA, Vaiciene-Magistris N, et al. Perampanel Study 207: long-term open-label evaluation in patients with epilepsy. Acta Neurol Scand. 2012;126:263–9.
Juhl S, Rubboli G. Perampanel as add-on treatment in refractory focal epilepsy. The Dianalund experience. Acta Neurol Scand. 2016;134:374–7.
Villanueva V, Garcés M, López-González FJ, Rodriguez-Osorio X, Toledo M, et al. Safety, efficacy and outcome-related factors of perampanel over 12 months in a real-world setting: the FYDATA study. Epilepsy Res. 2016;126:201–10.
Singh K, Shah YD, Luciano D, Friedman D, Devinsky O, Kothare SV. Safety and efficacy of perampanel in children and adults with various epilepsy syndromes: a single-center postmarketing study. Epilepsy Behav. 2016;61:41–5.
Snoeijen-Schouwenaars FM, van Ool JS, Tan IY, Schelhaas HJ, Majoie MHJM. Evaluation of perampanel in patients with intellectual disability and epilepsy. Epilepsy Behav. 2017;66:64–7.
Maurousset A, Limousin N, Praline J, Biberon J, Corcia P, De Toffol B. Adjunctive perampanel in refractory epilepsy: experience at tertiary epilepsy care center in Tours. Epilepsy Behav. 2016;61:237–41.
Tambucci R, Basti C, Maresca M, Coppola G, Verrotti A. Update on the role of eslicarbazepine acetate in the treatment of partial-onset epilepsy. Neuropsychiatr. Dis. Treat. 2016;12:1251–60.
Shirley M, Dhillon S. Eslicarbazepine acetate monotherapy: a review in partial-onset seizures. Drugs. 2016;76:707–17 (Springer International Publishing).
Gama H, Vieira M, Costa R, Graça J, Magalhães LM, Soares-da-Silva P. Safety profile of eslicarbazepine acetate as add-on therapy in adults with refractory focal-onset seizures: from clinical studies to 6 years of post-marketing experience. Drug Saf. 2017; vol 40, pp 1–10 (Springer International Publishing).
Gil-Nagel A, Elger C, Ben-Menachem E, Halász P, Lopes-Lima J, Gabbai AA, et al. Efficacy and safety of eslicarbazepine acetate as add-on treatment in patients with focal-onset seizures: integrated analysis of pooled data from double-blind phase III clinical studies. Epilepsia. 2013;54:98–107 (Blackwell Publishing Ltd).
Elger C, Halász P, Maia J, Almeida L, Soares-Da-Silva P. Efficacy and safety of eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures: a randomized, double-blind, placebo-controlled, parallel-group phase III study. Epilepsia. 2009;50:454–63.
Ben-Menachem E, Gabbai AA, Hufnagel A, Maia J, Almeida L, Soares-da-Silva P. Eslicarbazepine acetate as adjunctive therapy in adult patients with partial epilepsy. Epilepsy Res. 2010;89:278–85.
Gil-Nagel A, Lopes-Lima J, Almeida L, Maia J, Soares-da-Silva P. Efficacy and safety of 800 and 1200 mg eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures. Acta Neurol Scand. 2009;120:281–7 (Blackwell Publishing Ltd).
Sperling MR, Abou-Khalil B, Harvey J, Rogin JB, Biraben A, Galimberti CA, et al. Eslicarbazepine acetate as adjunctive therapy in patients with uncontrolled partial-onset seizures: results of a phase III, double-blind, randomized, placebo-controlled trial. Epilepsia. 2015;56:244–53.
Rocamora R. A review of the efficacy and safety of eslicarbazepine acetate in the management of partial-onset seizures. Ther Adv Neurol Disord. 2015;8:178–86 (SAGE Publications).
U.S. Food and Drug Administration/Center for Drug Evaluation and Research. NDA 22416 Aptiom/Eslicarbazepine Acetate. [Internet]. 2013. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/022416orig1s000medr.pdf. Accessed Aug 2017.
Jacobson MP, Pazdera L, Bhatia P, Grinnell T, Cheng H, Blum D, et al. Efficacy and safety of conversion to monotherapy with eslicarbazepine acetate in adults with uncontrolled partial-onset seizures: a historical-control phase III study. BMC Neurol. 2015;15:46.
Sperling MR, Harvey J, Grinnell T, Cheng H, Blum D. Efficacy and safety of conversion to monotherapy with eslicarbazepine acetate in adults with uncontrolled partial-onset seizures: a randomized historical-control phase III study based in North America. Epilepsia. 2015;56:546–55.
Halasz P, Cramer JA, Hodoba D, Czlonkowska A, Guekht A, Maia J, et al. Long-term efficacy and safety of eslicarbazepine acetate: results of a 1-year open-label extension study in partial-onset seizures in adults with epilepsy. Epilepsia. 2010;51:1963–9 (Blackwell Publishing Ltd).
Hufnagel A, Ben-Menachem E, Gabbai AA, Falcão A, Almeida L, Soares-da-Silva P. Long-term safety and efficacy of eslicarbazepine acetate as adjunctive therapy in the treatment of partial-onset seizures in adults with epilepsy: results of a 1-year open-label extension study. Epilepsy Res. 2013;103:262–9 (Elsevier B.V.).
Correia FD, Freitas J, Magalhães R, Lopes J, Ramalheira J, Lopes-Lima J, et al. Two-year follow-up with eslicarbazepine acetate: a consecutive, retrospective, observational study. Epilepsy Res. 2014;108:1399–405.
Villanueva V, Serratosa JM, Guillamón E, Garcés M, Giráldez BG, Toledo M, et al. Long-term safety and efficacy of eslicarbazepine acetate in patients with focal seizures: results of the 1-year ESLIBASE retrospective study. Epilepsy Res. 2014;108:1243–52.
Gupta DK, Bhoi SK, Kalita J, Misra UK. Hyponatremia following esclicarbazepine therapy. Seizure. 2015;29:11–4.
French JA, Costantini C, Brodsky A, Von Rosenstiel P. Adjunctive brivaracetam for refractory partial-onset seizures: a randomized, controlled trial. Neurology. 2010;75:519–25.
Van Paesschen W, Hirsch E, Johnson M, Falter U, von Rosenstiel P. Efficacy and tolerability of adjunctive brivaracetam in adults with uncontrolled partial-onset seizures: a phase IIb, randomized, controlled trial. Epilepsia. 2013;54:89–97.
Ryvlin P, Werhahn KJ, Blaszczyk B, Johnson ME, Lu S. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from a double-blind, randomized, placebo-controlled trial. Epilepsia. 2014;55:47–56.
Biton V, Berkovic SF, Abou-Khalil B, Sperling MR, Johnson ME, Lu S. Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: a phase III randomized, double-blind, placebo-controlled trial. Epilepsia. 2014;55:57–66.
Kwan P, Trinka E, Van Paesschen W, Rektor I, Johnson ME, Lu S. Adjunctive brivaracetam for uncontrolled focal and generalized epilepsies: results of a phase III, double-blind, randomized, placebo-controlled, flexible-dose trial. Epilepsia. 2014;55:38–46.
Klein P, Schiemann J, Sperling MR, Whitesides J, Liang W, Stalvey T, et al. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia. 2015;56:1890–8.
Toledo M, Whitesides J, Schiemann J, Johnson ME, Eckhardt K, McDonough B, et al. Safety, tolerability, and seizure control during long-term treatment with adjunctive brivaracetam for partial-onset seizures. Epilepsia. 2016;57:1139–51.
Steinig I, von Podewils F, Möddel G, Bauer S, Klein KM, Paule E, et al. Postmarketing experience with brivaracetam in the treatment of epilepsies: a multicenter cohort study from Germany. Epilepsia. 2017;58:1208–16.
Kramer LD, Satlin A, Krauss GL, French J, Perucca E, Ben-Menachem E, et al. Perampanel for adjunctive treatment of partial-onset seizures: a pooled dose-response analysis of phase III studies. Epilepsia. 2014;55:423–31.
Biton V, Rogin JB, Krauss G, Abou-Khalil B, Rocha JF, Moreira J, et al. Adjunctive eslicarbazepine acetate: a pooled analysis of three phase III trials. Epilepsy Behav. 2017;72:127–34 (United States).
Barcs G, Walker EB, Elger CE, Scaramelli A, Stefan H, Sturm Y, et al. Oxcarbazepine placebo-controlled, dose-ranging trial in refractory partial epilepsy. Epilepsia. 2000;41:1597–607 (United States).
Toledo M, Beale R, Evans JS, Steeves S, Elmoufti S, Townsend R, et al. Long-term retention rates for antiepileptic drugs: a review of long-term extension studies and comparison with brivaracetam. Epilepsy Res. 2017;138:53–61 (Netherlands).
Ben-Menachem E, Mameniskiene R, Quarato PP, Klein P, Gamage J, Schiemann J, et al. Efficacy and safety of brivaracetam for partial-onset seizures in 3 pooled clinical studies. Neurology. 2016;87:314–23 (United States).
Yates SL, Fakhoury T, Liang W, Eckhardt K, Borghs S, D’Souza J. An open-label, prospective, exploratory study of patients with epilepsy switching from levetiracetam to brivaracetam. Epilepsy Behav. 2015;52:165–8 (United States).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Charlotte S. Kwok, Emily L. Johnson, MD declare they have no conflicts to disclose. Gregory Krauss, MD is a consultant and investigator for Eisai Laboratory and SK Bios and an investigator for UCB Pharma.
Funding
No funding was received for this article.
Rights and permissions
About this article

Cite this article
Kwok, C.S., Johnson, E.L. & Krauss, G.L. Comparing Safety and Efficacy of “Third-Generation” Antiepileptic Drugs: Long-Term Extension and Post-marketing Treatment. CNS Drugs 31, 959–974 (2017). https://doi.org/10.1007/s40263-017-0480-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0480-6