
Abstract
Palbociclib, ribociclib, and abemaciclib are inhibitors of the cyclin-dependent kinases 4 and 6 approved for the treatment of locally advanced or metastatic breast cancer. In this review, we provide an overview of the available clinical pharmacokinetic and pharmacodynamic characteristics of these novel drugs, summarize the results of food–effect and drug–drug interaction studies, and highlight exposure–response and exposure–toxicity relationships. All three drugs exhibit a large inter-individual variability in exposure (coefficient of variation range 40–95% for minimum plasma concentration), are extensively metabolized by cytochrome P450 3A4, and have their brain penetration limited by efflux transporters. Abemaciclib has three active metabolites with similar potency that are clinically relevant (i.e., M2, M20, M18), whereas the metabolites of palbociclib and ribociclib are not of clinical significance. Pharmacokinetic exposure increases in a dose-proportional manner for palbociclib, whereas exposure increases under- and over-proportionally with an increasing dose for abemaciclib and ribociclib, respectively. High exposure is associated with an increased risk of neutropenia, and for ribociclib also to corrected QT prolongation. For abemaciclib, a clear exposure–efficacy relationship has been described, while for palbociclib and ribociclib exposure–response analyses remain inconclusive. Future studies are needed to address exposure–efficacy relationships to further improve dosing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Approved cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors palbociclib, ribociclib, and abemaciclib are characterized by a high inter-individual variability in exposure. |
All three CDK4/6 inhibitors are extensively metabolized by cytochrome P450 3A4, and their exposure is dramatically affected by strong cytochrome P450 3A4 modulators. |
Higher exposure is associated with an increased risk of neutropenia for all CDK4/6 inhibitors. In addition, an exposure–efficacy relationship has been demonstrated for abemaciclib, whereas these remain inconclusive so far for palbociclib and ribociclib. |
1 Introduction
Cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors have emerged as important targeted therapies in the treatment of patients with advanced breast cancer. Cyclin-dependent kinase 4 and 6 inhibitors act on the cell cycle and prevent G1-to-S-phase progression. For cells to proceed past this G1-to-S-phase checkpoint, retinoblastoma protein (Rb) needs to be phosphorylated, which is effectuated by CDK4/6 [1]. Aberrations in this pathway are often involved in carcinogenesis, resulting in persistent cell proliferation [2]. Treatment with CDK4/6 inhibitors prevents phosphorylation of Rb and thereby causes a G1 cell-cycle arrest, blocking cell division (Fig. 1).

Mechanism of action of cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors. For cells to progress from G1 to S phase in the cell cycle, retinoblastoma (Rb) needs to get phosphorylated, which is catalyzed by the complex formed by CDK4/6 and cyclin D. Upon phosphorylation of Rb, the transcription factor E2F is released, ultimately resulting in cells proceeding to S phase. CDK4/6 inhibitors prevent Rb from getting phosphorylated and thereby block cell-cycle progression. Created with BioRender.com. P phosphoryl (PO3−)
Currently, three CDK4/6 inhibitors are available in the clinic (i.e., palbociclib, ribociclib, and abemaciclib), and many more are in (pre)clinical development (Table 1). Although all three CDK4/6 inhibitors are approved for treatment in combination with endocrine therapies (i.e., aromatase inhibitors or fulvestrant), only abemaciclib is registered to use as monotherapy. In general, the efficacy of CDK4/6 inhibitors is strikingly consistent between endocrine partners and clinical settings with respect to improved progression-free survival (PFS), and emerging evidence of overall survival benefit, but their toxicity differs. The aim of this review is to summarize the available clinical pharmacokinetic and pharmacodynamic data on the currently approved CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib. In addition, we focus on exposure–response relationships and the potential for pharmacokinetically guided dose individualization.
2 Palbociclib
Palbociclib was the first CDK4/6 inhibitor to obtain approval by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) in 2015. In the pivotal PALOMA-2 study, the addition of palbociclib to letrozole as a first-line treatment for patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer resulted in a median progression-free survival (mPFS) of 24.8 months compared with 14.5 months for letrozole alone (hazard ratio [HR] 0.58 [95% confidence interval (CI) 0.46–0.72], p < 0.001) [3]. Similarly, the PALOMA-3 study demonstrated that palbociclib and fulvestrant were superior to fulvestrant alone in patients who progressed on one or more prior lines of treatment (mPFS 9.2 vs 3.8 months, HR 0.42 [95% CI 0.32–0.56], p < 0.001) [4].
The approved dose of palbociclib is 125 mg once daily (QD) in a 3-weeks-on/1-week-off dosing schedule. This was also the maximum tolerated dose (MTD), with neutropenia being the only dose-limiting toxicity [5].
2.1 Physiochemical Properties and Formulation
Palbociclib is a synthetic 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one, which belongs to the class of pyridopyrimidines (Fig. 2) [7]. Palbociclib is a weak base with two pKa values of 3.9 and 7.4, and a calculated log octanol–water partition coefficient (cLogP, which is an indicator of lipophilicity) of 2.7 [8, 9]. Palbociclib is highly soluble at pH < 4, but its solubility rapidly decreases at higher pH [9]. For drugs to be classified as high-solubility compounds, their highest approved dose needs to be soluble in ≤ 250 mL of aqueous media (i.e., ≥ 0.5 mg/mL for palbociclib) over the entire pH range of 1.0–6.8 [10]. Therefore, palbociclib is considered a low-solubility compound. Together with its high permeability, palbociclib is classified as a class II compound, according to the Biopharmaceutics Classification System [9]. Initially, palbociclib free base was formulated in capsules, but recently a bioequivalent tablet formulation was approved, containing the free base as well [11]. In vitro, palbociclib bound reversibly to its targets and the half-maximal inhibitory concentrations (IC50) were 0.011 and 0.016 µM for CDK4 and CDK6, respectively, corresponding to plasma concentrations of 33.5–48.7 ng/mL when corrected for protein binding [12].

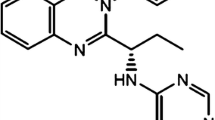
Chemical structures of cyclin-dependent kinase 4 and 6 inhibitors palbociclib (a), ribociclib (b), and abemaciclib (c) and their main metabolites. Chemical structures and metabolism were obtained from the US Food and Drug Administration and European Medicines Agency reviews [9, 46, 75]. This figure was created using ChemDraw Professional 15.0. CYP3A4 cytochrome P450 3A4, FMO flavin-containing monooxygenase, UGT uridine 5ʹ-diphospho-glucuronosyltransferase
2.2 Drug Transporters
In vitro assays demonstrated that palbociclib is a substrate of the efflux transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) [13, 14]. Although this only marginally affected the oral bioavailability in in vivo experiments with P-gp and/or BCRP knock-out mice, it has been demonstrated that the brain penetration was drastically restricted by these transporters [13].
In addition, in vitro and in vivo studies have shown that palbociclib inhibits the organic cation transporter 2 (OCT2) [15], which is involved in the renal tubular secretion of creatinine. Although this has not been studied in patients treated with palbociclib, inhibition of the OCT2 transporter has been associated with an increase in creatinine levels without affecting glomerular filtration [16].
2.3 Clinical Pharmacokinetics
Table 2 provides an overview of selected steady-state pharmacokinetic parameters of palbociclib. The bioavailability of palbociclib is low (46%) [9]. Palbociclib has a large volume of distribution of ~ 2800 L and the total plasma protein binding is 85.3%, with similar binding to albumin and α1-acid glycoprotein [5, 9]. Metabolism mainly takes place by cytochrome P450 (CYP) 3A4 and sulfotransferase 2A1 and results in the formation of many metabolites, of which M22 (i.e., palbociclib glucuronide) is the most abundant (14.8%) and M17 (i.e., a lactam of palbociclib) is pharmacologically active with a similar potency as palbociclib, but accounting for less than 10% of total plasma exposure (Fig. 2) [9]. Hepatic metabolism is the main route of elimination, as in the mass-balance study 74.1% of palbociclib was excreted in feces compared with 17.5% in urine, including both unchanged palbociclib and metabolites [9].
2.4 Pharmacokinetics in Special Populations
2.4.1 Patients with Pediatric Cancer
Currently, palbociclib is not approved for the treatment of pediatric cancer and hence no pharmacokinetic data are available in this subgroup [17]. Several phase I–II studies in pediatric patients are currently ongoing [18,19,20,21,22].
2.4.2 Patients with Renal Impairment
In a clinical study, subjects with mild (estimated glomerular filtration rate [eGFR] 60–90 mL/min/1.73 m2), moderate (eGFR 30–60 mL/min/1.73 m2), and severe (eGFR < 30 mL/min/1.73 m2) renal impairment showed an increase in palbociclib area under the plasma–concentration time curve from time zero to infinity (AUC0–∞) of 39%, 42%, and 31%, respectively, compared with patients with normal renal function. Similarly, maximum plasma concentration (Cmax) was 17%, 12%, and 15% higher, respectively [23]. In a population pharmacokinetic analysis (n = 183, of whom n = 73 and n = 29 with mild and moderate renal impairment, respectively), creatinine clearance did not significantly affect palbociclib exposure, which is consistent with renal clearance being a minor route of elimination [9]. No data are available for patients requiring hemodialysis, but based on the large fraction of palbociclib bound to plasma proteins (i.e., 85.3%), hemodialysis is expected to have limited effect on palbociclib exposure [9]. In conclusion, no dose adjustments are needed for patients with an eGFR ≥ 15 mL/min/1.73 m2 [23], but it should be kept in mind that exposure is 30–40% higher in patients with renal impairment.
2.4.3 Patients with Hepatic Impairment
Palbociclib unbound AUC0–∞ was 17% lower in subjects with mild hepatic impairment (Child–Pugh class A) and 34% and 77% higher in patients with moderate (Child–Pugh class B) and severe (Child–Pugh class C) hepatic impairment, respectively, compared with subjects with a normal hepatic function. Unbound Cmax was increased by 7%, 38%, and 72%, respectively [23]. These findings are in line with the fact that hepatic clearance is the major route of elimination, and were also supported by population pharmacokinetic analyses [9]. Based on the above, no dose adjustments are needed for patients with mild or moderate hepatic impairment, while a dose reduction from 125 mg (standard dose) to 75 mg QD is recommended for patients with severe hepatic impairment [23]. It has to be noted that interpretation of palbociclib plasma concentrations in this subgroup could be complicated by the increasing fraction unbound with worsening hepatic function because this might not be reflected in the total concentration, which is usually measured [23]. In addition, caution is warranted when using the Child–Pugh score in patients with cancer, as this score has not been developed nor validated for this population [24].
2.5 Other Factors Influencing Palbociclib Pharmacokinetics
The effect of other intrinsic factors on palbociclib exposure was investigated using a population pharmacokinetic model. Age and body weight were significant covariates on palbociclib clearance, which was higher in younger patients and in patients with a higher body weight (i.e., compared with a typical patient aged 61 years and 73.7 kg, clearance was increased by 14.7% and decreased by 8.33% in a 45-year-old subject and a 97-year-old subject, respectively, while for body weight, clearance was decreased by 13.2% at a weight of 55 kg and increased by 14.2% at a weight of 97 kg), although these small differences are not expected to be clinically relevant. Sex had no effect on palbociclib exposure [9].
In a subgroup analysis of the PALOMA-2 study, palbociclib exposure was higher in Japanese and other Asian patients compared with non-Asian patients (geometric mean minimum plasma concentration [Cmin] 95.4 ng/mL and 90.1 ng/mL vs 61.7 ng/mL), whereas in a similar analysis of the PALOMA-3 study no difference was found [25, 26]. In another study (n = 25), AUC0–∞ and Cmax were 30% and 35% higher, respectively, in Japanese subjects [27]. No dose adjustments are recommended based on ethnicity [9].
2.6 Food Effect
Food-effect studies of capsule and tablet formulations of palbociclib are summarized in Table 3. In a previous pooled analysis, it has been demonstrated that palbociclib exposure is substantially lower in a subset of patients (i.e., 13%), possibly due to a decreased absorption caused by an elevated stomach pH. This subgroup is classified as low-liers, defined as Cmax < 21.4 ng/mL [9]. In the food-effect study, when the patients who met the low-lier criteria were excluded, 90% CIs were within the bioequivalence margins, implying no food effect in patients with adequate absorption [28].
Concomitant intake with food resulted in a reduced inter-individual variability because the small subset of low-liers now leveled up to the exposure of the rest of the population, supporting the recommended ingestion of palbociclib capsules together with a meal [28]. While palbociclib capsules need to be administered with food, the recently approved tablet formulation can be taken with or without food, offering more flexibility to patients. Palbociclib exposure was not significantly altered as a result of food intake using the tablet formulation, showing it to be more robust to pH differences [11].
2.7 Drug–Drug Interactions
In Table 4, results of drug–drug interaction studies and recommendations for dose adjustments are shown. Overall, no (clinically relevant) interactions with fulvestrant, goserelin, or aromatase inhibitors were found. In contrast, palbociclib exposure was significantly altered by strong CYP3A4 modulators.
No clinical studies have been executed for moderate CYP3A4 inhibitors, but simulations predicted that they would increase palbociclib Cmax and AUC0–∞ by approximately 23% and 40%, respectively [29]. According to the label, no dose reduction is warranted although these results suggest that a dose reduction from 125 mg (standard dose) to 100 mg QD might be advised. To further substantiate this finding, we are currently performing a clinical study to investigate the effect of the moderate CYP3A4 inhibitor erythromycin on the pharmacokinetics of palbociclib [30]. In addition, relevant interactions between palbociclib and CYP3A4 substrates with a narrow therapeutic index could occur, as palbociclib can weakly inhibit CYP3A4 [31].
As the solubility of palbociclib is pH dependent, it could be expected that acid-reducing agents would decrease its exposure. Although palbociclib exposure was substantially reduced when administered concomitantly with rabeprazole under fasted conditions, this effect was only modest under fed conditions (Table 4) [32]. Therefore, no dose adjustments are indicated when palbociclib capsules are co-administered with acid-reducing agents, as they have to be administered under fed conditions. Exposure of palbociclib tablets was not affected by acid-reducing agents [11].
2.8 Pharmacokinetic–Pharmacodynamic Relationships
2.8.1 Exposure Response
Initial exposure–response analyses based on data of the PALOMA-1 study were inconclusive because of limited data (n = 81). Although a trend for prolonged PFS was observed in patients with an average palbociclib concentration (Cavg) above the median of 60 ng/mL (median PFS estimated from Kaplan–Meier curves were 17 months vs 24.5 months, p value not reported), multi-variable analyses yielded inconsistent results [9].
In the PALOMA-3 study, PFS was similar in patients with Cavg above and below the median of 78 ng/mL. It has to be noted, though, that exposure in this trial appeared to be higher than in PALOMA-1 (at the same dose, but with fulvestrant instead of aromatase inhibitors). Even in the group with low exposure, median Cavg was 63 ng/mL, which is higher than the cut-off value used in the PALOMA-1 study. Time-varying Cavg as a continuous variable was a significant predictor of PFS in a univariable analysis, although this did not remain significant in a multi-variable analysis [27, 33]. In the PALOMA-2 study, no exposure–response relationship has been identified [34, 35].
As exposure–response analyses have thus far not resulted in a clear answer and optimal data to perform them were not available, this needs to be further elucidated. Lower thresholds of Cmin may be related to efficacy. Preferably, these additional analyses should include palbociclib plasma concentrations measured at regular intervals throughout treatment and use median Cmin as a measure of exposure. Previously, it has been suggested that individual concentrations could be compared to the mean Cmin of 61 ng/mL of the PALOMA-1 study [36].
2.8.2 Exposure Toxicity
In phase I studies, higher area under the plasma–concentration time curve values were related to a greater reduction in absolute neutrophil count and platelet levels, with a wide range in EC50 values (estimated plasma exposure resulting in a 50% decrease from baseline) varying from 253 to 716 ng/mL*h for neutropenia and from 184 to 1370 ng/mL*h for thrombocytopenia [5, 6]. A semi-mechanistic pharmacokinetic-pharmacodynamic model has been developed to quantify the relationship between palbociclib exposure (i.e., plasma concentration) and neutropenia [37]. In this model, the maximum anti-proliferative effect on neutrophil precursor cells (Emax) was notably lower than for cytotoxic drugs (e.g., docetaxel and etoposide), and the reported EC50 value was 40.1 ng/mL.
Interestingly, patients with grade 3 or 4 neutropenia had a significantly longer PFS compared with patients with lower grade or no neutropenia (p = 0.0046). Multi-variable analysis resulted in a HR of 0.502 (95% CI 0.26–0.98, p = 0.046). This could be explained by either the hypothesis that tumor cells in patients with neutropenia are more sensitive to palbociclib or an underlying higher exposure in these patients [9]. As higher drug exposure causes more neutropenia and more neutropenia is related to a prolonged PFS, this suggests that an exposure–response relationship exists.
2.9 Population-Pharmacokinetic Models
In a population-pharmacokinetic model, palbociclib pharmacokinetics was best described by a two-compartment model with first-order absorption [9]. Two additional models have been developed to predict drug–drug interactions with CYP3A4 inhibitors and to quantify the exposure–response relationship for neutropenia [29, 37].
3 Ribociclib
In 2017, ribociclib has been approved by the FDA and EMA based on the results of a preplanned interim analysis of the MONALEESA-2 study [38]. In the second interim analysis of this randomized, placebo-controlled, phase III trial (n = 668) comparing first-line treatment with letrozole with or without ribociclib, mPFS was significantly longer in the ribociclib group compared with the control group (25.3 vs 16 months, HR 0.57 [95% CI 0.46–0.70], p < 0.001) [39]. In 2018, the indication was extended to combination treatment with fulvestrant, based on the MONALEESA-3 study. This study revealed that the addition of ribociclib to the treatment of fulvestrant improved mPFS from 12.8 to 20.5 months (HR 0.60 [95% CI 0.48– 0.73], p < 0.001) and resulted in a prolonged median overall survival (40.0 months vs not reached yet, HR 0.72 [95% CI 0.57–0.92], p = 0.005) [40, 41].
The approved dose of ribociclib is 600 mg QD in a 3-weeks-on/1-week-off dosing schedule. In the phase I, dose escalation study, doses from 50 to 1200 mg QD were explored [42]. The MTD was established as 900 mg QD, with neutropenia and thrombocytopenia being the most common dose-limiting toxicities [42]. The lower dose of 600 mg QD was selected for further development, as this resulted in a lower rate of corrected QT (QTc) prolongation, and clinical activity was already observed at this dose level [42].
3.1 Physiochemical Properties and Formulation
Ribociclib is a 7-cyclopentyl-N,N-dimethyl-2-{[5-(piperazin-1-yl)pyridine-2-yl]amino}-7H-pyrrolo[2,3-d]pyrimidine produced by chemical synthesis. It is formulated as a succinate salt in film-coated tablets containing 200 mg of ribociclib free base. Although initially a capsule formulation was used in clinical trials, the equivalence of both dosage forms was demonstrated in a bioequivalence study [43]. Ribociclib is a weak base with two pKa values of 5.5 and 8.6, with its succinate salt exhibiting high solubility at pH ≤ 4.5 (solubility > 2.4 mg/mL), but it decreases at higher pH. Thus, ribociclib succinate was classified as a low-solubility compound [43, 44]. The ribociclib Log P was reported to be 1.95, and its estimated effective human permeability was 0.9 × 10−4 cm/s. Based on these data, it was categorized as Biopharmaceutics Classification System class IV (low solubility, low permeability) [43, 45]. In vitro, IC50 values for ribociclib were 8 and 39 nM for CDK4 and CDK6, respectively, corresponding to plasma concentrations of 11.6–56.5 ng/mL when corrected for protein binding [46].
3.2 Drug Transporters
In vitro and in vivo studies have demonstrated that ribociclib is a transport substrate of P-gp [47, 48]. Interestingly, pharmacokinetic and tissue distribution studies in mouse models showed that this efflux transporter is responsible for limiting the ribociclib penetration into the brain, as the brain-to-plasma concentration ratio increased by at least 23-fold when the P-gp was knocked out or inhibited. Plasma pharmacokinetic parameters were not significantly affected, except for area under the plasma–concentration time curve from 0 to 24 h, which increased 2.3-fold in mice lacking P-gp and BCRP. This increase is likely due to P-gp activity, as ribociclib has not shown noticeable transport by BCRP [47].
Besides interacting as a substrate, ribociclib also inhibited P-gp [48]. Moreover, at clinically relevant concentrations it also inhibited BCRP, OCT2, multidrug and toxin extrusion protein (MATE) 1, and bile salt export pump [15, 46, 48, 49]. In a retrospective study in patients treated with ribociclib, creatinine levels increased 37% compared with baseline, probably due to OCT2 inhibition [50].
3.3 Clinical Pharmacokinetics
Selected steady-state pharmacokinetic parameters of ribociclib are displayed in Table 2. Exposure of ribociclib increased over-proportionally with dose in the range of 50–1200 mg, possibly caused by a lower clearance at higher doses [51].
The absolute oral bioavailability has not been determined, but using a physiologically based pharmacokinetic model it was predicted that 90% of the standard dose of ribociclib (600 mg) would be absorbed mainly in the small intestine [44]. Ribociclib has a moderate human protein binding (± 70%), and is equally distributed in plasma and red blood cells. The apparent volume of distribution was estimated at 1090 L, using a population pharmacokinetic analysis [46, 49].
Ribociclib is metabolized primarily by CYP3A4 with the formation of the active metabolite M4 (Fig. 2). It is also metabolized to a minor extent by flavin-containing monooxygenase 3 and flavin-containing monooxygenase 1, the last being involved in the formation of the metabolite M13. These two metabolites may be reactive by forming covalent adducts in hepatocytes. M4, M13, and M1 (a secondary glucuronide of ribociclib) were the major circulating metabolites, accounting for, respectively, 8.6%, 9.4%, and 7.8% of total radioactivity in a mass balance study. Considering these data, the contribution of the active metabolite M4 to the clinical activity was considered negligible [46, 49, 52]. Feces was the major route of excretion compared to urine, accounting for, respectively, 69.1% and 22.6% of the dose recovered, where ribociclib was the major entity found in excreta [46, 49].
3.4 Pharmacokinetics in Special Populations
3.4.1 Pediatric Patients with Cancer
Ribociclib was the first CDK4/6 inhibitor studied in pediatric patients in a phase I clinical trial, where patients with neuroblastoma, rhabdoid tumors, or solid tumors with alterations in the cyclin D-CDK4/6-INK4-Rb pathway were included. The MTD and recommended phase II dose were 470 and 350 mg/m2, respectively. The recommended phase II dose was selected based on overall safety and pharmacokinetic considerations, as the exposure at 350 mg/m2 was equivalent to that observed at 600 mg in adults. Pharmacokinetic parameters, including time to Cmax, Cmax, AUC0–τ, and terminal elimination half-life, were similar in adult and pediatric patients [53].
3.4.2 Patients with Renal Impairment
The effect of renal impairment on the pharmacokinetics of ribociclib was assessed in a population pharmacokinetic analysis, which included patients with normal renal function (n = 438), mild renal impairment (n = 488), and moderate renal impairment (n = 113). In this analysis, mild and moderate renal impairment had no effect on the exposure and clearance of ribociclib; therefore, no dose adjustments are recommended for patients with mild or moderate renal impairment [46, 49, 52].
Furthermore, a clinical trial showed that for patients with severe renal impairment and end-stage renal disease (eGFR < 15 mL/min/1.73 m2), AUC0–∞ increased 281% and 137%, and Cmax 168% and 110%, respectively, compared with subjects with normal renal function [54]. Based on these results, a starting dose of 200 mg daily is recommended by the FDA for patients with severe renal impairment, while the EMA recommends a starting dose of 400 mg in these cases [52, 55].
3.4.3 Patients with Hepatic Impairment
In a clinical study (n = 28), mild hepatic impairment had no effect on ribociclib exposure. In contrast, for moderate hepatic impairment, AUC0–∞ and Cmax increased 28% and 44%, respectively, while for severe hepatic impairment they increased 29% and 32%. A population pharmacokinetic analysis (n = 160 with normal hepatic function and n = 47 with mild hepatic impairment) further supported that ribociclib exposure was unaffected by mild hepatic impairment. Based on these results, a reduction of the starting dose to 400 mg is recommended for patients with moderate or severe hepatic impairment [46, 49].
3.5 Other Factors Influencing Ribociclib Pharmacokinetics
The effect of other intrinsic factors on ribociclib pharmacokinetics was evaluated by population pharmacokinetic analyses (n = 208). Body weight and age were statistically significant covariates for ribociclib clearance. Based on simulations, it was predicted that a change of body weight from the reference value of 70 kg to 50 or 100 kg would change steady-state Cmax, Cmin, and area under the plasma–concentration time curve from 0 to 24 h of ribociclib up to 22%, which was considered a small effect relative to the inherent pharmacokinetic variability. Age was predicted to have only a mild effect on exposure. Race and sex were statistically insignificant parameters [46].
Furthermore, a cross-study comparison exhibited that, on average, the exposure of ribociclib in Japanese patients was higher than in Caucasian patients, but the individual values were within the same range. In summary, the effects of body weight, age, sex, and race on ribociclib pharmacokinetics were considered not clinically relevant, and therefore, no dose adjustment is required [46, 49].
3.6 Food Effect
Table 3 summarizes the food-effect studies that have been performed for the capsule and tablet formulation, of which the latter is more relevant because this is the marketed formulation. As the geometric mean ratios were ≈ 1 for AUC0–∞ and Cmax, no effect of food intake was observed on the pharmacokinetics of ribociclib [44].
Additionally, the in vitro solubility of ribociclib was evaluated in biorelevant media, including simulated fed (pH 5.0) and fasted (pH 6.5) intestinal fluid, where the maximum dose (600 mg) was dissolved in 250 mL. This suggests that ribociclib absorption is unlikely to be affected by changes in the gastric pH due to food intake, among others. Physiologically based pharmacokinetic models also predicted that the exposure of ribociclib was independent of the gastric pH in the range of 1.0–8.0 [44]. Altogether, this information supports that ribociclib can be administered either with or without food [46, 49, 52].
3.7 Drug–Drug Interactions
An overview of all drug–drug interaction studies is provided in Table 4. Ribociclib had no (clinically relevant) interactions with fulvestrant and the aromatase inhibitors [46, 49, 52, 55]. Ribociclib is extensively metabolized via CYP3A4; therefore, its pharmacokinetics is strongly affected by strong inhibitors or inducers of this enzyme. Ribociclib can reversibly inhibit CYP3A4 and CYP1A2 [48]. Altogether, it is recommended that drugs with a narrow therapeutic index that are sensitive substrates of these drug-metabolizing enzymes or transporters that are inhibited by ribociclib (Sect. 3.2) should be cautiously monitored in concomitant treatments with ribociclib [46, 49].
Because ribociclib shows a pH-dependent solubility, drugs that alter the gastric pH could be expected to affect its exposure. However, ribociclib exposure was similar in patients with and without a proton pump inhibitor, and these drugs could thus be administered concomitantly [44].
3.8 Pharmacokinetic-Pharmacodynamic Relationships
3.8.1 Exposure Response
Because of very limited data, exposure–response analyses for ribociclib remain inconclusive. In the MONALEESA-2 study, only 44 out of 334 patients had progressive disease and available pharmacokinetic data. No indication for an exposure–efficacy relationship was found. Data on confirmed best response were available for 72 patients with pharmacokinetic data, and showed similar ribociclib exposure in responders vs non-responders. No exposure–response analyses have been performed for the MONALEESA-3 study [46, 56]. Future studies should establish exposure–efficacy relationships and identify an optimal threshold concentration.
3.8.2 Exposure Toxicity
Although higher Cmin levels were related to a greater decrease in absolute neutrophil count and platelet count in the phase I study, ribociclib is dosed at the flat ends of these plateauing curves [42]. Pooled data from four clinical studies (n = 196) were used to develop a logistic regression model for ≥ grade 2 neutropenia. Although a trend was found for an increased risk of neutropenia at higher ribociclib exposure, this was not statistically significant. For each 100-ng/mL increase in Cmin, the odds ratio for ≥ grade 2 neutropenia was 1.05 (95% CI 0.99–1.11, p = 0.087) [46]. In addition, a pharmacokinetic-pharmacodynamic model for neutropenia using data of 1052 patients from six clinical trials showed that the relationship between exposure and neutropenia was not influenced by age, race, or the use of anastrozole, letrozole, tamoxifen, or fulvestrant [56].
Furthermore, a relationship between ribociclib exposure and QTc prolongation has been established, which was described by a log-linear mixed-effect model. Mean QTc prolongation was 22.87 ms at the mean steady-state Cmax of 2237 ng/mL. No exposure–toxicity relationship could be demonstrated for hepatotoxicity because of the limited number of grade 3 or 4 events [46].
3.9 Population Pharmacokinetic Models
A population pharmacokinetic model has been developed based on pooled data of 208 patients of whom 4731 pharmacokinetic samples were available. The model was validated using a dataset consisting of 175 pharmacokinetic samples from 93 patients in the MONALEESA-2 study. A two-compartment model with delayed zero-order absorption and linear clearance best fitted the data, with dose and body weight being significant covariates on clearance. Clearance decreased with increasing dose, which is in line with the observed more than dose-proportional increase in exposure [46].
4 Abemaciclib
Abemaciclib was the third CDK4/6 inhibitor approved by the FDA and EMA in 2018. In the MONARCH-3 study, abemaciclib increased mPFS compared with placebo (14.7 months vs not reached, HR 0.54 [95% CI 0.41–0.72], p < 0.001) in the first-line setting combined with anastrozole or letrozole [57]. In the same manner, the MONARCH-2 study demonstrated that abemaciclib was superior to placebo in the second-line setting in combination with fulvestrant [58].
In contrast to palbociclib and ribociclib, abemaciclib is dosed twice daily (BID) and in a continuous dosing schedule. In the dose-escalation part of the phase I study, doses up to 275 mg BID have been evaluated with 200 mg BID being identified as MTD [59]. This is the recommended dose for abemaciclib monotherapy, whereas 150 mg BID is the recommended dose for combination therapy (i.e., with aromatase inhibitors or fulvestrant) because of better tolerability. Although fatigue was the most common dose-limiting toxicity, gastrointestinal and hematologic toxicities were also frequently observed [59].
4.1 Physiochemical Properties and Formulation
Abemaciclib is a synthetic N-(5-((4-ethylpiperazin-1-yl)methyl)pyrididin-2-yl)-5-fluoro-4-(4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole-6-yl)pyrimidin-2 amine. It is formulated in tablets containing 50, 100, 150, or 200 mg of the free base. As capsules were used in the pivotal MONARCH-1 and MONARCH-2 trials, bioequivalence between both formulations was tested and confirmed. Abemaciclib is a tribasic compound with pKa values of 3.80, 4.48, and 7.95, and a log P of 3.36. It is soluble over the pH range of 1.0–6.8 (solubility > 0.8 mg/mL), and classified as a highly soluble drug. Considering that abemaciclib showed a moderate permeability (predicted effective human permeability = 2.46 × 10−4 cm/s), it was classified as Biopharmaceutics Classification System class 3 (high-solubility, low-permeability) [60, 61].
Abemaciclib is a potent, ATP-competitive, reversible inhibitor of CDK4 and CDK6, with IC50 values of 2 and 10 nM, respectively, corresponding to plasma concentrations of 27.4–136.9 ng/mL after correcting for protein binding [62]. Abemaciclib has three active metabolites with similar potency: N-desethylabemaciclib (M2), hydroxyabemaciclib (M20), and hydroxy-N-desethylabemaciclib (M18) (Fig. 2). Their IC50 values (nM) for CDK4 and CDK6 are 1.2 and 1.3 for M2, 1.5 and 1.9 for M20, and 1.5 and 2.7 for M18 [63, 64].
4.2 Drug Transporters
Abemaciclib is a substrate of efflux transporters P-gp and BCRP. In vivo studies showed that abemaciclib penetration through the blood–brain barrier improved in P-gp-deficient mice [14]. The abemaciclib metabolite M2 is also a substrate of P-gp and BCRP, and its exposure increased significantly around 5.3-fold in P-gp/BCRP-deficient mice with respect to the wild type. Furthermore, in this mouse model, the brain penetration of both abemaciclib and M2 increased 25- and 4-fold, respectively, compared with the wild type [65]. Additionally, abemaciclib itself inhibits P-gp and BCRP [22, 66].
The renal transporters OCT2, MATE1, and MATE2-K are reversibly inhibited by abemaciclib and its active metabolites M2 and M20 at clinically relevant concentrations [67, 68]. In vitro studies have shown that OCT2, MATE1, and MATE-K metformin transport is inhibited in the presence of abemaciclib, M2, or M20. The clinical implications of this interaction were also determined (Table 4) [68]. This reversible inhibition of renal transporters has been related to elevated creatinine levels, without renal function being affected [68].
4.3 Clinical Pharmacokinetics
Abemaciclib pharmacokinetics is summarized in Table 2, and is characterized by high variability. It showed a relatively modest absolute oral bioavailability of 45% [59, 69, 70]. Abemaciclib and its active metabolites showed a high protein binding of 96.3% for abemaciclib, 93.4% for M2, 96.8% for M18, and 97.8% for M20. The mean volume of distribution is 750 L [67, 71]. Abemaciclib is cleared mainly by hepatic metabolism, primarily by CYP3A4 with the formation of M2, M20, and M18 (Fig. 2). The area under the plasma–concentration time curve of these metabolites represent 25%, 26%, and 13%, respectively, of the total circulating entities in plasma [67]. In a mass balance study, abemaciclib was excreted as metabolites mainly in feces, with 81% of the administered dose recovered in feces, and ≈ 3% in urine [67, 71].
4.4 Pharmacokinetics in Special Populations
4.4.1 Pediatric Patients with Cancer
Information on abemaciclib pharmacokinetics in the pediatric population is not available hitherto [72]. Currently, two phase I studies are ongoing [73, 74].
4.4.2 Patients with Renal Impairment
No dedicated study has evaluated the effect of renal impairment on the pharmacokinetics of abemaciclib. However, a population pharmacokinetic analysis, including patients with normal renal function (n = 483), mild renal impairment (n = 381), and moderate renal impairment (n = 126), showed no significant differences in abemaciclib exposure. Therefore, no dose adjustment is required in patients with mild or moderate renal impairment. This was expected because the renal clearance of abemaciclib and its active metabolites is minor. The effect of severe renal impairment has not been determined yet [64, 67, 71, 75].
4.4.3 Patients with Hepatic Impairment
In a clinical trial, the total exposure of abemaciclib plus M2, M20, and M18 was similar in participants with mild and moderate hepatic impairment, showing an increase of 20% and 10%, respectively, compared with participants with normal hepatic function. In contrast, severe hepatic impairment resulted in a 140% increase in exposure of abemaciclib active entities. Furthermore, the mean plasma terminal elimination half-life of abemaciclib was prolonged (55 h vs 24 h in healthy subjects), absorption was slower (time to Cmax = 24 h vs 7 h in healthy subjects), and protein binding decreased. Consequently, it is recommended to reduce the dose frequency to QD administration for patients with severe hepatic impairment (i.e., Child–Pugh class C) [64, 67, 71, 75].
4.5 Other Factors Influencing Abemaciclib Pharmacokinetics
The influence of intrinsic factors on abemaciclib pharmacokinetics was evaluated in a population pharmacokinetic analysis (n = 994), in which sex, age, race, and body weight were found to be insignificant covariates for the abemaciclib exposure [71, 76]. As a result, no special dose adjustments are required.
4.6 Food Effect
An overview of food-effect studies for abemaciclib using a capsule or tablet formulation is provided in Table 3. The food-effect study with the tablet formulation is the most relevant as abemaciclib is marketed in this formulation. The exposure of abemaciclib increased with concomitant administration of a high-fat meal, but this was deemed not clinically relevant considering the high inter-subject variability and the fact that changes in exposure were within the abemaciclib therapeutic window [77, 78]. Therefore, abemaciclib can be administered with or without food.
4.7 Drug–Drug Interactions
Drug–drug interaction studies for abemaciclib are summarized in Table 4. The potential pharmacokinetic interaction between abemaciclib and fulvestrant or aromatase inhibitors was not formally evaluated. However, historical comparisons indicated that these drugs had no clinically relevant effect on the pharmacokinetics of abemaciclib, or vice versa [67, 71, 75].
Because of the extensive metabolism of abemaciclib via CYP3A4, the exposure of abemaciclib plus its active metabolites M2, M20, and M18 is substantially affected when co-administered with strong CYP3A4 modulators. Additionally, interactions with abemaciclib as a perpetrator could occur with substrates of transporters inhibited by abemaciclib (i.e., P-gp and renal transporters, Table 4).
4.8 Pharmacokinetic–Pharmacodynamic Relationships
4.8.1 Exposure Response
In a preclinical pharmacokinetic-pharmacodynamic model of xenograft tumors, Cmin ≥ 200 ng/mL has been identified as a potential efficacy threshold. Simulations with this model indicated that a maximum decrease in phosphorylated Rb levels was attained at a dose of 50 mg/kg, corresponding to a Cmin of 200 ng/mL. A limitation of this study is that concentrations of the active metabolites M2 and M20 were not taken into account [79].
In all three MONARCH studies, exposure–response relationships were demonstrated. Although abemaciclib in the MONARCH-1 study (n = 132) could not be linked to the objective response rate and PFS, simulations with a pharmacokinetic-pharmacodynamic model found a positive relationship between exposure and tumor shrinkage. Additionally, these simulations suggested that the objective response rate would be higher at an abemaciclib dose of 200 mg BID compared with 150 mg BID (31% vs 25%, respectively) [64]. Using a similar approach, higher abemaciclib exposure was related to an increased tumor shrinkage in the MONARCH-2 study (n = 477) as well, with the effect being most pronounced in the first months after start of treatment [64]. Finally, in the MONARCH-3 study (n = 393), an exposure–response relationship was not only established for tumor size reduction, but also for PFS [63].
In summary, abemaciclib exposure was related to efficacy in several clinical trials. Therefore, it has been suggested that from an efficacy point of view, 200 mg BID would be a better starting dose than 150 mg BID. However, this higher starting dose is not deemed feasible, as 50% of patients need a dose reduction because of toxicity. Based on the available data, no specific target for TDM can be proposed yet, but the optimal target might be somewhere between 169 and 197 ng/mL (i.e., the median exposure at 150 mg and 200 mg, respectively). Future exposure–response analyses need to identify the optimal threshold for efficacy, which could be performed using data from the MONARCH studies or from a real-world patient cohort. Preferably, it should also be investigated whether abemaciclib has additional value to include the concentrations of the active metabolites in this threshold, or that abemaciclib concentrations alone could serve as a proxy.
4.8.2 Exposure Toxicity
Higher abemaciclib concentrations were related to an increased incidence and severity of neutropenia. Dynamic pharmacokinetic-pharmacodynamic models for neutrophil counts have been developed using data of the MONARCH-2 (n = 593) and MONARCH-3 (n = 477) studies. In these models, higher total Cmax of abemaciclib and its active metabolites was related to a greater decrease in neutrophil production rate, and thus an increased risk of neutropenia [63, 64].
4.9 Population Pharmacokinetic Models
In a population pharmacokinetic model based on data obtained from the phase I study (n = 224), abemaciclib pharmacokinetics was best described by a linear one-compartment model with time- and dose-dependent bioavailability. Relative bioavailability decreased with an increasing dose, being 10% lower at 200 mg compared with 150 mg. This could be attributed to a saturable absorption, which is in line with preclinical data [79]. Plasma exposure of abemaciclib also decreased over time with steady-state concentrations being attained after 70 days [76, 79].
5 Discussion
By providing an overview of the clinical pharmacokinetics and pharmacodynamics of the three licensed CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib, it becomes apparent that they share several characteristics. Similarities include the high inter-individual variability in exposure, the predominant metabolism by CYP3A4, the brain penetration being limited by efflux transporters, and the exposure–toxicity relationship for neutropenia. However, there are also substantial differences. First, abemaciclib has a divergent dosing schedule, as it is dosed BD and continuously, instead of QD and intermittently for palbociclib and ribociclib. Second, dose proportionality of pharmacokinetics varies between compounds. Palbociclib exposure increases linearly with an increasing dose, whereas ribociclib exhibits a more than dose-proportional dose–exposure relationship, and abemaciclib exposure, in contrast, increases less than proportionally with an increasing dose, owing to the lower fraction absorbed at higher doses. Third, abemaciclib has active and significantly abundant metabolites that should be taken into account when assessing its exposure (i.e., M2, M20, and M18), while this is not the case for palbociclib and ribociclib. Fourth, a clear exposure–efficacy relationship has been described for abemaciclib, while for palbociclib and ribociclib, exposure–response analyses remain inconclusive. This might be explained by the applied methodologies and sample sizes that were used in these exposure–response analyses. It is important to further elucidate the exposure–response relationship for all three CDK4/6 inhibitors. Finally, ribociclib frequently prolongs the QTc interval in an exposure-related manner, whereas this, to our current knowledge, has not been reported for palbociclib and abemaciclib. These particular characteristics may support the selection of the most appropriate CDK4/6 inhibitor for individual patients.
Interestingly, the incidence of neutropenia is much lower for abemaciclib than for palbociclib and ribociclib. This is possibly caused by the greater selectivity of abemaciclib for CDK4 compared with CDK6, its BID dosing schedule, or the conversion to metabolites with less hematologic toxicity [59, 62]. In general, the effect of CDK4/6 inhibitors on neutrophil progenitor cells is cytostatic rather than cytotoxic, and associated with a notably low incidence of febrile neutropenia, in contrast to chemotherapy [37].
Many patients require dose reductions because of neutropenia, which can remain problematic even at the lowest doses according to the label (i.e., 75 mg QD for palbociclib, 200 mg QD for ribociclib, and 50 mg BID for abemaciclib). If exposure in these patients is low, switching to an alternative treatment might be preferred, whereas in patients with adequate exposure prolonging the dose interval to every other day for palbociclib and ribociclib, or QD for abemaciclib, could be an option, as has previously been described for pazopanib [80]. Alternatively, the time off treatment could be prolonged (i.e., 2-weeks-on/2-weeks-off treatment, as was allowed in the PALOMA-3 study). From a pharmacological point of view, though, prolonging the dose interval would be more rational.
Although it is known that CDK4/6 inhibitors combined with endocrine therapy provide an effective treatment strategy, it is currently unclear whether CDK4/6 inhibitors can best be added to first- or second-line treatment. This paramount question is currently being addressed in the SONIA study, a nationwide study in The Netherlands that will randomize 1000 patients between first- and second-line treatment with a CDK4/6 inhibitor [81]. In an additional side study, pharmacokinetic samples are collected to further elucidate exposure–response relationships.
The currently approved CDK4/6 inhibitors are predominantly metabolized by CYP3A4. Therefore, increased exposure, and hence an increased risk of toxicity, can be expected in patients harboring mutations as in CYP3A4*22, as a result of lower levels of functional CYP3A4 and thus a decreased clearance. The reported prevalence of these mutations is up to 10% [82], and it could be argued that this subset of patients may benefit from a lower starting dose. This is currently being investigated in the STAR22 study [83]. Although CDK4/6 inhibitors are currently only approved for the treatment of breast cancer, they are in clinical development for many other indications.
6 Conclusions
Cyclin-dependent kinase 4 and 6 inhibitors are a new class of promising oral targeted therapies in oncology, with complex pharmacokinetic and pharmacodynamic profiles, which we summarized in this review. Future studies should focus on the further exploration of exposure–response relationships and the potential for pharmacokinetically guided dose individualization.
References
Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–69.
Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108.
Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–36.
Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209–19.
Flaherty KT, LoRusso PM, DeMichele A, Abramson VG, Courtney R, Randolph SS, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18:568–76.
Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011;104:1862–8.
Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–406.
Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Ther. 2016;15:2273–81.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review palbociclib. 2014. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207103Orig1s000ClinPharmR.pdf. Accessed 6 Aug 2020.
US Food and Drug Administration. Center for Drug Evaluation and Research. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system: guidance for industry. 2017. https://www.fda.gov/media/70963/download. Accessed 6 Aug 2020.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology review palbociclib. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212436Orig1s000ClinPharmR.pdf. Accessed 6 Aug 2020.
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade MA, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–37.
De Gooijer MC, Zhang P, Thota N, Mayayo-Peralta I, Buil LCM, Beijnen JH, et al. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Invest New Drugs. 2015;33:1012–9.
Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, et al. Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab Dispos. 2015;43:1360–71.
Pabla N, Gibson AA, Buege M, Ong SS, Li L, Hu S, et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci USA. 2015;112:5231–6.
Chu X, Bleasby K, Chan GH, Nunes I, Evers R. The complexities of interpreting reversible elevated serum creatinine levels in drug development: does a correlation with inhibition of renal transporters exist? Drug Metab Dispos. 2016;44:1498–509.
Janssen JM, Dorlo TPC, Steeghs N, Beijnen JH, Hanff LM, van Eijkelenburg NKA, et al. Pharmacokinetic targets for therapeutic drug monitoring of small molecule kinase inhibitors in pediatric oncology. Clin Pharmacol Ther. 2020. https://doi.org/10.1002/cpt.1808(Epub ahead of print).
ClinicalTrials.gov. Palbociclib isethionate in treating younger patients with recurrent, progressive, or refractory central nervous system tumors. https://clinicaltrials.gov/ct2/show/NCT02255461. Accessed 6 Aug 2020.
ClinicalTrials.gov. Palbociclib and sorafenib, decitabine, or dexamethasone in treating patients with recurrent or refractory leukemia. Available from: https://clinicaltrials.gov/ct2/show/NCT03132454. Accessed 6 Aug 2020.
ClinicalTrials.gov. Study of palbociclib combined with chemotherapy in pediatric patients with recurrent/refractory solid tumors. https://clinicaltrials.gov/ct2/show/NCT03709680. Accessed 6 Aug 2020.
ClinicalTrials.gov. Palbociclib in combination with chemotherapy in treating children with relapsed acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LL). https://clinicaltrials.gov/ct2/show/NCT03792256. Accessed 6 Aug 2020.
ClinicalTrials.gov. Palbociclib in treating patients with relapsed or refractory Rb positive advanced solid tumors, non-Hodgkin lymphoma, or histiocytic disorders with activating alterations in cell cycle genes (a pediatric MATCH treatment trial). https://clinicaltrials.gov/ct2/show/NCT03526250. Accessed 6 Aug 2020.
US Food and Drug Administration. Center for Drug Evaluation and Research. Highlights of prescribing information palbociclib. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207103s007lbl.pdf. Accessed 6 Aug 2020.
Palmieri C, Macpherson I. Use of the Child-Pugh score in anticancer drug dosing decision making: proceed with caution. Lancet Oncol. 2019;20:e289.
Mukai H, Shimizu C, Masuda N, Ohtani S, Ohno S, Takahashi M, et al. Palbociclib in combination with letrozole in patients with estrogen receptor-positive, human epidermal growth factor receptor 2–negative advanced breast cancer: PALOMA-2 subgroup analysis of Japanese patients. Int J Clin Oncol. 2019;24:274–87.
Masuda N, Inoue K, Nakamura R, Rai Y, Mukai H, Ohno S, et al. Palbociclib in combination with fulvestrant in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: PALOMA-3 subgroup analysis of Japanese patients. Int J Clin Oncol. 2019;24:262–73.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review palbociclib. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/207103Orig1s002.pdf. Accessed 6 Aug 2020.
Ruiz-Garcia A, Plotka A, O’Gorman M, Wang DD. Effect of food on the bioavailability of palbociclib. Cancer Chemother Pharmacol. 2017;79:1–7.
Yu Y, Loi C-M, Hoffman J, Wang D. Physiologically based pharmacokinetic modeling of palbociclib. J Clin Pharmacol. 2017;57:173–84.
Netherlands Trial Register. Effect of moderate CYP3A4 inhibitor erythromycin on the pharmacokinetics of palbociclib. https://www.trialregister.nl/trial/7549. Accessed 6 Aug 2020.
Hoffman JT, Plotka A, O’Gorman M, Loi C-M, Kirkovsky L, Gallo-Stampino C, et al. A phase 1 randomized, open-label, 2-sequence, 2-period crossover study of the effect of multiple doses of palbociclib (PD-0332991) on midazolam pharmacokinetics in healthy women of non-childbearing potential [abstract]. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; 5–9 April, 2014; San Diego (CA). Philadelphia (PA): AACR;2014. Cancer Res. 2014;74(19 Suppl.):abstract no. CT419.
Sun W, Klamerus KJ, Yuhas LM, Pawlak S, Plotka A, O’Gorman M, et al. Impact of acid-reducing agents on the pharmacokinetics of palbociclib, a weak base with pH-dependent solubility, with different food intake conditions. Clin Pharmacol Drug Dev. 2017;6:614–26.
Sun W, Yu Y, Hoffman J, Turner NC, Cristofanilli M, Wang DD. Palbociclib exposure-response analyses in second-line treatment of hormone-receptor positive advanced breast cancer (ABC). J Clin Oncol. 2017;35:1053.
Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology review palbociclib. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/207103Orig1s004.pdf. Accessed 6 Aug 2020.
Zheng J, Yu Y, Durairaj C, Amantea M, Dieras V, Finn R, et al. Palbociclib exposure-response analyses in the treatment of hormone-receptor positive (HR +), human epidermal growth factor receptor 2 negative (HER2–) advanced breast cancer (ABC) [abstract]. Proceedings of the 2017 San Antonio Breast Cancer Symposium; 5-9 December 2017; San Antonio (TX). Cancer Res. 2018;78:abstract no. P5-21-21.
Verheijen RB, Yu H, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin Pharmacol Ther. 2017;102:765–76.
Sun W, O’Dwyer PJ, Finn RS, Ruiz-Garcia A, Shapiro GI, Schwartz GK, et al. Characterization of neutropenia in advanced cancer patients following palbociclib treatment using a population pharmacokinetic-pharmacodynamic modeling and simulation approach. J Clin Pharmacol. 2017;57:1159–73.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738–48.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018;29:1541–7.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36:2465–72.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N Engl J Med. 2020;382:514–24.
Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, et al. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2016;22:5696–705.
US Food and Drug Administration. Center for Drug Evaluation and Research. Chemistry review ribociclib. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209092Orig1s000ChemR.pdf. Accessed 6 Aug 2020.
Samant TS, Dhuria S, Lu Y, Laisney M, Yang S, Grandeury A, et al. Ribociclib bioavailability is not affected by gastric pH changes or food intake: in silico and clinical evaluations. Clin Pharmacol Ther. 2018;104:374–83.
Mitra A, Parrott N, Miller N, Lloyd R, Tistaert C, Heimbach T, et al. Prediction of pH-dependent drug-drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharm Sci. 2020;109:1380–94.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology review ribociclib. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209092Orig1s000MultidisciplineR.pdf. Accessed 6 Aug 2020.
Martínez-Chávez A, Van Hoppe S, Rosing H, Lebre MC, Tibben M, Beijnen JH, et al. P-glycoprotein limits ribociclib brain exposure and CYP3A4 restricts its oral bioavailability. Mol Pharm. 2019;16:3842–52.
Sorf A, Hofman J, Kučera R, Staud F, Ceckova M. Ribociclib shows potential for pharmacokinetic drug-drug interactions being a substrate of ABCB1 and potent inhibitor of ABCB1, ABCG2 and CYP450 isoforms in vitro. Biochem Pharmacol. 2018;154:10–7.
Committee for Medicinal Products for Human Use. European Medicines Agency. European public assessment report ribociclib. 2017. https://www.ema.europa.eu/en/documents/assessment-report/kisqali-epar-public-assessment-report_en.pdf. Accessed 6 Aug 2020.
Wilson BE, Mok K, Kiely BE, Nguyen R, Moylan E. Association between ribociclib and changes in creatinine in patients with hormone receptor positive metastatic breast cancer. Intern Med J. 2019;49:1438–42.
Doi T, Hewes B, Kakizume T, Tajima T, Ishikawa N, Yamada Y. Phase I study of single-agent ribociclib in Japanese patients with advanced solid tumors. Cancer Sci. 2018;109:193–8.
US Food and Drug Administration. Center for Drug Evaluation and Research. Highlights of prescribing information ribociclib. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209092s003lbl.pdf. Accessed 6 Aug 2020.
Geoerger B, Bourdeaut F, DuBois SG, Fischer M, Geller JI, Gottardo NG, et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res. 2017;23:2433–41.
ClinicalTrials.gov. Evaluation of renal function impairment on the pharmacokinetics of LEE011. https://clinicaltrials.gov/ct2/show/NCT02431481. Accessed 6 Aug 2020.
Committee for Medicinal Products for Human Use. European Medicines Agency. Summary of product characteristics ribociclib. 2017. https://www.ema.europa.eu/en/documents/product-information/kisqali-epar-product-information_en.pdf. Accessed 6 Aug 2020.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology review ribociclib. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/209092Orig1s001.pdf. Accessed 6 Aug 2020.
Goetz MP, Toi M, Campone M, Trédan O, Bourayou N, Sohn J, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35:3638–46.
Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR +/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35:2875–84.
Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6:740–53.
US Food and Drug Administration. Center for Drug Evaluation and Research. Chemistry review abemaciclib. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208716Orig1s000ChemR.pdf. Accessed 6 Aug 2020.
Posada MM, Morse BL, Turner PK, Kulanthaivel P, Hall SD, Dickinson GL. Predicting clinical effects of CYP3A4 modulators on abemaciclib and active metabolites exposure using physiologically based pharmacokinetic modeling. J Clin Pharmacol. 2020;60(7):915–30.
Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32:825–37.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology review abemaciclib (in combination with aromatase inhibitors). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/208855Orig1s000MultidisciplineR.pdf. Accessed 6 Aug 2020.
US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical pharmacology review abemaciclib (in combination with fulvestrant). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208716Orig1s000MultidisciplineR.pdf. Accessed 6 Aug 2020.
Martínez-Chávez A, Tibben M, Lebre MC, Rosing H, Beijnen JH, Schinkel AH. The role of multidrug efflux transporters and CYP3A in the pharmacokinetics and tissue distribution of abemaciclib and its active metabolites [abstract no. 2258]. Proceedings of the 111th Annual Meeting of the American Association for Cancer Research; 2020 June 22-24. Philadelphia (PA): AACR; 2020.
Wu T, Chen Z, To KKW, Fang X, Wang F, Cheng B, et al. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem Pharmacol. 2017;124:29–42.
ClinicalTrials.gov. A Study of LY2835219 in Participants With Cancer. https://clinicaltrials.gov/ct2/show/results/NCT02117648.
Chappell JC, Turner PK, Pak YA, Bacon J, Chiang AY, Royalty J, et al. Abemaciclib inhibits renal tubular secretion without changing glomerular filtration rate. Clin Pharmacol Ther. 2019;105:1187–95.
Fujiwara Y, Tamura K, Kondo S, Tanabe Y, Iwasa S, Shimomura A, et al. Phase 1 study of abemaciclib, an inhibitor of CDK 4 and 6, as a single agent for Japanese patients with advanced cancer. Cancer Chemother Pharmacol. 2016;78:281–8.
Turner K, Chappell J, Kulanthaivel P, Ng WT, Royalty J. Food effect on the pharmacokinetics of 200-mg abemaciclib in healthy subject [abstract]. Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; 2016 Apr 16-20; New Orleans (LA). Philadelphia (PA): AACR;2016. Cancer Res. 2016;76(14 Suppl.):abstract no. CT152.
Committee for Medicinal Products for Human Use. European Medicines Agency. Summary of product characteristics abemaciclib. 2017. https://www.ema.europa.eu/en/documents/product-information/verzenios-epar-product-information_en.pdf. Accessed 6 Aug 2020.
Mills CC, Kolb EA, Sampson VB. Recent advances of cell-cycle inhibitor therapies for pediatric cancer. Cancer Res. 2017;77:6489–98.
ClinicalTrials.gov. Abemaciclib in children with DIPG or recurrent/refractory solid tumors (AflacST1501). https://clinicaltrials.gov/ct2/show/NCT02644460. Accessed 6 Aug 2020.
ClinicalTrials.gov. A study of abemaciclib (LY2835219) in combination with temozolomide and irinotecan and abemaciclib in combination with temozolomide in children and young adult participants with solid tumors. https://clinicaltrials.gov/ct2/show/NCT04238819. Accessed 6 Aug 2020.
Committee for Medicinal Products for Human Use. European Medicines Agency. Public assessment report abemaciclib. 2018. https://www.ema.europa.eu/en/documents/assessment-report/verzenios-epar-public-assessment-report_en.pdf. Accessed 6 Aug 2020.
Tate SC, Sykes AK, Kulanthaivel P, Chan EM, Turner PK, Cronier DM. A Population pharmacokinetic and pharmacodynamic analysis of abemaciclib in a phase I clinical trial in cancer patients. Clin Pharmacokinet. 2018;57:335–44.
Turner P, Chappell J, Aburub A, Ng W, Zhang W, Royalty J, et al. Abemaciclib tablet formulation is bioequivalent to capsules [abstract]. Proceedings of the 2017 San Antonio Breast Cancer Symposium; 5-9 December 2017; San Antonio (TX). Philadelphia (PA): AACR; 2018. Cancer Res. 2018;78(4 Suppl.):abstract no. P1-10-14.
ClinicalTrials.gov. A bioequivalence study comparing abemaciclib capsule and tablet formulations and effect of food on abemaciclib tablet pharmacokinetics in healthy subjects. https://clinicaltrials.gov/ct2/show/NCT02672423. Accessed 6 Aug 2020.
Tate SC, Cai S, Ajamie RT, Burke T, Beckmann RP, Chan EM, et al. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin Cancer Res. 2014;20:3763–74.
Groenland SL, Katz D, Huitema ADR, Steeghs N. Harnessing soft tissue sarcoma with low-dose pazopanib: a matter of blood levels. BMC Cancer. 2018;18:1–3.
Van Ommen-Nijhof A, Konings IR, Van Zeijl CJJ, Uyl-De Groot CA, Van Der Noort V, Jager A, et al. Selecting the optimal position of CDK4/6 inhibitors in hormone receptor-positive advanced breast cancer: the SONIA study: study protocol for a randomized controlled trial. BMC Cancer. 2018;18:1–7.
Zhou Y, Ingelman-Sundberg M, Lauschke VM. Worldwide distribution of cytochrome P450 alleles: a meta-analysis of population-scale sequencing projects. Clin Pharmacol Ther. 2017;102:688–700.
Netherlands Trial Register. CYP3A4*22 genotype-guided dosing of TKIs in cancer patients: a new way of personalized therapy. https://www.trialregister.nl/trial/7514. Accessed 6 Aug 2020.
Tamura K, Mukai H, Naito Y, Yonemori K, Kodaira M, Tanabe Y, et al. Phase I study of palbociclib, a cyclin-dependent kinase 4/6 inhibitor, in Japanese patients. Cancer Sci. 2016;107:755–63.
Curigliano G, Gómez Pardo P, Meric-Bernstam F, Conte P, Lolkema MP, Beck JT, et al. Ribociclib plus letrozole in early breast cancer: a presurgical, window-of-opportunity study. Breast. 2016;28:191–8.
Kim ES, Kelly K, Paz-Ares LG, Garrido P, Jalal S, Mahadevan D, et al. Abemaciclib in combination with single-agent options in patients with stage IV non–small cell lung cancer: a phase Ib study. Clin Cancer Res. 2018;24:5543–51.
ClinicalTrials.gov. A study of LY2835219 in healthy participants. https://www.clinicaltrials.gov/ct2/show/NCT02256267. Accessed 6 Aug 2020.
ClinicalTrials.gov. A study of abemaciclib (LY2835219) in healthy participants with and without food. https://clinicaltrials.gov/ct2/show/NCT02482935. Accessed 6 Aug 2020.
Hoffman JT, Loi C-M, O’Gorman M, Plotka A, Kosa M, Jakubowska A, et al. A phase I open-label, fixed-sequence, two-period crossover study of the effect of multiple doses of Itraconazole on Palbociclib (PD–0332991) pharmacokinetics in healthy volunteers [abstract]. Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; 16-20 April 2016; New Orleans (LA). Philadelphia (PA): AACR; 2016. Cancer Res. 2016;76 (14 Suppl.):abstract no. LB-196.
Hoffman JT, Plotka A, O’Gorman M, Chang A, Kosa M, Loi C-M, et al. A phase 1 randomized, open-label, fixed-sequence, 2-period study of the effect of multiple doses of rifampin on palbociclib (PD-0332991) pharmacokinetics in healthy volunteers [abstract]. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 18-22 Apr 2015; Philadelphia (PA). Philadelphia (PA): AACR; 2015. Cancer Res. 2015;75(15 Suppl.):abstract no. 4515.
Hoffman JT, Loi C-M, Plotka A, O’Gorman M, Shi H, Mori A, et al. A phase I open-label fixed-sequence two-period crossover study of the effect of multiple doses of modafinil on palbociclib (PD–0332991) pharmacokinetics in healthy volunteers [abstract]. Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; 16-20 April 2016; New Orleans (LA). Philadelphia (PA): AACR; 2016. Cancer Res. 2016;76 (14 Suppl.):abstract no. LB-198.
ClinicalTrials.gov. A study of abemaciclib in healthy participants (loperamide). https://clinicaltrials.gov/ct2/show/results/NCT02677844. Accessed 6 Aug 2020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding has been received for the preparation of this review.
Conflicts of interest
Jos H. Beijnen is a patent holder (in part), stock holder (indirectly), and a part-time employee of Modra Pharmaceuticals BV. Modra Pharmaceuticals BV is a small spin-off company clinically developing oral pharmaceutical formulations of taxanes; none of these positions are related to the current review. Stefanie L. Groenland, Alejandra Martínez-Chávez, Marloes G.J. van Dongen, Alfred H. Schinkel, Alwin D.R. Huitema, and Neeltje Steeghs have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
SG and AM reviewed the available literature and wrote the manuscript. MvD, JB, AS, AH, and NS critically reviewed the manuscript.
Rights and permissions
About this article

Cite this article
Groenland, S.L., Martínez-Chávez, A., van Dongen, M.G.J. et al. Clinical Pharmacokinetics and Pharmacodynamics of the Cyclin-Dependent Kinase 4 and 6 Inhibitors Palbociclib, Ribociclib, and Abemaciclib. Clin Pharmacokinet 59, 1501–1520 (2020). https://doi.org/10.1007/s40262-020-00930-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00930-x